
The Janus-Faced Role of Lipid Droplets in Aging: Insights from the Cellular Perspective
 ,
,  and
and 
Abstract
1. Introduction
2. Lipid Droplets in Saccharomyces cerevisiae
2.1. Replicative and Chronologic Lifespan
2.2. Lipid Droplets and Stress Adaptation
2.3. Lipid Droplets: Guardians of Mitochondrial Integrity
3. Lipid Droplets in Caenorhabditis elegans
3.1. The C. elegans “Dauer-Larva”
3.2. Detoxifying Role of Lipid Droplets
3.3. Lipid Droplets, Insulin Signaling, and Autophagy
3.4. Lipid Droplets and TGF-β Signaling
3.5. Significance of Lipid Droplet Accumulation to C. elegans Lifespan
4. Lipid Droplets in Drosophila melanogaster
4.1. Lipid Droplets and Drosophila Development
4.2. Control of the Lipid Droplet Pool in Drosophila Adipocytes
4.3. Lipid Droplets and Lifespan Extension in Drosophila
4.4. Lipid Droplets, Transsulfuration, and Cellular Antioxidant Defenses
4.5. Lipid Droplets and (Epi)Genetic Control
4.6. Intracellular Lipid Droplet Trafficking
5. LDs in Human Disease
5.1. Caloric Restriction, Lifespan Control, and Age-Related Disease
5.2. Bone Marrow Aging–Epigenetic Mechanisms
5.3. Lipid Droplets in Neurodegeneration
5.4. Lipid Droplets in Metabolic Disease
5.5. Lipid Droplets in Vascular Disease
5.6. LD Accumulation in Cardiomyocytes: Role of PPARs
5.7. Lipid Droplets and Cancer—A General Outline
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Murphy, D.J.; Vance, J. Mechanisms of lipid-body formation. Trends Biochem. Sci. 1999, 24, 109–115. [Google Scholar] [CrossRef]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Farese, R.V., Jr. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef]
- Geltinger, F.; Schartel, L.; Wiederstein, M.; Tevini, J.; Aigner, E.; Felder, T.K.; Rinnerthaler, M. Friend or foe: Lipid droplets as organelles for protein and lipid storage in cellular stress response, aging and disease. Molecules 2020, 25, 5053. [Google Scholar] [CrossRef] [PubMed]
- Plotz, T.; Hartmann, M.; Lenzen, S.; Elsner, M. The role of lipid droplet formation in the protection of unsaturated fatty acids against palmitic acid induced lipotoxicity to rat insulin-producing cells. Nutr. Metab. 2016, 13, 16. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Geltinger, F.; Tevini, J.; Briza, P.; Geiser, A.; Bischof, J.; Richter, K.; Felder, T.; Rinnerthaler, M. The transfer of specific mitochondrial lipids and proteins to lipid droplets contributes to proteostasis upon stress and aging in the eukaryotic model system Saccharomyces cerevisiae. Geroscience 2020, 42, 19–38. [Google Scholar] [CrossRef]
- Moldavski, O.; Amen, T.; Levin-Zaidman, S.; Eisenstein, M.; Rogachev, I.; Brandis, A.; Kaganovich, D.; Schuldiner, M. Lipid Droplets Are Essential for Efficient Clearance of Cytosolic Inclusion Bodies. Dev. Cell 2015, 33, 603–610. [Google Scholar] [CrossRef]
- Vevea, J.D.; Garcia, E.J.; Chan, R.B.; Zhou, B.; Schultz, M.; Di Paolo, G.; McCaffery, J.M.; Pon, L.A. Role for Lipid Droplet Biogenesis and Microlipophagy in Adaptation to Lipid Imbalance in Yeast. Dev. Cell 2015, 35, 584–599. [Google Scholar] [CrossRef]
- Garcia, E.J.; Liao, P.C.; Tan, G.; Vevea, J.D.; Sing, C.N.; Tsang, C.A.; McCaffery, J.M.; Boldogh, I.R.; Pon, L.A. Membrane dynamics and protein targets of lipid droplet microautophagy during ER stress-induced proteostasis in the budding yeast, Saccharomyces cerevisiae. Autophagy 2021, 17, 2363–2383. [Google Scholar] [CrossRef]
- Kumar, R.; Nawroth, P.P.; Tyedmers, J. Prion Aggregates Are Recruited to the Insoluble Protein Deposit (IPOD) via Myosin 2-Based Vesicular Transport. PLoS Genet. 2016, 12, e1006324. [Google Scholar] [CrossRef]
- Bersuker, K.; Peterson, C.W.H.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev. Cell 2018, 44, 97–112.e117. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Egan, J.J.; Wek, S.A.; Garty, N.B.; Blanchette-Mackie, E.J.; Londos, C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 1991, 266, 11341–11346. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Shin, E.S.; Park, P.J.; Shin, D.W.; Chang, H.K.; Kim, D.; Lee, H.H.; Lee, J.H.; Kim, S.H.; Song, M.J.; et al. Identification of mouse Prp19p as a lipid droplet-associated protein and its possible involvement in the biogenesis of lipid droplets. J. Biol. Chem. 2007, 282, 2456–2465. [Google Scholar] [CrossRef] [PubMed]
- Turro, S.; Ingelmo-Torres, M.; Estanyol, J.M.; Tebar, F.; Fernandez, M.A.; Albor, C.V.; Gaus, K.; Grewal, T.; Enrich, C.; Pol, A. Identification and characterization of associated with lipid droplet protein 1: A novel membrane-associated protein that resides on hepatic lipid droplets. Traffic 2006, 7, 1254–1269. [Google Scholar] [CrossRef]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef]
- Renne, M.F.; Hariri, H. Lipid Droplet-Organelle Contact Sites as Hubs for Fatty Acid Metabolism, Trafficking, and Metabolic Channeling. Front. Cell Dev. Biol. 2021, 9, 726261. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Thiel, K.; Thul, P.J.; Beller, M.; Kühnlein, R.P.; Welte, M.A. Lipid droplets control the maternal histone supply of Drosophila embryos. Curr. Biol. 2012, 22, 2104–2113. [Google Scholar] [CrossRef]
- Kovacs, M.; Geltinger, F.; Verwanger, T.; Weiss, R.; Richter, K.; Rinnerthaler, M. Lipid Droplets Protect Aging Mitochondria and Thus Promote Lifespan in Yeast Cells. Front. Cell Dev. Biol. 2021, 9, 774985. [Google Scholar] [CrossRef]
- Suriyalaksh, M.; Raimondi, C.; Mains, A.; Segonds-Pichon, A.; Mukhtar, S.; Murdoch, S.; Aldunate, R.; Krueger, F.; Guimera, R.; Andrews, S.; et al. Gene regulatory network inference in long-lived C. elegans reveals modular properties that are predictive of novel aging genes. Iscience 2022, 25, 103663. [Google Scholar] [CrossRef]
- Zhao, X.; Li, X.; Shi, X.; Karpac, J. Diet-MEF2 interactions shape lipid droplet diversification in muscle to influence Drosophila lifespan. Aging Cell 2020, 19, e13172. [Google Scholar] [CrossRef]
- Zimmermann, A.; Hofer, S.; Pendl, T.; Kainz, K.; Madeo, F.; Carmona-Gutierrez, D. Yeast as a tool to identify anti-aging compounds. Fems Yeast Res. 2018, 18, foy020. [Google Scholar] [CrossRef]
- Stefanini, I.; De Filippo, C.; Cavalieri, D. Yeast as a Model in High-Throughput Screening of Small-Molecule Libraries. In Diversity-Oriented Synthesis; Wiley Online Library: Hoboken, NJ, USA, 2013; pp. 455–482. [Google Scholar]
- Steinkraus, K.A.; Kaeberlein, M.; Kennedy, B.K. Replicative aging in yeast: The means to the end. Annu. Rev. Cell Dev. Biol. 2008, 24, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Rockenfeller, P.; Madeo, F. Apoptotic death of ageing yeast. Exp. Gerontol. 2008, 43, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, P.; Longo, V.D. The chronological life span of Saccharomyces cerevisiae. Methods Mol. Biol. 2007, 371, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, K.J.; Medvedik, O.; Sinclair, D.A. Longevity regulation in Saccharomyces cerevisiae: Linking metabolism, genome stability, and heterochromatin. Microbiol. Mol. Biol. Rev. 2003, 67, 376–399. [Google Scholar] [CrossRef]
- Pringle, J.R. Staining of bud scars and other cell wall chitin with calcofluor. Methods Enzymol. 1991, 194, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Klinger, H.; Rinnerthaler, M.; Lam, Y.T.; Laun, P.; Heeren, G.; Klocker, A.; Simon-Nobbe, B.; Dickinson, J.R.; Dawes, I.W.; Breitenbach, M. Quantitation of (a)symmetric inheritance of functional and of oxidatively damaged mitochondrial aconitase in the cell division of old yeast mother cells. Exp. Gerontol. 2010, 45, 533–542. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Sinclair, D.A. Studying the Replicative Life Span of Yeast Cells. In Biological Aging: Methods and Protocols, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 1048, pp. 49–63. [Google Scholar] [CrossRef]
- Lin, S.J.; Defossez, P.A.; Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128. [Google Scholar] [CrossRef]
- Goldberg, A.A.; Bourque, S.D.; Kyryakov, P.; Boukh-Viner, T.; Gregg, C.; Beach, A.; Burstein, M.T.; Machkalyan, G.; Richard, V.; Rampersad, S.; et al. A novel function of lipid droplets in regulating longevity. Biochem. Soc. Trans. 2009, 37, 1050–1055. [Google Scholar] [CrossRef]
- Hiltunen, J.K.; Mursula, A.M.; Rottensteiner, H.; Wierenga, R.K.; Kastaniotis, A.J.; Gurvitz, A. The biochemistry of peroxisomal beta-oxidation in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2003, 27, 35–64. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.; Salzmann, M.; Streubel, M.K.; Hasek, J.; Geltinger, F.; Duschl, J.; Bresgen, N.; Briza, P.; Haskova, D.; Lejskova, R.; et al. Clearing the outer mitochondrial membrane from harmful proteins via lipid droplets. Cell Death Discov. 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Beas, A.O.; Gordon, P.B.; Prentiss, C.L.; Olsen, C.P.; Kukurugya, M.A.; Bennett, B.D.; Parkhurst, S.M.; Gottschling, D.E. Independent regulation of age associated fat accumulation and longevity. Nat. Commun. 2020, 11, 2790. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Duran-Aniotz, C.; Cabral-Miranda, F.; Vivar, J.P.; Hetz, C. Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 2017, 16, 615–623. [Google Scholar] [CrossRef]
- Cui, H.-J.; Liu, X.-G.; McCormick, M.; Wasko, B.M.; Zhao, W.; He, X.; Yuan, Y.; Fang, B.-X.; Sun, X.-R.; Kennedy, B.K.; et al. PMT1 deficiency enhances basal UPR activity and extends replicative lifespan of Saccharomyces cerevisiae. Age 2015, 37, 46. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Kopito, R.R.; Sitia, R. Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep. 2000, 1, 225–231. [Google Scholar] [CrossRef]
- Celik, C.; Lee, S.Y.T.; Yap, W.S.; Thibault, G. Endoplasmic reticulum stress and lipids in health and diseases. Prog. Lipid Res. 2022, 89, 101198. [Google Scholar] [CrossRef]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hanelt, I.; Hummer, G.; Ernst, R. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol. Cell 2017, 67, 673–684.e678. [Google Scholar] [CrossRef]
- Rubio, C.; Pincus, D.; Korennykh, A.; Schuck, S.; El-Samad, H.; Walter, P. Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J. Cell Biol. 2011, 193, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, H.; Ren, L.; Lu, Z.; Zheng, Q.; Ding, L.; Xie, H.; Wang, R.; Yu, C.; Lin, Y.; et al. Adding fuel to the fire: The lipid droplet and its associated proteins in cancer progression. Int. J. Biol. Sci. 2022, 18, 6020–6034. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Weindling, E.; Rabinovich, E.; Nachman, I.; Fuchs, S.; Chuartzman, S.; Gal, L.; Schuldiner, M.; Bar-Nun, S. Water-Transfer Slows Aging in Saccharomyces cerevisiae. PLoS ONE 2016, 11, e0148650. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Binns, D.D.; Kinch, L.N.; Grishin, N.V.; Ortiz, N.; Chen, X.; Goodman, J.M. Pet10p is a yeast perilipin that stabilizes lipid droplets and promotes their assembly. J. Cell Biol. 2017, 216, 3199–3217. [Google Scholar] [CrossRef]
- Di Gregorio, S.E.; Duennwald, M.L. Yeast as a model to study protein misfolding in aged cells. FEMS Yeast Res. 2018, 18, foy054. [Google Scholar] [CrossRef]
- Currie, E.; Guo, X.; Christiano, R.; Chitraju, C.; Kory, N.; Harrison, K.; Haas, J.; Walther, T.C.; Farese, R.V. High confidence proteomic analysis of yeast LDs identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation. J. Lipid Res. 2014, 55, 1465–1477. [Google Scholar] [CrossRef]
- Grillitsch, K.; Connerth, M.; Köfeler, H.; Arrey, T.N.; Rietschel, B.; Wagner, B.; Karas, M.; Daum, G. Lipid particles/droplets of the yeast Saccharomyces cerevisiae revisited: Lipidome meets proteome. Biochim. Biophys. Acta 2011, 1811, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-W.; Lee, S.-C. The ubiquitin-like (UBX)-domain-containing protein Ubx2/Ubxd8 regulates lipid droplet homeostasis. J. Cell Sci. 2012, 125, 2930–2939. [Google Scholar] [CrossRef] [PubMed]
- Neuber, O.; Jarosch, E.; Volkwein, C.; Walter, J.; Sommer, T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Laun, P.; Büttner, S.; Rinnerthaler, M.; Burhans, W.C.; Breitenbach, M. Yeast Aging and Apoptosis. In Aging Research in Yeast; Breitenbach, M., Jazwinski, S.M., Laun, P., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 207–232. [Google Scholar]
