

Identification of Exosome-Related Genes Associated with Prognosis and Immune Infiltration Features in Head-Neck Squamous Cell Carcinoma
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
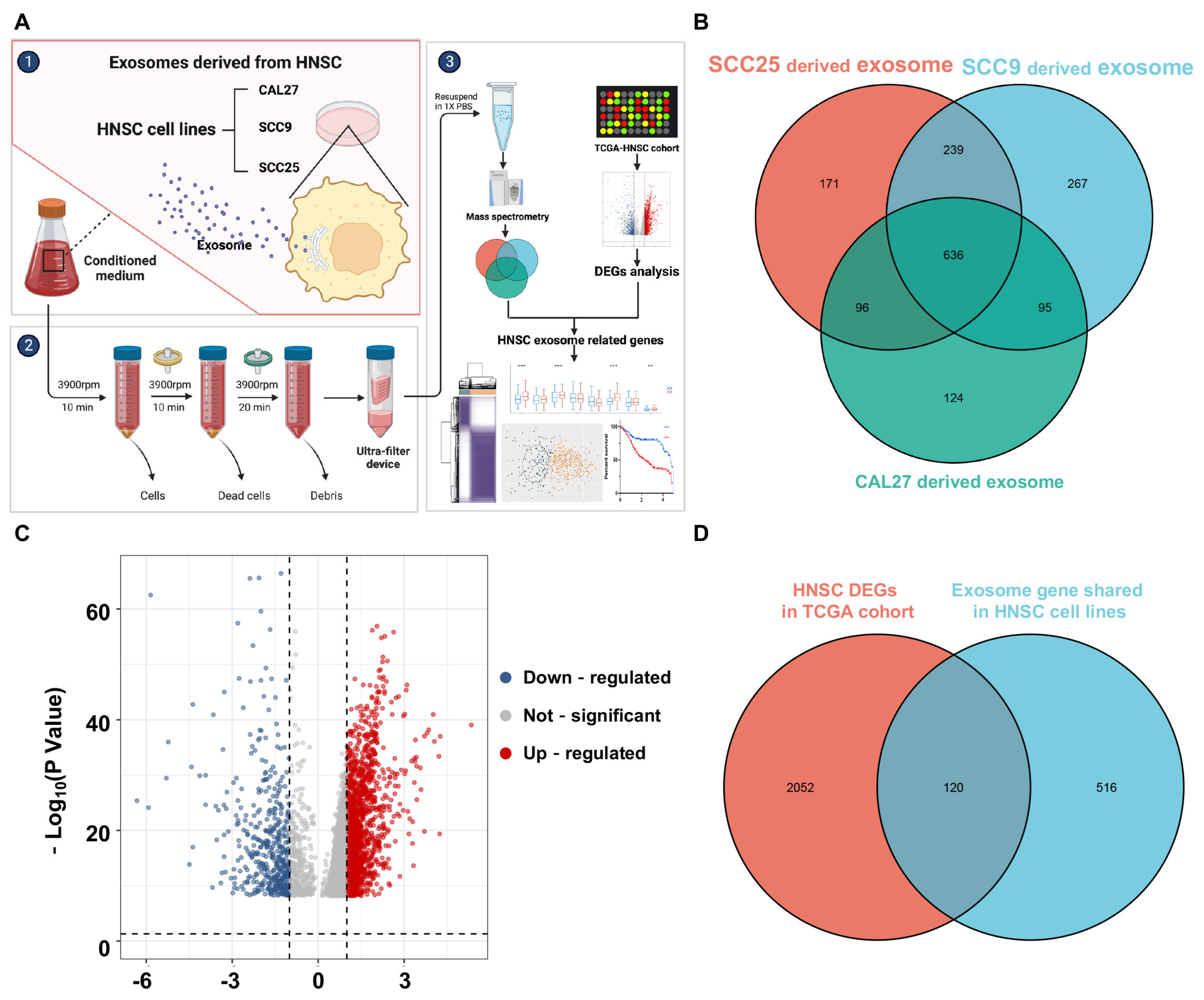
2.1. Differently Expressed Genes of HNSCC Samples
2.2. Isolation of HNSCC Exosome-Related Genes