- Côrte-Real, M.; Madeo, F. Yeast Programed Cell Death and Aging. Front. Oncol. 2013, 3, 283. [Google Scholar] [CrossRef] [PubMed]
- Tower, J. Programmed cell death in aging. Ageing Res. Rev. 2015, 23, 90–100. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef]
- Dadsena, S.; King, L.E.; García-Sáez, A.J. Apoptosis regulation at the mitochondria membrane level. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183716. [Google Scholar] [CrossRef]
- Subburaj, Y.; Cosentino, K.; Axmann, M.; Pedrueza-Villalmanzo, E.; Hermann, E.; Bleicken, S.; Spatz, J.; García-Sáez, A.J. Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat. Commun. 2015, 6, 8042. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Lejskova, R.; Grousl, T.; Stradalova, V.; Heeren, G.; Richter, K.; Breitenbach-Koller, L.; Malinsky, J.; Hasek, J.; Breitenbach, M. Mmi1, the yeast homologue of mammalian TCTP, associates with stress granules in heat-shocked cells and modulates proteasome activity. PLoS ONE 2013, 8, e77791. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Jarolim, S.; Heeren, G.; Palle, E.; Perju, S.; Klinger, H.; Bogengruber, E.; Madeo, F.; Braun, R.J.; Breitenbach-Koller, L.; et al. MMI1 (YKL056c, TMA19), the yeast orthologue of the translationally controlled tumor protein (TCTP) has apoptotic functions and interacts with both microtubules and mitochondria. Biochim. Biophys. Acta 2006, 1757, 631–638. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Kissová, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef]
- Kim, E.H.; Choi, K.S. A critical role of superoxide anion in selenite-induced mitophagic cell death. Autophagy 2008, 4, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Zhu, J.; Dagda, R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: Implications for neurodegeneration and cell death. Autophagy 2007, 3, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Nowikovsky, K.; Reipert, S.; Devenish, R.J.; Schweyen, R.J. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 2007, 14, 1647–1656. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef]
- Bergamini, E. Autophagy: A cell repair mechanism that retards ageing and age-associated diseases and can be intensified pharmacologically. Mol. Asp. Med. 2006, 27, 403–410. [Google Scholar] [CrossRef]
- Stevens, M.; Oltean, S. Modulation of the Apoptosis Gene Bcl-x Function Through Alternative Splicing. Front. Genet. 2019, 10, 804. [Google Scholar] [CrossRef]
- Kanagavijayan, D.; Rajasekharan, R.; Srinivasan, M. Yeast MRX deletions have short chronological life span and more triacylglycerols. Fems Yeast Res. 2016, 16, fov109. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Aung-Htut, M.T.; Lam, Y.T.; Lim, Y.L.; Rinnerthaler, M.; Gelling, C.L.; Yang, H.; Breitenbach, M.; Dawes, I.W. Maintenance of mitochondrial morphology by autophagy and its role in high glucose effects on chronological lifespan of Saccharomyces cerevisiae. Oxidative Med. Cell. Longev. 2013, 2013, 636287. [Google Scholar] [CrossRef]
- Sorger, D.; Athenstaedt, K.; Hrastnik, C.; Daum, G. A yeast strain lacking lipid particles bears a defect in ergosterol formation. J. Biol. Chem. 2004, 279, 31190–31196. [Google Scholar] [CrossRef] [PubMed]
- Valachovic, M.; Hronska, L.; Hapala, I. Anaerobiosis induces complex changes in sterol esterification pattern in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Lett. 2001, 197, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Leber, R.; Zinser, E.; Zellnig, G.; Paltauf, F.; Daum, G. Characterization of lipid particles of the yeast, Saccharomyces cerevisiae. Yeast 1994, 10, 1421–1428. [Google Scholar] [CrossRef]
- Ruan, L.H.; Zhou, C.K.; Jin, E.L.; Kucharavy, A.; Zhang, Y.; Wen, Z.H.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017, 543, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Erjavec, N.; Bayot, A.; Gareil, M.; Camougrand, N.; Nystrom, T.; Friguet, B.; Bulteau, A.L. Deletion of the mitochondrial Pim1/Lon protease in yeast results in accelerated aging and impairment of the proteasome. Free Radic. Biol. Med. 2013, 56, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Kaeberlein, M.; Andalis, A.A.; Sturtz, L.A.; Defossez, P.A.; Culotta, V.C.; Fink, G.R.; Guarente, L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 2002, 418, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Powers, R.W., 3rd; Kaeberlein, M.; Caldwell, S.D.; Kennedy, B.K.; Fields, S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006, 20, 174–184. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Madeira, J.B.; Masuda, C.A.; Maya-Monteiro, C.M.; Matos, G.S.; Montero-Lomeli, M.; Bozaquel-Morais, B.L. TORC1 Inhibition Induces Lipid Droplet Replenishment in Yeast. Mol. Cell. Biol. 2015, 35, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Vall-llaura, N.; Mir, N.; Garrido, L.; Vived, C.; Cabiscol, E. Redox control of yeast Sir2 activity is involved in acetic acid resistance and longevity. Redox. Biol. 2019, 24, 101229. [Google Scholar] [CrossRef] [PubMed]
- Boender, L.G.; Almering, M.J.; Dijk, M.; van Maris, A.J.; de Winde, J.H.; Pronk, J.T.; Daran-Lapujade, P. Extreme calorie restriction and energy source starvation in Saccharomyces cerevisiae represent distinct physiological states. Biochim. Biophys. Acta 2011, 1813, 2133–2144. [Google Scholar] [CrossRef]
- Schurmanns, L.; Hamann, A.; Osiewacz, H.D. Lifespan Increase of Podospora anserina by Oleic Acid Is Linked to Alterations in Energy Metabolism, Membrane Trafficking and Autophagy. Cells 2022, 11, 519. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.B.; Johnson, T.E. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 1988, 118, 75–86. [Google Scholar] [CrossRef]
- Iser, W.B.; Wolkow, C.A. DAF-2/insulin-like signaling in C. elegans modifies effects of dietary restriction and nutrient stress on aging, stress and growth. PLoS ONE 2007, 2, e1240. [Google Scholar] [CrossRef]
- Jia, K.; Chen, D.; Riddle, D.L. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 2004, 131, 3897–3906. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef]
- Lakowski, B.; Hekimi, S. The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1998, 95, 13091–13096. [Google Scholar] [CrossRef]
- Luo, S.; Kleemann, G.A.; Ashraf, J.M.; Shaw, W.M.; Murphy, C.T. TGF-beta and insulin signaling regulate reproductive aging via oocyte and germline quality maintenance. Cell 2010, 143, 299–312. [Google Scholar] [CrossRef]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef]
- Leiser, S.F.; Kaeberlein, M. The hypoxia-inducible factor HIF-1 functions as both a positive and negative modulator of aging. Biol. Chem. 2010, 391, 1131–1137. [Google Scholar] [CrossRef]
- Golden, J.W.; Riddle, D.L. The Caenorhabditis elegans dauer larva: Developmental effects of pheromone, food, and temperature. Dev. Biol. 1984, 102, 368–378. [Google Scholar] [CrossRef]
- Klass, M.; Hirsh, D. Non-ageing developmental variant of Caenorhabditis elegans. Nature 1976, 260, 523–525. [Google Scholar] [CrossRef]
- Zhang, S.; Li, F.; Zhou, T.; Wang, G.; Li, Z. Caenorhabditis elegans as a Useful Model for Studying Aging Mutations. Front. Endocrinol. 2020, 11, 554994. [Google Scholar] [CrossRef]
- Ewald, C.Y.; Castillo-Quan, J.I.; Blackwell, T.K. Untangling Longevity, Dauer, and Healthspan in Caenorhabditis elegans Insulin/IGF-1-Signalling. Gerontology 2018, 64, 96–104. [Google Scholar] [CrossRef]
- Zhang, S.O.; Trimble, R.; Guo, F.; Mak, H.Y. Lipid droplets as ubiquitous fat storage organelles in C. elegans. BMC Cell Biol. 2010, 11, 96. [Google Scholar] [CrossRef]
- Wang, P.; Liu, B.; Zhang, D.; Belew, M.Y.; Tissenbaum, H.A.; Cheng, J.X. Imaging lipid metabolism in live Caenorhabditis elegans using fingerprint vibrations. Angew. Chem. Int. Ed. 2014, 53, 11787–11792. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S. Regulation of body fat in Caenorhabditis elegans. Annu. Rev. Physiol. 2015, 77, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Mak, H.Y. Lipid droplets as fat storage organelles in Caenorhabditis elegans: Thematic Review Series: Lipid Droplet Synthesis and Metabolism: From Yeast to Man. J. Lipid Res. 2012, 53, 28–33. [Google Scholar] [CrossRef] [PubMed]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. J. Nutr. 1935, 10, 63–79. [Google Scholar] [CrossRef]
- Partridge, L.; Piper, M.D.; Mair, W. Dietary restriction in Drosophila. Mech. Ageing Dev. 2005, 126, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.D. The C. elegans intestine. In WormBook: The Online Review of C. elegans Biology; WormBook: Pasadena, CA, USA, 2007. [Google Scholar]
- Klass, M.R. Aging in Nematode Caenorhabditis-elegans—Major Biological and Environmental-Factors Influencing Life-Span. Mech. Ageing Dev. 1977, 6, 413–429. [Google Scholar] [CrossRef]
- Hosono, R.; Nishimoto, S.; Kuno, S. Alterations of life span in the nematode Caenorhabditis elegans under monoxenic culture conditions. Exp. Gerontol. 1989, 24, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, B.C.; Ashrafi, K. C. elegans fat storage and metabolic regulation. Biochim. Biophys. Acta 2009, 1791, 474–478. [Google Scholar] [CrossRef]
- Wu, Z.; Isik, M.; Moroz, N.; Steinbaugh, M.J.; Zhang, P.; Blackwell, T.K. Dietary Restriction Extends Lifespan through Metabolic Regulation of Innate Immunity. Cell Metab. 2019, 29, 1192–1205.e1198. [Google Scholar] [CrossRef] [PubMed]
- Houthoofd, K.; Gems, D.; Johnson, T.E.; Vanfleteren, J.R. Dietary restriction in the nematode Caenorhabditis elegans. Interdiscip. Top. Gerontol. 2007, 35, 98–114. [Google Scholar] [CrossRef]
- Walker, G.; Houthoofd, K.; Vanfleteren, J.R.; Gems, D. Dietary restriction in C. elegans: From rate-of-living effects to nutrient sensing pathways. Mech. Ageing Dev. 2005, 126, 929–937. [Google Scholar] [CrossRef]
- McElwee, J.; Bubb, K.; Thomas, J.H. Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell 2003, 2, 111–121. [Google Scholar] [CrossRef]
- Sun, X.; Chen, W.D.; Wang, Y.D. DAF-16/FOXO Transcription Factor in Aging and Longevity. Front. Pharmacol. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, R.; Schaffitzel, E.; Hertweck, M. Endocrine signaling in Caenorhabditis elegans controls stress response and longevity. J. Endocrinol. 2006, 190, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans Mutant That Lives Twice as Long as Wild-Type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef]
- Paradis, S.; Ailion, M.; Toker, A.; Thomas, J.H.; Ruvkun, G. A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Gene Dev. 1999, 13, 1438–1452. [Google Scholar] [CrossRef]
- Ogg, S.; Paradis, S.; Gottlieb, S.; Patterson, G.I.; Lee, L.; Tissenbaum, H.A.; Ruvkun, G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389, 994–999. [Google Scholar] [CrossRef]
- Zecic, A.; Braeckman, B.P. DAF-16/FoxO in Caenorhabditis elegans and Its Role in Metabolic Remodeling. Cells 2020, 9, 109. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Sewell, A.K.; Wu, Z.; Han, M. TOR Signaling in Caenorhabditis elegans Development, Metabolism, and Aging. Genetics 2019, 213, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.Q.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Lapierre, L.R.; Hansen, M. Lessons from C. elegans: Signaling pathways for longevity. Trends Endocrinol. Metab. 2012, 23, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Powers, T. TOR signaling and S6 kinase 1: Yeast catches up. Cell Metab. 2007, 6, 1–2. [Google Scholar] [CrossRef]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Muller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. TOR Signaling and Rapamycin Influence Longevity by Regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012, 15, 713–724. [Google Scholar] [CrossRef]
- Honjoh, S.; Yamamoto, T.; Uno, M.; Nishida, E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 2009, 457, 726–730. [Google Scholar] [CrossRef]
- Hansen, M.; Taubert, S.; Crawford, D.; Libina, N.; Lee, S.J.; Kenyon, C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 2007, 6, 95–110. [Google Scholar] [CrossRef]
- Jones, K.T.; Greer, E.R.; Pearce, D.; Ashrafi, K. Rictor/TORC2 regulates Caenorhabditis elegans fat storage, body size, and development through sgk-1. PLoS Biol. 2009, 7, e60. [Google Scholar] [CrossRef]
- Soukas, A.A.; Kane, E.A.; Carr, C.E.; Melo, J.A.; Ruvkun, G. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev. 2009, 23, 496–511. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Long, L.; Liu, W.; Ruan, P.; Yang, X.; Chen, X.; Li, L.; Yuan, F.; He, D.; Huang, P.; Gong, A.; et al. Visualizing the Interplay of Lipid Droplets and Protein Aggregates During Aging via a Dual-Functional Fluorescent Probe. Anal. Chem. 2022, 94, 2803–2811. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, M.; Kim, D.H. Age-Dependent Neuroendocrine Signaling from Sensory Neurons Modulates the Effect of Dietary Restriction on Longevity of Caenorhabditis elegans. PLoS Genet. 2017, 13, e1006544. [Google Scholar] [CrossRef] [PubMed]
- Gumienny, T.L.; Savage-Dunn, C. TGF-Beta Signaling in C. elegans; WormBook: Pasadena, CA, USA, 2013; pp. 1–34. [Google Scholar] [CrossRef]
- Liu, T.; Zimmerman, K.K.; Patterson, G.I. Regulation of signaling genes by TGFbeta during entry into dauer diapause in C. elegans. BMC Dev. Biol. 2004, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.H.; Crossman, D.; Prasain, J.K.; Miller, M.A.; Serra, R.A. Transcriptomic Profiling of DAF-7/TGF beta Pathway Mutants in C. elegans. Genes 2020, 11, 288. [Google Scholar] [CrossRef]
- Lant, B.; Storey, K.B. An Overview of Stress Response and Hypometabolic Strategies in Caenorhabditis elegans: Conserved and Contrasting Signals with the Mammalian System. Int. J. Biol. Sci. 2010, 6, 9–50. [Google Scholar] [CrossRef]
- Shaw, W.M.; Luo, S.; Landis, J.; Ashraf, J.; Murphy, C.T. The C. elegans TGF-beta Dauer pathway regulates longevity via insulin signaling. Curr. Biol. 2007, 17, 1635–1645. [Google Scholar] [CrossRef]
- Greer, E.R.; Perez, C.L.; Van Gilst, M.R.; Lee, B.H.; Ashrafi, K. Neural and molecular dissection of a C. elegans sensory circuit that regulates fat and feeding. Cell Metab. 2008, 8, 118–131. [Google Scholar] [CrossRef]
- Rashid, S.; Pho, K.B.; Mesbahi, H.; MacNeil, L.T. Nutrient Sensing and Response Drive Developmental Progression in Caenorhabditis elegans. Bioessays 2020, 42, e1900194. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Parker, W.M.; Leitão, J.A.; Rodriguez, D.I.; Ng, C.; Patel, R.; Aguilera, J.L.; Johnson, J.R.; Wong, S.Q.; et al. Lipid droplets modulate proteostasis, SQST-1/SQSTM1 dynamics, and lifespan in C. elegans. bioRxiv 2022, 2021.04.22.440991. [Google Scholar] [CrossRef]
- Castillo-Quan, J.I.; Steinbaugh, M.J.; Fernández-Cárdenas, L.P.; Pohl, N.K.; Wu, Z.; Zhu, F.; Moroz, N.; Teixeira, V.; Bland, M.S.; Lehrbach, N.J.; et al. An anti-steatosis response regulated by oleic acid through lipid droplet-mediated ERAD enhancement. bioRxiv 2022, 2022.06.15.496302. [Google Scholar] [CrossRef]
- Na, H.; Zhang, P.; Chen, Y.; Zhu, X.; Liu, Y.; Liu, Y.; Xie, K.; Xu, N.; Yang, F.; Yu, Y.; et al. Identification of lipid droplet structure-like/resident proteins in Caenorhabditis elegans. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2015, 1853, 2481–2491. [Google Scholar] [CrossRef]
- Chughtai, A.A.; Kaššák, F.; Kostrouchová, M.; Novotný, J.P.; Krause, M.W.; Saudek, V.; Kostrouch, Z.; Kostrouchová, M. Perilipin-related protein regulates lipid metabolism in C. elegans. PeerJ 2015, 3, e1213. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, V.; Ojha, N.; Golden, A.; Prinz, W.A. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J. Cell Biol. 2015, 211, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Papsdorf, K.; Miklas, J.W.; Hosseini, A.; Cabruja, M.; Morrow, C.S.; Savini, M.; Yu, Y.; Silva-García, C.G.; Haseley, N.R.; Murphy, L.M.; et al. Lipid droplets and peroxisomes are co-regulated to drive lifespan extension in response to mono-unsaturated fatty acids. Nat. Cell Biol. 2023, 25, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.V.; Bacher, M.C.; Priess, J.R. Nuclear lipid droplets and nuclear damage in Caenorhabditis elegans. PLoS Genet. 2021, 17, e1009602. [Google Scholar] [CrossRef]
- He, Y.; Jasper, H. Studying aging in Drosophila. Methods 2014, 68, 129–133. [Google Scholar] [CrossRef]
- Piper, M.D.W.; Partridge, L. Drosophila as a model for ageing. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2707–2717. [Google Scholar] [CrossRef]
- Proshkina, E.N.; Shaposhnikov, M.V.; Sadritdinova, A.F.; Kudryavtseva, A.V.; Moskalev, A.A. Basic mechanisms of longevity: A case study of Drosophila pro-longevity genes. Ageing Res. Rev. 2015, 24, 218–231. [Google Scholar] [CrossRef]
- Liao, S.; Amcoff, M.; Nässel, D.R. Impact of high-fat diet on lifespan, metabolism, fecundity and behavioral senescence in Drosophila. Insect Biochem. Mol. Biol. 2021, 133, 103495. [Google Scholar] [CrossRef]
- Hofbauer, H.F.; Heier, C.; Sen Saji, A.K.; Kühnlein, R.P. Lipidome remodeling in aging normal and genetically obese Drosophila males. Insect Biochem. Mol. Biol. 2021, 133, 103498. [Google Scholar] [CrossRef]
- Buszczak, M.; Lu, X.; Segraves, W.A.; Chang, T.Y.; Cooley, L. Mutations in the midway gene disrupt a Drosophila acyl coenzyme A: Diacylglycerol acyltransferase. Genetics 2002, 160, 1511–1518. [Google Scholar] [CrossRef]
- Gronke, S.; Mildner, A.; Fellert, S.; Tennagels, N.; Petry, S.; Muller, G.; Jackle, H.; Kuhnlein, R.P. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 2005, 1, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Park, J.O.; Tanner, L.; Nagano, Y.; Rabinowitz, J.D.; Shvartsman, S.Y. Energy budget of Drosophila embryogenesis. Curr. Biol. 2019, 29, R566–R567. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.M.; Barry, W.E.; Cox, J.; Thummel, C.S. Methods for studying metabolism in Drosophila. Methods 2014, 68, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Bickel, P.E.; Tansey, J.T.; Welte, M.A. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 2009, 1791, 419–440. [Google Scholar] [CrossRef]
- Miura, S.; Gan, J.W.; Brzostowski, J.; Parisi, M.J.; Schultz, C.J.; Londos, C.; Oliver, B.; Kimmel, A.R. Functional conservation for lipid storage droplet association among Perilipin, ADRP, and TIP47 (PAT)-related proteins in mammals, Drosophila, and Dictyostelium. J. Biol. Chem. 2002, 277, 32253–32257. [Google Scholar] [CrossRef]
- Teixeira, L.; Rabouille, C.; Rørth, P.; Ephrussi, A.; Vanzo, N.F. Drosophila Perilipin/ADRP homologue Lsd2 regulates lipid metabolism. Mech. Dev. 2003, 120, 1071–1081. [Google Scholar] [CrossRef]
- Beller, M.; Bulankina, A.V.; Hsiao, H.H.; Urlaub, H.; Jackle, H.; Kuhnlein, R.P. PERILIPIN-dependent control of lipid droplet structure and fat storage in Drosophila. Cell Metab. 2010, 12, 521–532. [Google Scholar] [CrossRef]
- Grönke, S.; Beller, M.; Fellert, S.; Ramakrishnan, H.; Jäckle, H.; Kühnlein, R.P. Control of fat storage by a Drosophila PAT domain protein. Curr. Biol. 2003, 13, 603–606. [Google Scholar] [CrossRef]
- Kayukawa, T.; Jouraku, A.; Ito, Y.; Shinoda, T. Molecular mechanism underlying juvenile hormone-mediated repression of precocious larval-adult metamorphosis. Proc. Natl. Acad. Sci. USA 2017, 114, 1057–1062. [Google Scholar] [CrossRef]
- Mirth, C.K.; Shingleton, A.W. Integrating body and organ size in Drosophila: Recent advances and outstanding problems. Front. Endocrinol. 2012, 3, 49. [Google Scholar] [CrossRef]
- Mirth, C.K.; Tang, H.Y.; Makohon-Moore, S.C.; Salhadar, S.; Gokhale, R.H.; Warner, R.D.; Koyama, T.; Riddiford, L.M.; Shingleton, A.W. Juvenile hormone regulates body size and perturbs insulin signaling in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, 7018–7023. [Google Scholar] [CrossRef]
- Texada, M.J.; Malita, A.; Christensen, C.F.; Dall, K.B.; Faergeman, N.J.; Nagy, S.; Halberg, K.A.; Rewitz, K. Autophagy-Mediated Cholesterol Trafficking Controls Steroid Production. Dev. Cell 2019, 48, 659–671.e654. [Google Scholar] [CrossRef] [PubMed]
- Werthebach, M.; Stewart, F.A.; Gahlen, A.; Mettler-Altmann, T.; Akhtar, I.; Maas-Enriquez, K.; Droste, A.; Eichmann, T.O.; Poschmann, G.; Stuhler, K.; et al. Control of Drosophila Growth and Survival by the Lipid Droplet-Associated Protein CG9186/Sturkopf. Cell Rep. 2019, 26, 3726–3740.e3727. [Google Scholar] [CrossRef] [PubMed]
- Thiel, K.; Heier, C.; Haberl, V.; Thul, P.J.; Oberer, M.; Lass, A.; Jäckle, H.; Beller, M. The evolutionarily conserved protein CG9186 is associated with lipid droplets, required for their positioning and for fat storage. J. Cell Sci. 2013, 126, 2198–2212. [Google Scholar] [CrossRef]
- Ugrankar, R.; Bowerman, J.; Hariri, H.; Chandra, M.; Chen, K.; Bossanyi, M.F.; Datta, S.; Rogers, S.; Eckert, K.M.; Vale, G.; et al. Drosophila Snazarus Regulates a Lipid Droplet Population at Plasma Membrane-Droplet Contacts in Adipocytes. Dev. Cell 2019, 50, 557–572.e555. [Google Scholar] [CrossRef] [PubMed]
- Blumrich, A.; Vogler, G.; Dresen, S.; Diop, S.B.; Jaeger, C.; Leberer, S.; Grune, J.; Wirth, E.K.; Hoeft, B.; Renko, K.; et al. Fat-body brummer lipase determines survival and cardiac function during starvation in Drosophila melanogaster. Iscience 2021, 24, 102288. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Xiang, Y.; Chen, H.; Liu, Z.; Grönke, S.; Kühnlein, R.P.; Huang, X. Opposite and redundant roles of the two Drosophila perilipins in lipid mobilization. J. Cell Sci. 2012, 125, 3568–3577. [Google Scholar] [CrossRef]
- Binh, T.D.; Nguyen, Y.D.H.; Pham, T.L.A.; Komori, K.; Nguyen, T.Q.C.; Taninaka, M.; Kamei, K. Dysfunction of lipid storage droplet-2 suppresses endoreplication and induces JNK pathway-mediated apoptotic cell death in Drosophila salivary glands. Sci. Rep. 2022, 12, 4302. [Google Scholar] [CrossRef]
- Fauny, J.D.; Silber, J.; Zider, A. Drosophila Lipid Storage Droplet 2 gene (Lsd-2) is expressed and controls lipid storage in wing imaginal discs. Dev. Dyn. 2005, 232, 725–732. [Google Scholar] [CrossRef]
- Binh, T.D.; Pham, T.L.A.; Men, T.T.; Dang, T.T.P.; Kamei, K. LSD-2 dysfunction induces dFoxO-dependent cell death in the wing of Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2019, 509, 491–497. [Google Scholar] [CrossRef]
- Goyal, L.; McCall, K.; Agapite, J.; Hartwieg, E.; Steller, H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 2000, 19, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Hanifeh, M.; Ataei, F. XIAP as a multifaceted molecule in Cellular Signaling. Apoptosis 2022, 27, 441–453. [Google Scholar] [CrossRef]
- Lu, M.; Lin, S.C.; Huang, Y.; Kang, Y.J.; Rich, R.; Lo, Y.C.; Myszka, D.; Han, J.; Wu, H. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol. Cell. 2007, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, H.; Hu, M.; Jiang, L.; Wang, Y.; Liu, P.; Liang, X.; Liu, J.; Li, C.; Lindström-Battle, A.; et al. HDAC6 Suppresses Age-Dependent Ectopic Fat Accumulation by Maintaining the Proteostasis of PLIN2 in Drosophila. Dev. Cell 2017, 43, 99–111.e115. [Google Scholar] [CrossRef] [PubMed]
- Katewa, S.D.; Akagi, K.; Bose, N.; Rakshit, K.; Camarella, T.; Zheng, X.; Hall, D.; Davis, S.; Nelson, C.S.; Brem, R.B.; et al. Peripheral Circadian Clocks Mediate Dietary Restriction-Dependent Changes in Lifespan and Fat Metabolism in Drosophila. Cell Metab. 2016, 23, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Katewa, S.D.; Kapahi, P. Dietary restriction and aging, 2009. Aging Cell 2010, 9, 105–112. [Google Scholar] [CrossRef]
- Skorupa, D.A.; Dervisefendic, A.; Zwiener, J.; Pletcher, S.D. Dietary composition specifies consumption, obesity, and lifespan in Drosophila melanogaster. Aging Cell 2008, 7, 478–490. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef]
- Puig, O.; Marr, M.T.; Ruhf, M.L.; Tjian, R. Control of cell number by Drosophila FOXO: Downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 2003, 17, 2006–2020. [Google Scholar] [CrossRef] [PubMed]