2.3. Mass Spectrometry (MS)-Based Label-Free Quantitative Proteomics
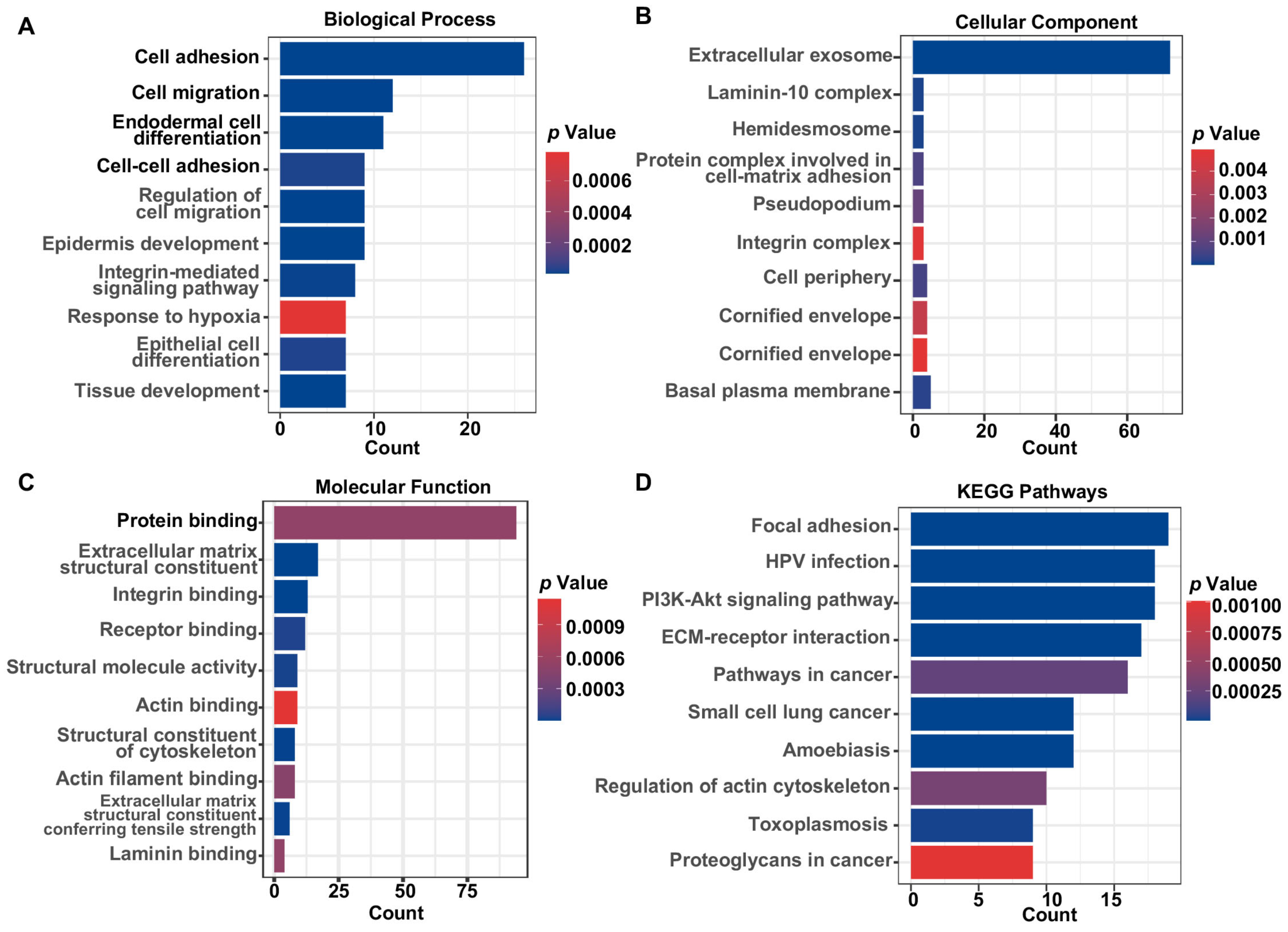
2.4. Enrichment Analysis and ssGSEA Analysis
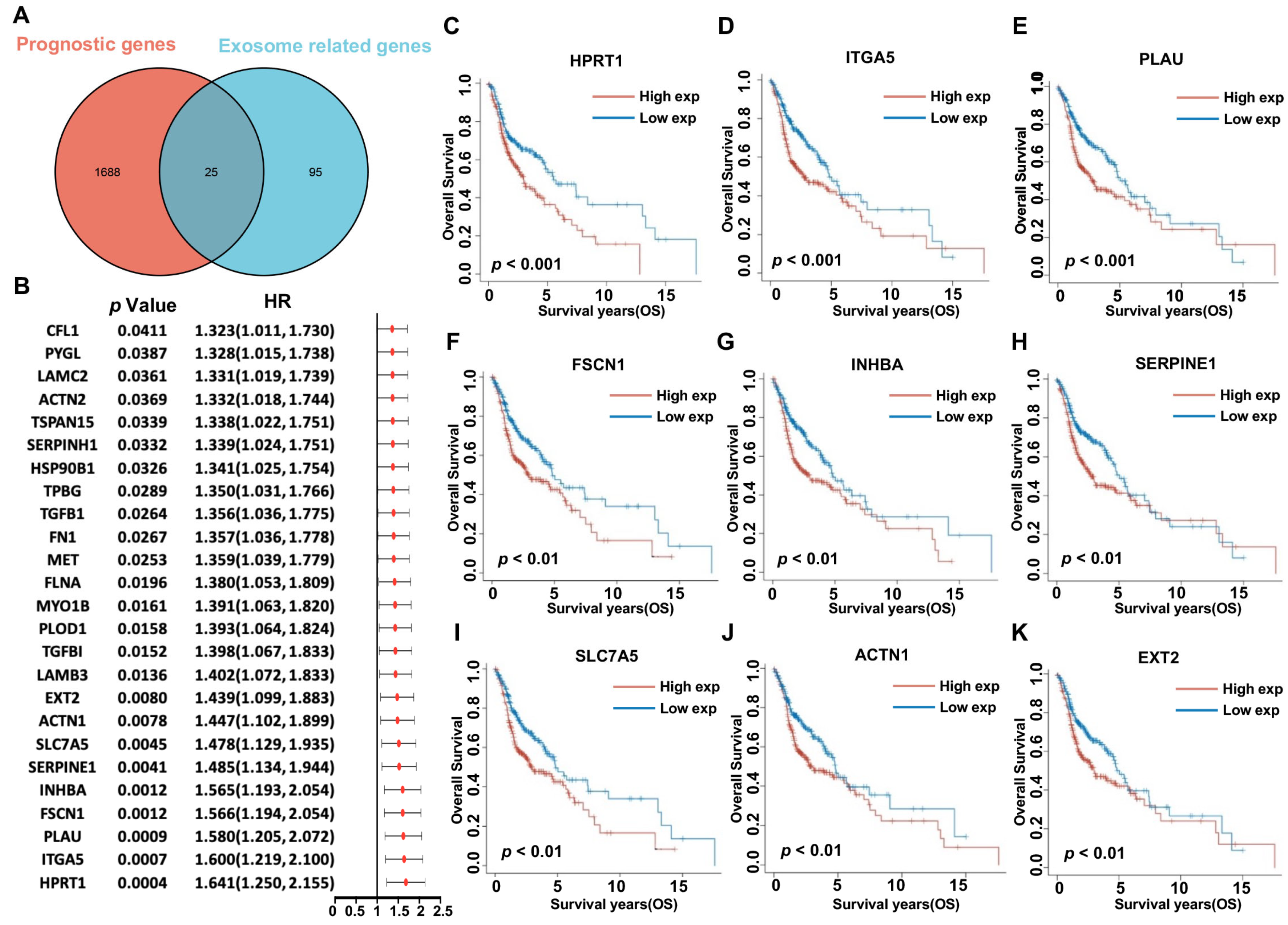