- Slack, C.; Giannakou, M.E.; Foley, A.; Goss, M.; Partridge, L. dFOXO-independent effects of reduced insulin-like signaling in Drosophila. Aging Cell 2011, 10, 735–748. [Google Scholar] [CrossRef]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.C.; Bohmann, D.; Jasper, H. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev. Cell 2003, 5, 811–816. [Google Scholar] [CrossRef]
- Yamamoto, R.; Tatar, M. Insulin receptor substrate chico acts with the transcription factor FOXO to extend Drosophila lifespan. Aging Cell 2011, 10, 729–732. [Google Scholar] [CrossRef]
- Alic, N.; Giannakou, M.E.; Papatheodorou, I.; Hoddinott, M.P.; Andrews, T.D.; Bolukbasi, E.; Partridge, L. Interplay of dFOXO and two ETS-family transcription factors determines lifespan in Drosophila melanogaster. PLoS Genet. 2014, 10, e1004619. [Google Scholar] [CrossRef] [PubMed]
- Min, K.J.; Yamamoto, R.; Buch, S.; Pankratz, M.; Tatar, M. Drosophila lifespan control by dietary restriction independent of insulin-like signaling. Aging Cell 2008, 7, 199–206. [Google Scholar] [CrossRef]
- Gronke, S.; Clarke, D.F.; Broughton, S.; Andrews, T.D.; Partridge, L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 2010, 6, e1000857. [Google Scholar] [CrossRef]
- Vereshchagina, N.; Wilson, C. Cytoplasmic activated protein kinase Akt regulates lipid-droplet accumulation in Drosophila nurse cells. Development 2006, 133, 4731–4735. [Google Scholar] [CrossRef]
- DiAngelo, J.R.; Birnbaum, M.J. Regulation of fat cell mass by insulin in Drosophila melanogaster. Mol. Cell. Biol. 2009, 29, 6341–6352. [Google Scholar] [CrossRef]
- Wang, B.; Moya, N.; Niessen, S.; Hoover, H.; Mihaylova, M.M.; Shaw, R.J.; Yates, J.R.; Fischer, W.H.; Thomas, J.B.; Montminy, M. A Hormone-Dependent Module Regulating Energy Balance. Cell 2011, 145, 596–606. [Google Scholar] [CrossRef]
- Biteau, B.; Karpac, J.; Supoyo, S.; Degennaro, M.; Lehmann, R.; Jasper, H. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 2010, 6, e1001159. [Google Scholar] [CrossRef]
- Wang, L.; Zeng, X.; Ryoo, H.D.; Jasper, H. Integration of UPRER and oxidative stress signaling in the control of intestinal stem cell proliferation. PLoS Genet. 2014, 10, e1004568. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ryoo, H.D.; Qi, Y.; Jasper, H. PERK Limits Drosophila Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress. PLoS Genet. 2015, 11, e1005220. [Google Scholar] [CrossRef] [PubMed]
- Luis, N.M.; Wang, L.F.; Ortega, M.; Deng, H.S.; Katewa, S.D.; Li, P.W.L.; Karpac, J.; Jasper, H.; Kapahi, P. Intestinal IRE1 Is Required for Increased Triglyceride Metabolism and Longer Lifespan under Dietary Restriction. Cell Rep. 2016, 17, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef]
- Cermelli, S.; Guo, Y.; Gross, S.P.; Welte, M.A. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr. Biol. 2006, 16, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Riedel, D.; Jansch, L.; Dieterich, G.; Wehland, J.; Jackle, H.; Kuhnlein, R.P. Characterization of the Drosophila lipid droplet subproteome. Mol. Cell. Proteom. 2006, 5, 1082–1094. [Google Scholar] [CrossRef]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef]
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef]
- Marshall, L.; Rideout, E.J.; Grewal, S.S. Nutrient/TOR-dependent regulation of RNA polymerase III controls tissue and organismal growth in Drosophila. EMBO J. 2012, 31, 1916–1930. [Google Scholar] [CrossRef] [PubMed]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef]
- Reeg, S.; Grune, T. Protein Oxidation in Toxicology. In Studies on Experimental Toxicology and Pharmacology; Oxidative Stress in Applied Basic Research and Clinical Practice; Humana Press: Cham, Switzerland, 2015; pp. 81–102. [Google Scholar] [CrossRef]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef]
- Kovacs, T.; Billes, V.; Komlos, M.; Hotzi, B.; Manzeger, A.; Tarnoci, A.; Papp, D.; Szikszai, F.; Szinyakovics, J.; Racz, A.; et al. The small molecule AUTEN-99 (autophagy enhancer-99) prevents the progression of neurodegenerative symptoms. Sci. Rep. 2017, 7, 42014. [Google Scholar] [CrossRef] [PubMed]
- Papp, D.; Kovács, T.; Billes, V.; Varga, M.; Tarnóci, A.; Hackler, L., Jr.; Puskás, L.G.; Liliom, H.; Tárnok, K.; Schlett, K.; et al. AUTEN-67, an autophagy-enhancing drug candidate with potent antiaging and neuroprotective effects. Autophagy 2016, 12, 273–286. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Pang, X.; Zhang, X.; Jiang, Y.; Su, Q.; Li, Q.; Li, Z. Autophagy: Mechanisms and Therapeutic Potential of Flavonoids in Cancer. Biomolecules 2021, 11, 135. [Google Scholar] [CrossRef]
- Fantin, M.; Garelli, F.; Napoli, B.; Forgiarini, A.; Gumeni, S.; De Martin, S.; Montopoli, M.; Vantaggiato, C.; Orso, G. Flavonoids Regulate Lipid Droplets Biogenesis in Drosophila melanogaster. Nat. Prod. Commun. 2019, 14, 1934578X19852430. [Google Scholar] [CrossRef]
- Wongchum, N.; Dechakhamphu, A. Xanthohumol prolongs lifespan and decreases stress-induced mortality in Drosophila melanogaster. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 244, 108994. [Google Scholar] [CrossRef]
- Gajender; Mazumder, A.; Sharma, A.; Azad, M.A.K. A Comprehensive Review of the Pharmacological Importance of Dietary Flavonoids as Hepatoprotective Agents. Evid.-Based Complement. Altern. Med. 2023, 2023, 4139117. [Google Scholar] [CrossRef]
- Heijnen, C.G.; Haenen, G.R.; Oostveen, R.M.; Stalpers, E.M.; Bast, A. Protection of flavonoids against lipid peroxidation: The structure activity relationship revisited. Free Radic. Res. 2002, 36, 575–581. [Google Scholar] [CrossRef]
- Partridge, L.; Alic, N.; Bjedov, I.; Piper, M.D. Ageing in Drosophila: The role of the insulin/Igf and TOR signalling network. Exp. Gerontol. 2011, 46, 376–381. [Google Scholar] [CrossRef]
- Zid, B.M.; Rogers, A.N.; Katewa, S.D.; Vargas, M.A.; Kolipinski, M.C.; Lu, T.A.; Benzer, S.; Kapahi, P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 2009, 139, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Vasudevan, D.; Kang, K.; Kim, K.; Park, J.E.; Zhang, N.; Zeng, X.; Neubert, T.A.; Marr, M.T., 2nd; Ryoo, H.D. 4E-BP is a target of the GCN2-ATF4 pathway during Drosophila development and aging. J. Cell Biol. 2017, 216, 115–129. [Google Scholar] [CrossRef]
- Srivastava, A.; Lu, J.; Gadalla, D.S.; Hendrich, O.; Grönke, S.; Partridge, L. The Role of GCN2 Kinase in Mediating the Effects of Amino Acids on Longevity and Feeding Behaviour in Drosophila. Front. Aging 2022, 3, 944466. [Google Scholar] [CrossRef] [PubMed]
- Mair, W.; Morantte, I.; Rodrigues, A.P.; Manning, G.; Montminy, M.; Shaw, R.J.; Dillin, A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 2011, 470, 404–408. [Google Scholar] [CrossRef]
- Stenesen, D.; Suh, J.M.; Seo, J.; Yu, K.; Lee, K.S.; Kim, J.S.; Min, K.J.; Graff, J.M. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 2013, 17, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.; Kazgan, N.; Bretz, C.A.; Forsberg, L.J.; Hector, C.E.; Worthen, R.J.; Onyenwoke, R.; Brenman, J.E. Altered metabolism and persistent starvation behaviors caused by reduced AMPK function in Drosophila. PLoS ONE 2010, 5, e12799. [Google Scholar] [CrossRef]
- Gutierrez, E.; Wiggins, D.; Fielding, B.; Gould, A.P. Specialized hepatocyte-like cells regulate Drosophila lipid metabolism. Nature 2007, 445, 275–280. [Google Scholar] [CrossRef]
- Triggle, C.R.; Mohammed, I.; Bshesh, K.; Marei, I.; Ye, K.; Ding, H.; MacDonald, R.; Hollenberg, M.D.; Hill, M.A. Metformin: Is it a drug for all reasons and diseases? Metabolism 2022, 133, 155223. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Berstein, L.M.; Egormin, P.A.; Piskunova, T.S.; Popovich, I.G.; Zabezhinski, M.A.; Tyndyk, M.L.; Yurova, M.V.; Kovalenko, I.G.; Poroshina, T.E.; et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle 2008, 7, 2769–2773. [Google Scholar] [CrossRef]
- Cabreiro, F.; Au, C.; Leung, K.Y.; Vergara-Irigaray, N.; Cocheme, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.; Gems, D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef]
- Slack, C.; Foley, A.; Partridge, L. Activation of AMPK by the putative dietary restriction mimetic metformin is insufficient to extend lifespan in Drosophila. PLoS ONE 2012, 7, e47699. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, M.; Tsuda, M.; Muramatsu, K.; Hatsuda, H.; Morishita, S.; Aigaki, T. A gain-of-function screen identifies wdb and lkb1 as lifespan-extending genes in Drosophila. Biochem. Biophys. Res. Commun. 2011, 405, 667–672. [Google Scholar] [CrossRef]
- Choi, S.; Lim, D.S.; Chung, J. Feeding and Fasting Signals Converge on the LKB1-SIK3 Pathway to Regulate Lipid Metabolism in Drosophila. PLoS Genet. 2015, 11, e1005263. [Google Scholar] [CrossRef]
- Grandison, R.C.; Piper, M.D.W.; Partridge, L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 2009, 462, 1061–1064. [Google Scholar] [CrossRef]
- Kabil, H.; Kabil, O.; Banerjee, R.; Harshman, L.G.; Pletcher, S.D. Increased transsulfuration mediates longevity and dietary restriction in Drosophila. Proc. Natl. Acad. Sci. USA 2011, 108, 16831–16836. [Google Scholar] [CrossRef]
- Jonsson, W.O.; Margolies, N.S.; Anthony, T.G. Dietary Sulfur Amino Acid Restriction and the Integrated Stress Response: Mechanistic Insights. Nutrients 2019, 11, 1349. [Google Scholar] [CrossRef]
- Richie, J.P., Jr.; Leutzinger, Y.; Parthasarathy, S.; Malloy, V.; Orentreich, N.; Zimmerman, J.A. Methionine restriction increases blood glutathione and longevity in F344 rats. Faseb J. 1994, 8, 1302–1307. [Google Scholar] [CrossRef]
- Miller, R.A.; Buehner, G.; Chang, Y.; Harper, J.M.; Sigler, R.; Smith-Wheelock, M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 2005, 4, 119–125. [Google Scholar] [CrossRef]
- Ruckenstuhl, C.; Netzberger, C.; Entfellner, I.; Carmona-Gutierrez, D.; Kickenweiz, T.; Stekovic, S.; Gleixner, C.; Schmid, C.; Klug, L.; Sorgo, A.G.; et al. Lifespan Extension by Methionine Restriction Requires Autophagy-Dependent Vacuolar Acidification. PLoS Genet. 2014, 10, e1004347. [Google Scholar] [CrossRef]
- Malloy, V.L.; Perrone, C.E.; Mattocks, D.A.; Ables, G.P.; Caliendo, N.S.; Orentreich, D.S.; Orentreich, N. Methionine restriction prevents the progression of hepatic steatosis in leptin-deficient obese mice. Metabolism 2013, 62, 1651–1661. [Google Scholar] [CrossRef]
- Perrone, C.E.; Mattocks, D.A.; Jarvis-Morar, M.; Plummer, J.D.; Orentreich, N. Methionine restriction effects on mitochondrial biogenesis and aerobic capacity in white adipose tissue, liver, and skeletal muscle of F344 rats. Metabolism 2010, 59, 1000–1011. [Google Scholar] [CrossRef]
- Nakata, K.; Kawase, M.; Ogino, S.; Kinoshita, C.; Murata, H.; Sakaue, T.; Ogata, K.; Ohmori, S. Effects of age on levels of cysteine, glutathione and related enzyme activities in livers of mice and rats and an attempt to replenish hepatic glutathione level of mouse with cysteine derivatives. Mech. Ageing Dev. 1996, 90, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.K.; Xiong, X.; Mustafi, S.B.; Saha, S.; Dhanasekaran, D.; Mandal, N.A.; McMeekin, S.; Bhattacharya, R.; Mukherjee, P. Role of cystathionine beta synthase in lipid metabolism in ovarian cancer. Oncotarget 2015, 6, 37367–37384. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Auge, N.; Ayala, V.; Basaga, H.; Boada, J.; Brenke, R.; Chapple, S.; Cohen, G.; Feher, J.; Grune, T.; et al. Pathological aspects of lipid peroxidation. Free Radic. Res. 2010, 44, 1125–1171. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef]
- Boal, A.K.; Yavin, E.; Barton, J.K. DNA repair glycosylases with a [4Fe-4S] cluster: A redox cofactor for DNA-mediated charge transport? J. Inorg. Biochem. 2007, 101, 1913–1921. [Google Scholar] [CrossRef]
- Rouault, T.A. Mammalian iron-sulphur proteins: Novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Crooks, D.R.; Maio, N.; Lane, A.N.; Jarnik, M.; Higashi, R.M.; Haller, R.G.; Yang, Y.; Fan, T.W.; Linehan, W.M.; Rouault, T.A. Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J. Biol. Chem. 2018, 293, 8297–8311. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Napierala, M.; Puccio, H. Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis. Model. Mech. 2012, 5, 165–176. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Navarro, J.A.; Ohmann, E.; Sanchez, D.; Botella, J.A.; Liebisch, G.; Moltó, M.D.; Ganfornina, M.D.; Schmitz, G.; Schneuwly, S. Altered lipid metabolism in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 2010, 19, 2828–2840. [Google Scholar] [CrossRef]
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M. Biologically relevant metal ion-dependent hydroxyl radical generation. An update. FEBS Lett. 1992, 307, 108–112. [Google Scholar] [CrossRef]
- Schaur, R.J.; Siems, W.; Bresgen, N.; Eckl, P.M. 4-Hydroxy-nonenal-A Bioactive Lipid Peroxidation Product. Biomolecules 2015, 5, 2247–2337. [Google Scholar] [CrossRef]
- Girard, V.; Goubard, V.; Querenet, M.; Seugnet, L.; Pays, L.; Nataf, S.; Dufourd, E.; Cluet, D.; Mollereau, B.; Davoust, N. Spen modulates lipid droplet content in adult Drosophila glial cells and protects against paraquat toxicity. Sci. Rep. 2020, 10, 20023. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; McWilliams, T.G. Lipid droplets promote efficient mitophagy. Autophagy 2022, 19, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.S.; Colombani, J.; Palmerini, V.; Chakrabandhu, K.; Boone, E.; Röthlisberger, M.; Toggweiler, J.; Basler, K.; Mapelli, M.; Hueber, A.O.; et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature 2015, 522, 482–486. [Google Scholar] [CrossRef]
- Muliyil, S.; Levet, C.; Düsterhöft, S.; Dulloo, I.; Cowley, S.A.; Freeman, M. ADAM17-triggered TNF signalling protects the ageing Drosophila retina from lipid droplet-mediated degeneration. Embo J. 2020, 39, e104415. [Google Scholar] [CrossRef] [PubMed]
- Lagrutta, L.C.; Layerenza, J.P.; Bronsoms, S.; Trejo, S.A.; Ves-Losada, A. Nuclear-lipid-droplet proteome: Carboxylesterase as a nuclear lipase involved in lipid-droplet homeostasis. Heliyon 2021, 7, e06539. [Google Scholar] [CrossRef]
- Lian, J.; Nelson, R.; Lehner, R. Carboxylesterases in lipid metabolism: From mouse to human. Protein Cell 2018, 9, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, E.; Quiroga, A.D.; Sun, X.; Touret, N.; Lehner, R. Altered lipid droplet dynamics in hepatocytes lacking triacylglycerol hydrolase expression. Mol. Biol. Cell 2010, 21, 1991–2000. [Google Scholar] [CrossRef]
- Lian, J.H.; Wei, E.H.; Wang, S.P.; Quiroga, A.D.; Li, L.N.; Di Pardo, A.; van der Veen, J.; Sipione, S.; Mitchell, G.A.; Lehner, R. Liver Specific Inactivation of Carboxylesterase 3/Triacylglycerol Hydrolase Decreases Blood Lipids Without Causing Severe Steatosis in Mice. Hepatology 2012, 56, 2154–2162. [Google Scholar] [CrossRef]
- Fujimoto, T. Nuclear lipid droplets—How are they different from their cytoplasmic siblings? J. Cell Sci. 2022, 135, jcs259253. [Google Scholar] [CrossRef]
- McPhee, M.J.; Salsman, J.; Foster, J.; Thompson, J.; Mathavarajah, S.; Dellaire, G.; Ridgway, N.D. Running ‘LAPS’ around nLD: Nuclear Lipid Droplet Form and Function. Front. Cell Dev. Biol. 2022, 10, 837406. [Google Scholar] [CrossRef]
- Chen, H.; Zheng, X.; Xiao, D.; Zheng, Y. Age-associated de-repression of retrotransposons in the Drosophila fat body, its potential cause and consequence. Aging Cell 2016, 15, 542–552. [Google Scholar] [CrossRef]
- Wood, J.G.; Jones, B.C.; Jiang, N.; Chang, C.; Hosier, S.; Wickremesinghe, P.; Garcia, M.; Hartnett, D.A.; Burhenn, L.; Neretti, N.; et al. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, 11277–11282. [Google Scholar] [CrossRef] [PubMed]
- Czech, B.; Hannon, G.J. Small RNA sorting: Matchmaking for Argonautes. Nat. Rev. Genet. 2011, 12, 19–31. [Google Scholar] [CrossRef]
- Li, W.; Prazak, L.; Chatterjee, N.; Grüninger, S.; Krug, L.; Theodorou, D.; Dubnau, J. Activation of transposable elements during aging and neuronal decline in Drosophila. Nat. Neurosci. 2013, 16, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Johnson, M.R.; Ke, Z.; Chen, L.; Welte, M.A. Drosophila lipid droplets buffer the H2Av supply to protect early embryonic development. Curr. Biol. 2014, 24, 1485–1491. [Google Scholar] [CrossRef]
- Loyola, A.C.; Zhang, L.; Shang, R.; Dutta, P.; Li, J.; Li, W.X. Identification of methotrexate as a heterochromatin-promoting drug. Sci. Rep. 2019, 9, 11673. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Hillenmeyer, S.; Lawrence, C.; Chang, C.; Hosier, S.; Lightfoot, W.; Mukherjee, E.; Jiang, N.; Schorl, C.; Brodsky, A.S.; et al. Chromatin remodeling in the aging genome of Drosophila. Aging Cell 2010, 9, 971–978. [Google Scholar] [CrossRef]
- Yang, L.; Ma, Z.; Wang, H.; Niu, K.; Cao, Y.; Sun, L.; Geng, Y.; Yang, B.; Gao, F.; Chen, Z.; et al. Ubiquitylome study identifies increased histone 2A ubiquitylation as an evolutionarily conserved aging biomarker. Nat. Commun. 2019, 10, 2191. [Google Scholar] [CrossRef]
- Maleszewska, M.; Mawer, J.S.P.; Tessarz, P. Histone Modifications in Ageing and Lifespan Regulation. Curr. Mol. Biol. Rep. 2016, 2, 26–35. [Google Scholar] [CrossRef]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated histone expression promotes life span extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Najar, U.; Sedivy, J.M. Epigenetic control of aging. Antioxid. Redox Signal. 2011, 14, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef]
- Toiber, D.; Erdel, F.; Bouazoune, K.; Silberman, D.M.; Zhong, L.; Mulligan, P.; Sebastian, C.; Cosentino, C.; Martinez-Pastor, B.; Giacosa, S.; et al. SIRT6 Recruits SNF2H to DNA Break Sites, Preventing Genomic Instability through Chromatin Remodeling. Mol. Cell 2013, 51, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Xiao, C.Y.; Wang, R.H.; Lahusen, T.; Xu, X.L.; Vassilopoulos, A.; Vazquez-Ortiz, G.; Jeong, W.I.; Park, O.; Ki, S.H.; et al. Hepatic-Specific Disruption of SIRT6 in Mice Results in Fatty Liver Formation Due to Enhanced Glycolysis and Triglyceride Synthesis. Cell Metab. 2010, 12, 224–236. [Google Scholar] [CrossRef]
- Penrose, H.; Heller, S.; Cable, C.; Makboul, R.; Chadalawada, G.; Chen, Y.; Crawford, S.E.; Savkovic, S.D. Epidermal growth factor receptor mediated proliferation depends on increased lipid droplet density regulated via a negative regulatory loop with FOXO3/Sirtuin6. Biochem. Biophys. Res. Commun. 2016, 469, 370–376. [Google Scholar] [CrossRef]
- Eckl, P.M.; Bresgen, N. Genotoxicity of lipid oxidation compounds. Free Radic. Biol. Med. 2017, 111, 244–252. [Google Scholar] [CrossRef]
- Shubeita, G.T.; Tran, S.L.; Xu, J.; Vershinin, M.; Cermelli, S.; Cotton, S.L.; Welte, M.A.; Gross, S.P. Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell 2008, 135, 1098–1107. [Google Scholar] [CrossRef]
- Welte, M.A. Proteins under new management: Lipid droplets deliver. Trends Cell Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef]
- Welte, M.A.; Gross, S.P.; Postner, M.; Block, S.M.; Wieschaus, E.F. Developmental regulation of vesicle transport in Drosophila embryos: Forces and kinetics. Cell 1998, 92, 547–557. [Google Scholar] [CrossRef]
- Bartsch, T.F.; Longoria, R.A.; Florin, E.L.; Shubeita, G.T. Lipid droplets purified from Drosophila embryos as an endogenous handle for precise motor transport measurements. Biophys. J. 2013, 105, 1182–1191. [Google Scholar] [CrossRef]
- Larsen, K.S.; Xu, J.; Cermelli, S.; Shu, Z.; Gross, S.P. BicaudalD actively regulates microtubule motor activity in lipid droplet transport. PLoS ONE 2008, 3, e3763. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A.; Cermelli, S.; Griner, J.; Viera, A.; Guo, Y.; Kim, D.H.; Gindhart, J.G.; Gross, S.P. Regulation of lipid-droplet transport by the perilipin homolog LSD2. Curr. Biol. 2005, 15, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Jäckle, H.; Jahn, R. Vesicle transport: Klarsicht clears up the matter. Curr. Biol. 1998, 8, R542–R544. [Google Scholar] [CrossRef]
- Myat, M.M.; Andrew, D.J. Epithelial tube morphology is determined by the polarized growth and delivery of apical membrane. Cell 2002, 111, 879–891. [Google Scholar] [CrossRef]
- Yu, Y.V.; Li, Z.; Rizzo, N.P.; Einstein, J.; Welte, M.A. Targeting the motor regulator Klar to lipid droplets. BMC Cell Biol. 2011, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jangi, S.; Welte, M.A. Organelle-specific control of intracellular transport: Distinctly targeted isoforms of the regulator Klar. Mol. Biol. Cell 2005, 16, 1406–1416. [Google Scholar] [CrossRef]
- Ingle, L.; Wood, T.R.; Banta, A.M. A study of longevity, growth, reproduction and heart rate in Daphnia longispina as influenced by limitations in quantity of food. J. Exp. Zool. 1937, 76, 325–352. [Google Scholar] [CrossRef]
- Fanestil, D.D.; Barrows, C.H., Jr. Aging in the rotifer. J. Gerontol. 1965, 20, 462–469. [Google Scholar]
- Comfort, A. Effect of Delayed and resumed growth on the longevity of a fish (Lebistes reticulatus, peters) in captivity. Gerontologia 1963, 49, 150–155. [Google Scholar] [CrossRef]
- Dilan, C.-B.; Begun, E.; Ahmet Tugrul, O.; Hulusi, K.; Michelle, A. Zebrafish Aging Models and Possible Interventions. In Recent Advances in Zebrafish Researches; Yusuf, B., Ed.; IntechOpen: Rijeka, Croatia, 2018; pp. 3–26. [Google Scholar]
- Rudzinska, M.A. The influence of amount of food on the reproduction rate and longevity of a sectarian. (Tokophyra infusionum). Science 1951, 113, 10–11. [Google Scholar] [CrossRef]
- Sutphin, G.L.; Kaeberlein, M. Dietary restriction by bacterial deprivation increases life span in wild-derived nematodes. Exp. Gerontol. 2008, 43, 130–135. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose Restriction Extends Caenorhabditis elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef]
- Suckow, B.K.; Suckow, M.A. Lifespan extension by the antioxidant curcumin in Drosophila melanogaster. Int. J. Biomed. Sci. 2006, 2, 402–405. [Google Scholar]
- Mohammed, I.; Hollenberg, M.D.; Ding, H.; Triggle, C.R. A Critical Review of the Evidence That Metformin Is a Putative Anti-Aging Drug That Enhances Healthspan and Extends Lifespan. Front. Endocrinol. 2021, 12, 718942. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Racette, F.G.; Bogan, K.L.; McClure, J.M.; Smith, J.S.; Brenner, C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 2007, 129, 473–484. [Google Scholar] [CrossRef]
- Hofer, S.J.; Davinelli, S.; Bergmann, M.; Scapagnini, G.; Madeo, F. Caloric Restriction Mimetics in Nutrition and Clinical Trials. Front. Nutr. 2021, 8, 717343. [Google Scholar] [CrossRef]
- Ross, M.H. Length of life and nutrition in the rat. J. Nutr. 1961, 75, 197–210. [Google Scholar] [CrossRef]
- Weindruch, R.; Walford, R.L. Dietary restriction in mice beginning at 1 year of age: Effect on life-span and spontaneous cancer incidence. Science 1982, 215, 1415–1418. [Google Scholar] [CrossRef]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol.-Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Darcy, J.; Tseng, Y.-H. ComBATing aging—Does increased brown adipose tissue activity confer longevity? GeroScience 2019, 41, 285–296. [Google Scholar] [CrossRef]
- Zoico, E.; Rubele, S.; De Caro, A.; Nori, N.; Mazzali, G.; Fantin, F.; Rossi, A.; Zamboni, M. Brown and Beige Adipose Tissue and Aging. Front. Endocrinol. 2019, 10, 368. [Google Scholar] [CrossRef]
- Fabbiano, S.; Suárez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Baumeier, C.; Kaiser, D.; Heeren, J.; Scheja, L.; John, C.; Weise, C.; Eravci, M.; Lagerpusch, M.; Schulze, G.; Joost, H.-G.; et al. Caloric restriction and intermittent fasting alter hepatic lipid droplet proteome and diacylglycerol species and prevent diabetes in NZO mice. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2015, 1851, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Lizardo, D.Y.; Lin, Y.L.; Gokcumen, O.; Atilla-Gokcumen, G.E. Regulation of lipids is central to replicative senescence. Mol. Biosyst. 2017, 13, 498–509. [Google Scholar] [CrossRef]
- Bartke, A.; Darcy, J. GH and ageing: Pitfalls and new insights. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 113–125. [Google Scholar] [CrossRef]
- Vitale, G.; Salvioli, S.; Franceschi, C. Oxidative stress and the ageing endocrine system. Nat. Rev. Endocrinol. 2013, 9, 228–240. [Google Scholar] [CrossRef]
- Vitale, G.; Cesari, M.; Mari, D. Aging of the endocrine system and its potential impact on sarcopenia. Eur. J. Intern. Med. 2016, 35, 10–15. [Google Scholar] [CrossRef]
- Sadria, M.; Layton, A.T. Interactions among mTORC, AMPK and SIRT: A computational model for cell energy balance and metabolism. Cell Commun. Signal. 2021, 19, 57. [Google Scholar] [CrossRef] [PubMed]
- Ralhan, I.; Chang, C.L.; Lippincott-Schwartz, J.; Ioannou, M.S. Lipid droplets in the nervous system. J. Cell Biol. 2021, 220, e202102136. [Google Scholar] [CrossRef]
- Yang, D.S.; Stavrides, P.; Saito, M.; Kumar, A.; Rodriguez-Navarro, J.A.; Pawlik, M.; Huo, C.; Walkley, S.U.; Saito, M.; Cuervo, A.M.; et al. Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: Prevention by correcting lysosomal proteolytic deficits. Brain 2014, 137, 3300–3318. [Google Scholar] [CrossRef]
- Derk, J.; Bermudez Hernandez, K.; Rodriguez, M.; He, M.; Koh, H.; Abedini, A.; Li, H.; Fenyo, D.; Schmidt, A.M. Diaphanous 1 (DIAPH1) is Highly Expressed in the Aged Human Medial Temporal Cortex and Upregulated in Myeloid Cells during Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, L.K.; Dufresne, M.; Joppe, S.E.; Petryszyn, S.; Aumont, A.; Calon, F.; Barnabe-Heider, F.; Furtos, A.; Parent, M.; Chaurand, P.; et al. Aberrant Lipid Metabolism in the Forebrain Niche Suppresses Adult Neural Stem Cell Proliferation in an Animal Model of Alzheimer’s Disease. Cell Stem Cell 2015, 17, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef]
- Brekk, O.R.; Honey, J.R.; Lee, S.; Hallett, P.J.; Isacson, O. Cell type-specific lipid storage changes in Parkinson’s disease patient brains are recapitulated by experimental glycolipid disturbance. Proc. Natl. Acad. Sci. USA 2020, 117, 27646–27654. [Google Scholar] [CrossRef]
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef] [PubMed]
- Yako, T.; Otsu, W.; Nakamura, S.; Shimazawa, M.; Hara, H. Lipid Droplet Accumulation Promotes RPE Dysfunction. Int. J. Mol. Sci. 2022, 23, 1790. [Google Scholar] [CrossRef]
- Arbaizar-Rovirosa, M.; Gallizioli, M.; Pedragosa, J.; Lozano, J.J.; Casal, C.; Pol, A.; Planas, A.M. Age-dependent lipid droplet-rich microglia worsen stroke outcome in old mice. bioRxiv 2022, 2022.2003.2014.484305. [Google Scholar] [CrossRef]
- Plakkal Ayyappan, J.; Paul, A.; Goo, Y.H. Lipid droplet-associated proteins in atherosclerosis (Review). Mol. Med. Rep. 2016, 13, 4527–4534. [Google Scholar] [CrossRef]
- Sukhorukov, V.N.; Khotina, V.A.; Chegodaev, Y.S.; Ivanova, E.; Sobenin, I.A.; Orekhov, A.N. Lipid Metabolism in Macrophages: Focus on Atherosclerosis. Biomedicines 2020, 8, 262. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Reue, K.; Abumrad, N.A.; Bickel, P.E.; Cohen, S.; Fisher, E.A.; Galis, Z.S.; Granneman, J.G.; Lewandowski, E.D.; Murphy, R.; et al. Deciphering the Role of Lipid Droplets in Cardiovascular Disease: A Report from the 2017 National Heart, Lung, and Blood Institute Workshop. Circulation 2018, 138, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Gao, F.; Zhang, Y.; Chen, T.; Xu, C. Lipid Droplet-Associated Proteins in Cardiomyopathy. Ann. Nutr. Metab. 2022, 78, 1–13. [Google Scholar] [CrossRef]
- Al Saedi, A.; Debruin, D.A.; Hayes, A.; Hamrick, M. Lipid metabolism in sarcopenia. Bone 2022, 164, 116539. [Google Scholar] [CrossRef]
- Conte, M.; Vasuri, F.; Trisolino, G.; Bellavista, E.; Santoro, A.; Degiovanni, A.; Martucci, E.; D’Errico-Grigioni, A.; Caporossi, D.; Capri, M.; et al. Increased Plin2 expression in human skeletal muscle is associated with sarcopenia and muscle weakness. PLoS ONE 2013, 8, e73709. [Google Scholar] [CrossRef]
- Weyand, C.M.; Wu, B.; Goronzy, J.J. The metabolic signature of T cells in rheumatoid arthritis. Curr. Opin. Rheumatol. 2020, 32, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Naiff, P.F.; Kuckelhaus, S.A.S.; Corazza, D.; Leite, L.M.; Couto, S.; deOliveira, M.S.; Santiago, L.M.; Silva, L.F.; Oliveira, L.A.; Grisi, D.C.; et al. Quantification of lipid bodies in monocytes from patients with periodontitis. Clin. Exp. Dent. Res. 2021, 7, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.H.; Kim, H.J.; Joo, J.Y.; Lee, J.Y.; Lee, J.H.; Park, H.R. Periodontal Pathogens Promote Foam Cell Formation by Blocking Lipid Efflux. J. Dent. Res. 2021, 100, 1367–1377. [Google Scholar] [CrossRef]
- McGee-Lawrence, M.E.; Carpio, L.R.; Schulze, R.J.; Pierce, J.L.; McNiven, M.A.; Farr, J.N.; Khosla, S.; Oursler, M.J.; Westendorf, J.J. Hdac3 Deficiency Increases Marrow Adiposity and Induces Lipid Storage and Glucocorticoid Metabolism in Osteochondroprogenitor Cells. J. Bone Miner. Res. 2016, 31, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Rendina-Ruedy, E.; Rosen, C.J. Lipids in the Bone Marrow: An Evolving Perspective. Cell Metab. 2020, 31, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Lippiello, L.; Walsh, T.; Fienhold, M. The association of lipid abnormalities with tissue pathology in human osteoarthritic articular cartilage. Metabolism 1991, 40, 571–576. [Google Scholar] [CrossRef]
- Lee, S.W.; Rho, J.H.; Lee, S.Y.; Chung, W.T.; Oh, Y.J.; Kim, J.H.; Yoo, S.H.; Kwon, W.Y.; Bae, J.Y.; Seo, S.Y.; et al. Dietary fat-associated osteoarthritic chondrocytes gain resistance to lipotoxicity through PKCK2/STAMP2/FSP27. Bone Res. 2018, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, A.M.; Nieminen, P. Fatty Acids and Oxylipins in Osteoarthritis and Rheumatoid Arthritis-a Complex Field with Significant Potential for Future Treatments. Curr. Rheumatol. Rep. 2021, 23, 41. [Google Scholar] [CrossRef]
- Tong, X.; Liu, S.; Stein, R.; Imai, Y. Lipid Droplets’ Role in the Regulation of beta-Cell Function and beta-Cell Demise in Type 2 Diabetes. Endocrinology 2022, 163, bqac007. [Google Scholar] [CrossRef]
- Tong, X.; Stein, R. Lipid Droplets Protect Human beta-Cells From Lipotoxicity-Induced Stress and Cell Identity Changes. Diabetes 2021, 70, 2595–2607. [Google Scholar] [CrossRef]
- He, Y.; Su, Y.; Duan, C.; Wang, S.; He, W.; Zhang, Y.; An, X.; He, M. Emerging role of aging in the progression of NAFLD to HCC. Ageing Res. Rev. 2023, 84, 101833. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.; Macdonald, A.J.; Greig, C.A.; Ross, J.A.; Fearon, K.C. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J. Cachexia Sarcopenia Muscle 2011, 2, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Nardi, F.; Fitchev, P.; Brooks, K.M.; Franco, O.E.; Cheng, K.; Hayward, S.W.; Welte, M.A.; Crawford, S.E. Lipid droplet velocity is a microenvironmental sensor of aggressive tumors regulated by V-ATPase and PEDF. Lab. Investig. 2019, 99, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Hamsanathan, S.; Gurkar, A.U. Lipids as Regulators of Cellular Senescence. Front. Physiol. 2022, 13, 796850. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Kruger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077.e1068. [Google Scholar] [CrossRef]
- Chee, W.Y.; Kurahashi, Y.; Kim, J.; Miura, K.; Okuzaki, D.; Ishitani, T.; Kajiwara, K.; Nada, S.; Okano, H.; Okada, M. beta-catenin-promoted cholesterol metabolism protects against cellular senescence in naked mole-rat cells. Commun. Biol. 2021, 4, 357. [Google Scholar] [CrossRef]
- Justesen, J.; Stenderup, K.; Ebbesen, E.N.; Mosekilde, L.; Steiniche, T.; Kassem, M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2001, 2, 165–171. [Google Scholar] [CrossRef]
- Verma, S.; Rajaratnam, J.H.; Denton, J.; Hoyland, J.A.; Byers, R.J. Adipocytic proportion of bone marrow is inversely related to bone formation in osteoporosis. J. Clin. Pathol. 2002, 55, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.J.; Bouxsein, M.L. Mechanisms of disease: Is osteoporosis the obesity of bone? Nat. Clin. Pract. Rheumatol. 2006, 2, 35–43. [Google Scholar] [CrossRef]
- Rendina-Ruedy, E.; Guntur, A.R.; Rosen, C.J. Intracellular lipid droplets support osteoblast function. Adipocyte 2017, 6, 250–258. [Google Scholar] [CrossRef]
- Diascro, D.D.; Vogel, R.L.; Johnson, T.E.; Witherup, K.M.; Pitzenberger, S.M.; Rutledge, S.J.; Prescott, D.J.; Rodan, G.A.; Schmidt, A. High fatty acid content in rabbit serum is responsible for the differentiation of osteoblasts into adipocyte-like cells. J. Bone Miner. Res. 1998, 13, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Sebo, Z.L.; Rendina-Ruedy, E.; Ables, G.P.; Lindskog, D.M.; Rodeheffer, M.S.; Fazeli, P.K.; Horowitz, M.C. Bone Marrow Adiposity: Basic and Clinical Implications. Endocr. Rev. 2019, 40, 1187–1206. [Google Scholar] [CrossRef]
- Maurin, A.C.; Chavassieux, P.M.; Frappart, L.; Delmas, P.D.; Serre, C.M.; Meunier, P.J. Influence of mature adipocytes on osteoblast proliferation in human primary cocultures. Bone 2000, 26, 485–489. [Google Scholar] [CrossRef]
- Elbaz, A.; Wu, X.; Rivas, D.; Gimble, J.M.; Duque, G. Inhibition of fatty acid biosynthesis prevents adipocyte lipotoxicity on human osteoblasts in vitro. J. Cell. Mol. Med. 2010, 14, 982–991. [Google Scholar] [CrossRef]
- Backesjo, C.M.; Li, Y.; Lindgren, U.; Haldosen, L.A. Activation of Sirt1 decreases adipocyte formation during osteoblast differentiation of mesenchymal stem cells. Cells Tissues Organs 2009, 189, 93–97. [Google Scholar] [CrossRef]
- Najt, C.P.; Khan, S.A.; Heden, T.D.; Witthuhn, B.A.; Perez, M.; Heier, J.L.; Mead, L.E.; Franklin, M.P.; Karanja, K.K.; Graham, M.J.; et al. Lipid Droplet-Derived Monounsaturated Fatty Acids Traffic via PLIN5 to Allosterically Activate SIRT1. Mol. Cell 2020, 77, 810–824 e818. [Google Scholar] [CrossRef]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPARgamma-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef]
- Moerman, E.J.; Teng, K.; Lipschitz, D.A.; Lecka-Czernik, B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: The role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 2004, 3, 379–389. [Google Scholar] [CrossRef]
- Gong, J.; Sun, Z.; Wu, L.; Xu, W.; Schieber, N.; Xu, D.; Shui, G.; Yang, H.; Parton, R.G.; Li, P. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J. Cell Biol. 2011, 195, 953–963. [Google Scholar] [CrossRef]
- Wang, F.; Ren, S.Y.; Chen, J.F.; Liu, K.; Li, R.X.; Li, Z.F.; Hu, B.; Niu, J.Q.; Xiao, L.; Chan, J.R.; et al. Myelin degeneration and diminished myelin renewal contribute to age-related deficits in memory. Nat. Neurosci. 2020, 23, 481–486. [Google Scholar] [CrossRef]
- Farokhian, F.; Yang, C.; Beheshti, I.; Matsuda, H.; Wu, S. Age-Related Gray and White Matter Changes in Normal Adult Brains. Aging Dis. 2017, 8, 899–909. [Google Scholar] [CrossRef]
- Capilla-Gonzalez, V.; Cebrian-Silla, A.; Guerrero-Cazares, H.; Garcia-Verdugo, J.M.; Quiñones-Hinojosa, A. Age-related changes in astrocytic and ependymal cells of the subventricular zone. Glia 2014, 62, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Kabaso, D.; Coskren, P.J.; Henry, B.I.; Hof, P.R.; Wearne, S.L. The electrotonic structure of pyramidal neurons contributing to prefrontal cortical circuits in macaque monkeys is significantly altered in aging. Cereb. Cortex 2009, 19, 2248–2268. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Sampson, E.L. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid Res. 1965, 6, 537–544. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X.; Wang, J.; Wang, X.; Chen, W.; Lu, N.; Siniossoglou, S.; Yao, Z.; Liu, K. Rewiring Neuronal Glycerolipid Metabolism Determines the Extent of Axon Regeneration. Neuron 2020, 105, 276–292.e275. [Google Scholar] [CrossRef]
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011, 14, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e1514. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Shimabukuro, M.K.; Langhi, L.G.; Cordeiro, I.; Brito, J.M.; Batista, C.M.; Mattson, M.P.; Mello Coelho, V. Lipid-laden cells differentially distributed in the aging brain are functionally active and correspond to distinct phenotypes. Sci. Rep. 2016, 6, 23795. [Google Scholar] [CrossRef]
- Bresgen, N.; Jaksch, H.; Bauer, H.C.; Eckl, P.; Krizbai, I.; Tempfer, H. Astrocytes are more resistant than cerebral endothelial cells toward geno- and cytotoxicity mediated by short-term oxidative stress. J. Neurosci. Res. 2006, 84, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, M.L.; Mertsch, K.; Giese, H.; Muller, S.; Sporbert, A.; Hickel, B.; Blasig, I.E. Astrocytes enhance radical defence in capillary endothelial cells constituting the blood-brain barrier. FEBS Lett. 1999, 449, 241–244. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletic-Savatic, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737.e716. [Google Scholar] [CrossRef]
- Xu, L.; Pu, J. Alpha-Synuclein in Parkinson’s Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Park. Dis. 2016, 2016, 1720621. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Loov, C.; Scherzer, C.R.; Hyman, B.T.; Breakefield, X.O.; Ingelsson, M. alpha-Synuclein in Extracellular Vesicles: Functional Implications and Diagnostic Opportunities. Cell. Mol. Neurobiol. 2016, 36, 437–448. [Google Scholar] [CrossRef]
- Gasser, T.; Hardy, J.; Mizuno, Y. Milestones in PD genetics. Mov. Disord. 2011, 26, 1042–1048. [Google Scholar] [CrossRef]
- Islimye, E.; Girard, V.; Gould, A.P. Functions of Stress-Induced Lipid Droplets in the Nervous System. Front. Cell Dev. Biol. 2022, 10, 863907. [Google Scholar] [CrossRef]
- Taran, A.S.; Shuvalova, L.D.; Lagarkova, M.A.; Alieva, I.B. Huntington’s Disease-An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components. Cells 2020, 9, 1514. [Google Scholar] [CrossRef]
- Churkina Taran, A.S.; Shakhov, A.S.; Kotlobay, A.A.; Alieva, I.B. Huntingtin and Other Neurodegeneration-Associated Proteins in the Development of Intracellular Pathologies: Potential Target Search for Therapeutic Intervention. Int. J. Mol. Sci. 2022, 23, 5533. [Google Scholar] [CrossRef]
- Gruber, A.; Hornburg, D.; Antonin, M.; Krahmer, N.; Collado, J.; Schaffer, M.; Zubaite, G.; Lüchtenborg, C.; Sachsenheimer, T.; Brügger, B.; et al. Molecular and structural architecture of polyQ aggregates in yeast. Proc. Natl. Acad. Sci. USA 2018, 115, E3446–E3453. [Google Scholar] [CrossRef]
- Cyske, Z.; Gaffke, L.; Pierzynowska, K.; Węgrzyn, G. Tubulin Cytoskeleton in Neurodegenerative Diseases-not Only Primary Tubulinopathies. Cell. Mol. Neurobiol. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Ketut, S.; Pande, D.; Made Siswadi, S.; Kuswardhani, R.A.T. Age is an Important Risk Factor for Type 2 Diabetes Mellitus and Cardiovascular Diseases. In Glucose Tolerance; Sureka, C., Ed.; IntechOpen: Rijeka, Croatia, 2012; p. Ch. 5. [Google Scholar]
- Han, S.K.; Baik, S.K.; Kim, M.Y. Non-alcoholic fatty liver disease: Definition and subtypes. Clin. Mol. Hepatol. 2022, 29, S5–S16. [Google Scholar] [CrossRef]
- Guo, X.; Yin, X.; Liu, Z.; Wang, J. Non-Alcoholic Fatty Liver Disease (NAFLD) Pathogenesis and Natural Products for Prevention and Treatment. Int. J. Mol. Sci. 2022, 23, 5489. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Lau, L.F. CCN1/CYR61: The very model of a modern matricellular protein. Cell. Mol. Life Sci. 2011, 68, 3149–3163. [Google Scholar] [CrossRef]
- Kim, K.H.; Won, J.H.; Cheng, N.; Lau, L.F. The matricellular protein CCN1 in tissue injury repair. J. Cell Commun. Signal. 2018, 12, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Sun, Y.; Xue, H.; Chen, L.; Gu, C.; Shao, J.; Lu, R.; Luo, X.; Wei, J.; Ma, X.; et al. CCN1 promotes hepatic steatosis and inflammation in non-alcoholic steatohepatitis. Sci. Rep. 2020, 10, 3201. [Google Scholar] [CrossRef]
- Kim, K.H.; Chen, C.C.; Monzon, R.I.; Lau, L.F. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell. Biol. 2013, 33, 2078–2090. [Google Scholar] [CrossRef]
- Quan, T.; Qin, Z.; Voorhees, J.J.; Fisher, G.J. Cysteine-rich protein 61 (CCN1) mediates replicative senescence-associated aberrant collagen homeostasis in human skin fibroblasts. J. Cell Biochem. 2012, 113, 3011–3018. [Google Scholar] [CrossRef]
- Qin, Z.; Robichaud, P.; He, T.; Fisher, G.J.; Voorhees, J.J.; Quan, T. Oxidant exposure induces cysteine-rich protein 61 (CCN1) via c-Jun/AP-1 to reduce collagen expression in human dermal fibroblasts. PLoS ONE 2014, 9, e115402. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; Steffen, B.T.; Van de Leur, E.; Haas, U.; Tihaa, L.; Friedman, S.L.; Weiskirchen, R. CCN1/CYR61 overexpression in hepatic stellate cells induces ER stress-related apoptosis. Cell. Signal. 2016, 28, 34–42. [Google Scholar] [CrossRef]
- Cheng, N.Y.; Kim, K.H.; Lau, L.F. Senescent hepatic stellate cells promote liver regeneration through IL-6 and ligands of CXCR2. JCI Insight 2022, 7, e158207. [Google Scholar] [CrossRef]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef]
- Duvigneau, J.C.; Luis, A.; Gorman, A.M.; Samali, A.; Kaltenecker, D.; Moriggl, R.; Kozlov, A.V. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine 2019, 124, 154577. [Google Scholar] [CrossRef]
- Long, Z.; Cao, M.; Su, S.H.; Wu, G.Y.; Meng, F.S.; Wu, H.; Liu, J.Z.; Yu, W.H.; Atabai, K.; Wang, X. Inhibition of hepatocyte nuclear factor 1b induces hepatic steatosis through DPP4/NOX1-mediated regulation of superoxide. Free Radic. Biol. Med. 2017, 113, 71–83. [Google Scholar] [CrossRef]
- Bellanné-Chantelot, C.; Clauin, S.; Chauveau, D.; Collin, P.; Daumont, M.; Douillard, C.; Dubois-Laforgue, D.; Dusselier, L.; Gautier, J.F.; Jadoul, M.; et al. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 2005, 54, 3126–3132. [Google Scholar] [CrossRef]
- Bonnycastle, L.L.; Willer, C.J.; Conneely, K.N.; Jackson, A.U.; Burrill, C.P.; Watanabe, R.M.; Chines, P.S.; Narisu, N.; Scott, L.J.; Enloe, S.T.; et al. Common variants in maturity-onset diabetes of the young genes contribute to risk of type 2 diabetes in Finns. Diabetes 2006, 55, 2534–2540. [Google Scholar] [CrossRef]
- Barak, P.; Rai, A.; Rai, P.; Mallik, R. Quantitative optical trapping on single organelles in cell extract. Nat. Methods 2013, 10, 68–70. [Google Scholar] [CrossRef]
- Rai, P.; Kumar, M.; Sharma, G.; Barak, P.; Das, S.; Kamat, S.S.; Mallik, R. Kinesin-dependent mechanism for controlling triglyceride secretion from the liver. Proc. Natl. Acad. Sci. USA 2017, 114, 12958–12963. [Google Scholar] [CrossRef]
- Thiam, A.R.; Antonny, B.; Wang, J.; Delacotte, J.; Wilfling, F.; Walther, T.C.; Beck, R.; Rothman, J.E.; Pincet, F. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc. Natl. Acad. Sci. USA 2013, 110, 13244–13249. [Google Scholar] [CrossRef]
- Kumar, M.; Ojha, S.; Rai, P.; Joshi, A.; Kamat, S.S.; Mallik, R. Insulin activates intracellular transport of lipid droplets to release triglycerides from the liver. J. Cell Biol. 2019, 218, 3697–3713. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef]
- Korovila, I.; Höhn, A.; Jung, T.; Grune, T.; Ott, C. Reduced Liver Autophagy in High-Fat Diet Induced Liver Steatosis in New Zealand Obese Mice. Antioxidants 2021, 10, 501. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.W.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef]
- Unger, R.H.; Orci, L. Lipoapoptosis: Its mechanism and its diseases. Biochim. Biophys. Acta 2002, 1585, 202–212. [Google Scholar] [CrossRef]
- Akazawa, Y.; Nakao, K. Lipotoxicity pathways intersect in hepatocytes: Endoplasmic reticulum stress, c-Jun N-terminal kinase-1, and death receptors. Hepatol. Res. 2016, 46, 977–984. [Google Scholar] [CrossRef]
- Gaggini, M.; Ndreu, R.; Michelucci, E.; Rocchiccioli, S.; Vassalle, C. Ceramides as Mediators of Oxidative Stress and Inflammation in Cardiometabolic Disease. Int. J. Mol. Sci. 2022, 23, 2719. [Google Scholar] [CrossRef]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef]
- Gueraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Zhou, Q.; Huang, X.; Cao, P.; Wang, Y. Ferroptosis plays a novel role in nonalcoholic steatohepatitis pathogenesis. Front. Pharmacol. 2022, 13, 1055793. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef]
- Matsumoto, A.; Naito, M.; Itakura, H.; Ikemoto, S.; Asaoka, H.; Hayakawa, I.; Kanamori, H.; Aburatani, H.; Takaku, F.; Suzuki, H.; et al. Human macrophage scavenger receptors: Primary structure, expression, and localization in atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1990, 87, 9133–9137. [Google Scholar] [CrossRef]
- Moore, K.J.; Kunjathoor, V.V.; Koehn, S.L.; Manning, J.J.; Tseng, A.A.; Silver, J.M.; McKee, M.; Freeman, M.W. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J. Clin. Investig. 2005, 115, 2192–2201. [Google Scholar] [CrossRef]
- Gerrity, R.G. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am. J. Pathol. 1981, 103, 181–190. [Google Scholar] [PubMed]
- Tabas, I. Nonoxidative modifications of lipoproteins in atherogenesis. Annu. Rev. Nutr. 1999, 19, 123–139. [Google Scholar] [CrossRef]
- Pohl, A.; Devaux, P.F.; Herrmann, A. Function of prokaryotic and eukaryotic ABC proteins in lipid transport. Biochim. Biophys. Acta 2005, 1733, 29–52. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Rader, D.J. Macrophage reverse cholesterol transport: Key to the regression of atherosclerosis? Circulation 2006, 113, 2548–2555. [Google Scholar] [CrossRef]
- Chang, T.Y.; Chang, C.C.; Lin, S.; Yu, C.; Li, B.L.; Miyazaki, A. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and -2. Curr. Opin. Lipidol. 2001, 12, 289–296. [Google Scholar] [CrossRef]
- Yano, H.; Fujiwara, Y.; Horlad, H.; Pan, C.; Kai, K.; Niino, D.; Ohsawa, K.; Higashi, M.; Nosaka, K.; Okuno, Y.; et al. Blocking cholesterol efflux mechanism is a potential target for antilymphoma therapy. Cancer Sci. 2022, 113, 2129–2143. [Google Scholar] [CrossRef]
- Hynynen, R.; Suchanek, M.; Spandl, J.; Bäck, N.; Thiele, C.; Olkkonen, V.M. OSBP-related protein 2 is a sterol receptor on lipid droplets that regulates the metabolism of neutral lipids. J. Lipid Res. 2009, 50, 1305–1315. [Google Scholar] [CrossRef]
- Guyard, V.; Monteiro-Cardoso, V.F.; Omrane, M.; Sauvanet, C.; Houcine, A.; Boulogne, C.; Ben Mbarek, K.; Vitale, N.; Faklaris, O.; El Khallouki, N.; et al. ORP5 and ORP8 orchestrate lipid droplet biogenesis and maintenance at ER-mitochondria contact sites. J. Cell Biol. 2022, 221, 2107. [Google Scholar] [CrossRef]
- Anderson, A.; Campo, A.; Fulton, E.; Corwin, A.; Jerome, W.G., 3rd; O’Connor, M.S. 7-Ketocholesterol in disease and aging. Redox Biol. 2020, 29, 101380. [Google Scholar] [CrossRef]
- Ghzaiel, I.; Nury, T.; Zarrouk, A.; Vejux, A.; Lizard, G. Oxiapoptophagy in Age-Related Diseases. Comment on Ouyang et al. 7-Ketocholesterol Induces Oxiapoptophagy and Inhibits Osteogenic Differentiation in MC3T3-E1 Cells. Cells 2022, 11, 2882. [Google Scholar] [CrossRef]
- Monier, S.; Samadi, M.; Prunet, C.; Denance, M.; Laubriet, A.; Athias, A.; Berthier, A.; Steinmetz, E.; Jürgens, G.; Nègre-Salvayre, A.; et al. Impairment of the cytotoxic and oxidative activities of 7 beta-hydroxycholesterol and 7-ketocholesterol by esterification with oleate. Biochem. Biophys. Res. Commun. 2003, 303, 814–824. [Google Scholar] [CrossRef]
- Nury, T.; Zarrouk, A.; Yammine, A.; Mackrill, J.J.; Vejux, A.; Lizard, G. Oxiapoptophagy: A type of cell death induced by some oxysterols. Br. J. Pharmacol. 2021, 178, 3115–3123. [Google Scholar] [CrossRef]
- Ouyang, J.; Xiao, Y.; Ren, Q.; Huang, J.; Zhou, Q.; Zhang, S.; Li, L.; Shi, W.; Chen, Z.; Wu, L. 7-Ketocholesterol Induces Oxiapoptophagy and Inhibits Osteogenic Differentiation in MC3T3-E1 Cells. Cells 2022, 11, 2882. [Google Scholar] [CrossRef]
- Samadi, A.; Sabuncuoglu, S.; Samadi, M.; Isikhan, S.Y.; Chirumbolo, S.; Peana, M.; Lay, I.; Yalcinkaya, A.; Bjørklund, G. A Comprehensive Review on Oxysterols and Related Diseases. Curr. Med. Chem. 2021, 28, 110–136. [Google Scholar] [CrossRef]
- Lee-Rueckert, M.; Lappalainen, J.; Kovanen, P.T.; Escola-Gil, J.C. Lipid-Laden Macrophages and Inflammation in Atherosclerosis and Cancer: An Integrative View. Front. Cardiovasc. Med. 2022, 9, 777822. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Chawla, A. Alternative macrophage activation and metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef]
- Lathe, R.; Sapronova, A.; Kotelevtsev, Y. Atherosclerosis and Alzheimer--diseases with a common cause? Inflammation, oxysterols, vasculature. BMC Geriatr. 2014, 14, 36. [Google Scholar] [CrossRef]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef]
- Robichaud, S.; Rasheed, A.; Pietrangelo, A.; Kim, A.D.; Boucher, D.M.; Emerton, C.; Vijithakumar, V.; Gharibeh, L.; Fairman, G.; Mak, E.; et al. Autophagy Is Differentially Regulated in Leukocyte and Nonleukocyte Foam Cells during Atherosclerosis. Circ. Res. 2022, 130, 831–847. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Banke, N.H.; Wende, A.R.; Leone, T.C.; O’Donnell, J.M.; Abel, E.D.; Kelly, D.P.; Lewandowski, E.D. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ. Res. 2010, 107, 233–241. [Google Scholar] [CrossRef]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef]
- Ding, G.; Fu, M.; Qin, Q.; Lewis, W.; Kim, H.W.; Fukai, T.; Bacanamwo, M.; Chen, Y.E.; Schneider, M.D.; Mangelsdorf, D.J.; et al. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc. Res. 2007, 76, 269–279. [Google Scholar] [CrossRef]
- Amen, T.; Kaganovich, D. Small Molecule Screen Reveals Joint Regulation of Stress Granule Formation and Lipid Droplet Biogenesis. Front. Cell Dev. Biol. 2020, 8, 606111. [Google Scholar] [CrossRef]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef]
- Cui, X.; Wang, J.; Zhang, Y.; Wei, J.; Wang, Y. Plin5, a New Target in Diabetic Cardiomyopathy. Oxidative Med. Cell. Longev. 2022, 2022, 2122856. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, W.; Xu, R.; Wang, Z.; Zhang, X.; Wang, P.; Peng, K.; Li, M.; Li, J.; Tan, Y.; et al. Plin5 Bidirectionally Regulates Lipid Metabolism in Oxidative Tissues. Oxidative Med. Cell. Longev. 2022, 2022, 4594956. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef]
- Holzem, K.M.; Vinnakota, K.C.; Ravikumar, V.K.; Madden, E.J.; Ewald, G.A.; Dikranian, K.; Beard, D.A.; Efimov, I.R. Mitochondrial structure and function are not different between nonfailing donor and end-stage failing human hearts. Faseb J. 2016, 30, 2698–2707. [Google Scholar] [CrossRef]
- Pollak, N.M.; Schweiger, M.; Jaeger, D.; Kolb, D.; Kumari, M.; Schreiber, R.; Kolleritsch, S.; Markolin, P.; Grabner, G.F.; Heier, C.; et al. Cardiac-specific overexpression of perilipin 5 provokes severe cardiac steatosis via the formation of a lipolytic barrier. J. Lipid Res. 2013, 54, 1092–1102. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Gong, D.W.; O’Connell, K.A.; Dabkowski, E.R.; Hecker, P.A.; Ionica, N.; Konig, M.; Mahurkar, A.; Sun, Y.; et al. Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J. Lipid Res. 2013, 54, 953–965. [Google Scholar] [CrossRef]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, X.; Zhang, L.; Zhang, M.; Li, L.; Luo, D.; Zhong, Y. Perilipin5 protects against lipotoxicity and alleviates endoplasmic reticulum stress in pancreatic beta-cells. Nutr. Metab. 2019, 16, 50. [Google Scholar] [CrossRef]
- Cinato, M.; Mardani, I.; Miljanovic, A.; Drevinge, C.; Laudette, M.; Bollano, E.; Henricsson, M.; Tolo, J.; Bauza Thorbrugge, M.; Levin, M.; et al. Cardiac Plin5 interacts with SERCA2 and promotes calcium handling and cardiomyocyte contractility. Life Sci. Alliance 2023, 6, e202201690. [Google Scholar] [CrossRef]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef]
- Ukraintseva, S.V.; Yashin, A.I. Opposite Phenotypes of Cancer and Aging Arise from Alternative Regulation of Common Signaling Pathways. Ann. N. Y. Acad. Sci. 2003, 1010, 489–492. [Google Scholar] [CrossRef]
- Antunes, P.; Cruz, A.; Barbosa, J.; Bonifácio, V.D.B.; Pinto, S.N. Lipid Droplets in Cancer: From Composition and Role to Imaging and Therapeutics. Molecules 2022, 27, 991. [Google Scholar] [CrossRef]
- Li, Z.; Liu, H.; Luo, X. Lipid droplet and its implication in cancer progression. Am. J. Cancer Res. 2020, 10, 4112–4122. [Google Scholar]
- Lung, J.; Hung, M.-S.; Wang, T.-Y.; Chen, K.-L.; Luo, C.-W.; Jiang, Y.-Y.; Wu, S.-Y.; Lee, L.-W.; Lin, P.-Y.; Chen, F.-F.; et al. Lipid Droplets in Lung Cancers Are Crucial for the Cell Growth and Starvation Survival. Int. J. Mol. Sci. 2022, 23, 12533. [Google Scholar] [CrossRef]
- Shyu, P.; Wong, X.F.A.; Crasta, K.; Thibault, G. Dropping in on lipid droplets: Insights into cellular stress and cancer. Biosci. Rep. 2018, 38, BSR20180764. [Google Scholar] [CrossRef]
- Castelli, S.; De Falco, P.; Ciccarone, F.; Desideri, E.; Ciriolo, M.R. Lipid Catabolism and ROS in Cancer: A Bidirectional Liaison. Cancers 2021, 13, 5484. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Lian, Z.; Liao, R.; Chen, Y.; Qin, Y.; Wang, J.; Jiang, Q.; Wang, X.; Gong, J. Monoacylglycerol Lipase: A Novel Potential Therapeutic Target and Prognostic Indicator for Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 35784. [Google Scholar] [CrossRef]
- Patel, D.; Salloum, D.; Saqcena, M.; Chatterjee, A.; Mroz, V.; Ohh, M.; Foster, D.A. A Late G1 Lipid Checkpoint That Is Dysregulated in Clear Cell Renal Carcinoma Cells. J. Biol. Chem. 2017, 292, 936–944. [Google Scholar] [CrossRef]
- Qi, W.; Weber, C.R.; Wasland, K.; Roy, H.; Wali, R.; Joshi, S.; Savkovic, S.D. Tumor suppressor FOXO3 mediates signals from the EGF receptor to regulate proliferation of colonic cells. Am. J. Physiol. -Gastrointest. Liver Physiol. 2011, 300, G264–G272. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Bozza, P.T.; Tzizik, D.M.; Gray, J.P.; Cassara, J.; Dvorak, A.M.; Weller, P.F. Co-compartmentalization of MAP kinases and cytosolic phospholipase A2 at cytoplasmic arachidonate-rich lipid bodies. Am. J. Pathol. 1998, 152, 759–769. [Google Scholar] [PubMed]
- Yu, W.; Cassara, J.; Weller, P.F. Phosphatidylinositide 3-kinase localizes to cytoplasmic lipid bodies in human polymorphonuclear leukocytes and other myeloid-derived cells. Blood 2000, 95, 1078–1085. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Bozza, P.T.; Bakker-Abreu, I.; Navarro-Xavier, R.A.; Bandeira-Melo, C. Lipid body function in eicosanoid synthesis: An update. Prostaglandins Leukot. Essent. Fat. Acids (PLEFA) 2011, 85, 205–213. [Google Scholar] [CrossRef]
- Accioly, M.T.; Pacheco, P.; Maya-Monteiro, C.M.; Carrossini, N.; Robbs, B.K.; Oliveira, S.S.; Kaufmann, C.; Morgado-Diaz, J.A.; Bozza, P.T.; Viola, J.P. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008, 68, 1732–1740. [Google Scholar] [CrossRef]
- Johnson, A.M.; Kleczko, E.K.; Nemenoff, R.A. Eicosanoids in Cancer: New Roles in Immunoregulation. Front. Pharmacol. 2020, 11, 595498. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef]
- Hakumaki, J.M.; Kauppinen, R.A. 1H NMR visible lipids in the life and death of cells. Trends Biochem. Sci. 2000, 25, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, F.G. In vivo detection of apoptosis. J. Nucl. Med. 2008, 49 (Suppl. S2), 81S–95S. [Google Scholar] [CrossRef] [PubMed]
- Henique, C.; Mansouri, A.; Fumey, G.; Lenoir, V.; Girard, J.; Bouillaud, F.; Prip-Buus, C.; Cohen, I. Increased mitochondrial fatty acid oxidation is sufficient to protect skeletal muscle cells from palmitate-induced apoptosis. J. Biol. Chem. 2010, 285, 36818–36827. [Google Scholar] [CrossRef]
- Choi, S.E.; Jung, I.R.; Lee, Y.J.; Lee, S.J.; Lee, J.H.; Kim, Y.; Jun, H.S.; Lee, K.W.; Park, C.B.; Kang, Y. Stimulation of lipogenesis as well as fatty acid oxidation protects against palmitate-induced INS-1 beta-cell death. Endocrinology 2011, 152, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Brindle, K.M. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 2012, 19, 1561–1570. [Google Scholar] [CrossRef]
- Wang, J.-B.; Qi, L.-L.; Zheng, S.-D.; Wu, T.-X. Curcumin induces apoptosis through the mitochondria-mediated apoptotic pathway in HT-29 cells. J. Zhejiang Univ. Sci. B 2009, 10, 93–102. [Google Scholar] [CrossRef]
- Roy, M.; Chakraborty, S.; Siddiqi, M.; Bhattacharya, R.K. Induction of Apoptosis in Tumor Cells by Natural Phenolic Compounds. Asian Pac. J. Cancer Prev. APJCP 2002, 3, 61–67. [Google Scholar]
- Jiang, M.C.; Yang-Yen, H.F.; Yen, J.J.; Lin, J.K. Curcumin induces apoptosis in immortalized NIH 3T3 and malignant cancer cell lines. Nutr. Cancer 1996, 26, 111–120. [Google Scholar] [CrossRef]
- Cao, J.; Liu, Y.; Jia, L.; Zhou, H.M.; Kong, Y.; Yang, G.; Jiang, L.P.; Li, Q.J.; Zhong, L.F. Curcumin induces apoptosis through mitochondrial hyperpolarization and mtDNA damage in human hepatoma G2 cells. Free Radic. Biol. Med. 2007, 43, 968–975. [Google Scholar] [CrossRef]
- Zhang, I.; Cui, Y.; Amiri, A.; Ding, Y.; Campbell, R.E.; Maysinger, D. Pharmacological inhibition of lipid droplet formation enhances the effectiveness of curcumin in glioblastoma. Eur. J. Pharm. Biopharm. 2016, 100, 66–76. [Google Scholar] [CrossRef]
- Jin, C.; Yuan, P. Implications of lipid droplets in lung cancer: Associations with drug resistance. Oncol. Lett. 2020, 20, 2091–2104. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L. The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef]
- Song, J.H.; Tse, M.C.L.; Bellail, A.; Phuphanich, S.; Khuri, F.; Kneteman, N.M.; Hao, C. Lipid Rafts and Nonrafts Mediate Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Induced Apoptotic and Nonapoptotic Signals in Non–Small Cell Lung Carcinoma Cells. Cancer Res. 2007, 67, 6946–6955. [Google Scholar] [CrossRef]
- Zembroski, A.S.; Andolino, C.; Buhman, K.K.; Teegarden, D. Proteomic Characterization of Cytoplasmic Lipid Droplets in Human Metastatic Breast Cancer Cells. Front. Oncol. 2021, 11, 576326. [Google Scholar] [CrossRef] [PubMed]
- Sofi, F.; Abbate, R.; Gensini, G.F.; Casini, A. Accruing evidence on benefits of adherence to the Mediterranean diet on health: An updated systematic review and meta-analysis. Am. J. Clin. Nutr. 2010, 92, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.A.; Sansone, L.; Polletta, L.; Runci, A.; Rashid, M.M.; De Santis, E.; Vernucci, E.; Carnevale, I.; Tafani, M. Sirtuins and resveratrol-derived compounds: A model for understanding the beneficial effects of the Mediterranean diet. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 300–308. [Google Scholar] [CrossRef]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]




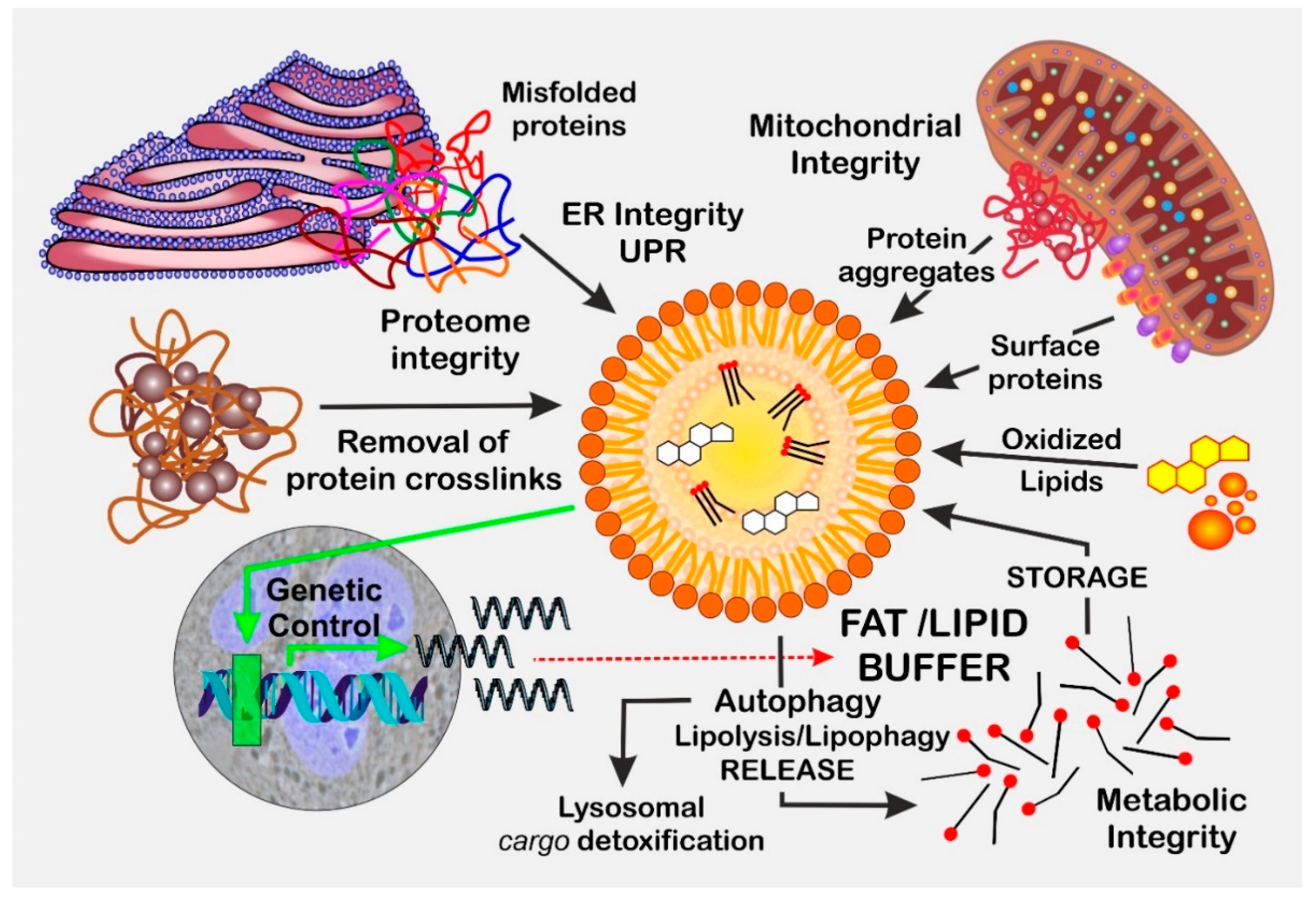{kind=link}
{kind=link}
{kind=link}
| Disease | Main Affected Cell Type/Tissue | References |
|---|---|---|
| Alzheimer’s disease | neurons, glia, myeloid cells, ependymal cells, astrocytes | [319,320,321,322,323] |
| Parkinson’s disease | neurons, microglia | [319,324,325] |
| Age-related macular degeneration | retinal pigment epithelium | [326] |
| Stroke | microglia | [327] |
| Atherosclerosis | Foam cells | [328,329] |
| Cardiovascular disease | myocardium | [330,331] |
| Sarcopenia | muscle cells | [332,333] |
| Rheumatoid arthritis | T-cells | [334] |
| Chronic obstructive pulmonary disease (COPD) | macrophages | [327] |
| Periodontitis | monocytes, macrophages | [335,336] |
| Osteopenia | osteoblasts, osteocytes | [337,338] |
| Osteoarthritis | chondrocytes, cartilage | [339,340,341] |
| Diabetes | β-cells | [342,343] |
| Liver disease (NAFLD) 1 | parenchymal hepatocytes | [344] 2 |
| Cancer | several | [343,345,346] |
| Senescence | several | [347,348,349] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bresgen, N.; Kovacs, M.; Lahnsteiner, A.; Felder, T.K.; Rinnerthaler, M. The Janus-Faced Role of Lipid Droplets in Aging: Insights from the Cellular Perspective. Biomolecules 2023, 13, 912. https://doi.org/10.3390/biom13060912
Bresgen N, Kovacs M, Lahnsteiner A, Felder TK, Rinnerthaler M. The Janus-Faced Role of Lipid Droplets in Aging: Insights from the Cellular Perspective. Biomolecules. 2023; 13(6):912. https://doi.org/10.3390/biom13060912
Chicago/Turabian StyleBresgen, Nikolaus, Melanie Kovacs, Angelika Lahnsteiner, Thomas Klaus Felder, and Mark Rinnerthaler. 2023. "The Janus-Faced Role of Lipid Droplets in Aging: Insights from the Cellular Perspective" Biomolecules 13, no. 6: 912. https://doi.org/10.3390/biom13060912
APA StyleBresgen, N., Kovacs, M., Lahnsteiner, A., Felder, T. K., & Rinnerthaler, M. (2023). The Janus-Faced Role of Lipid Droplets in Aging: Insights from the Cellular Perspective. Biomolecules, 13(6), 912. https://doi.org/10.3390/biom13060912