2.5. Identification of Prognostic HNSCC Exosome-Related Genes
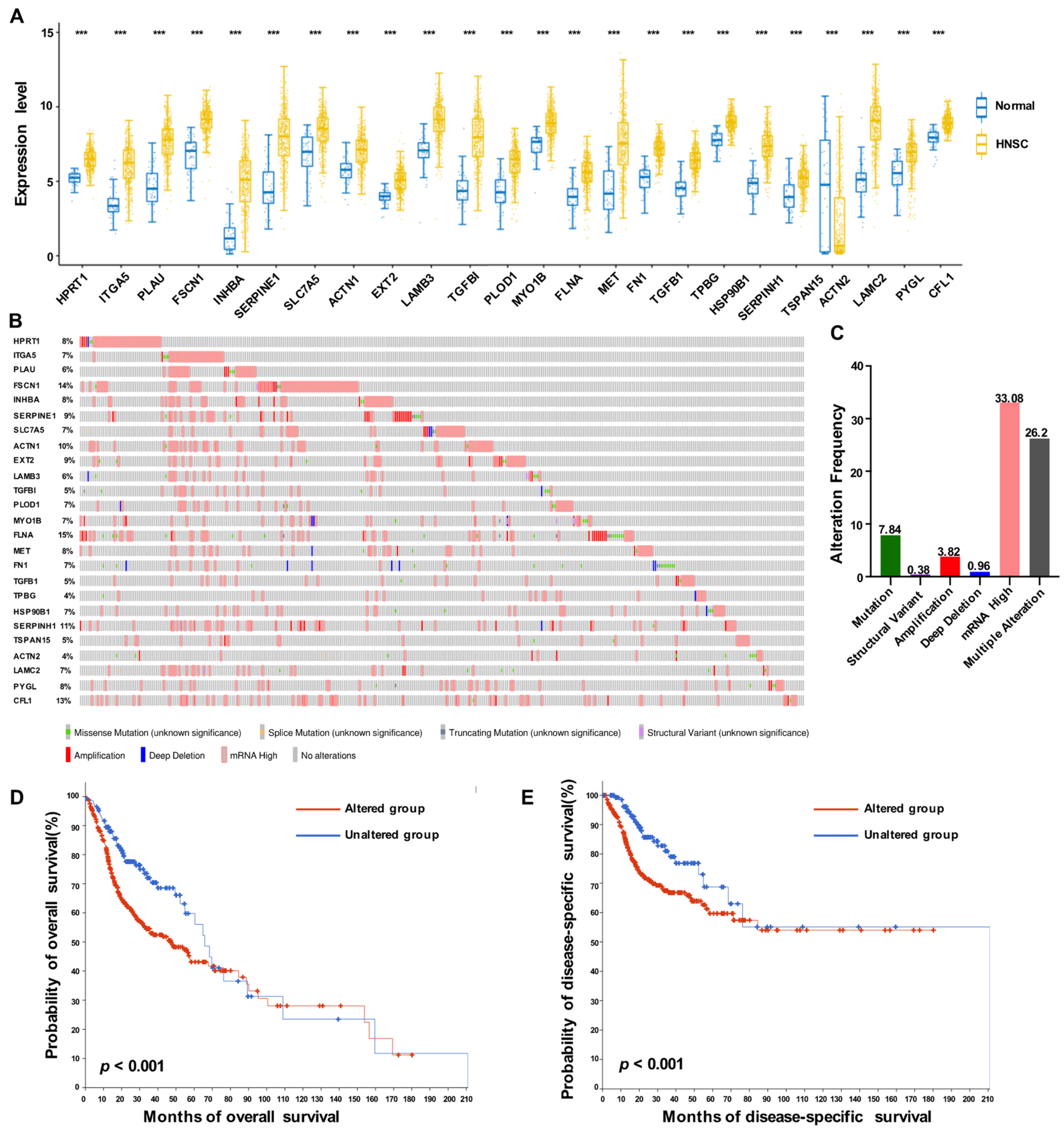
2.6. Exploration of Gene Alteration Landscape of Prognostic ERGs
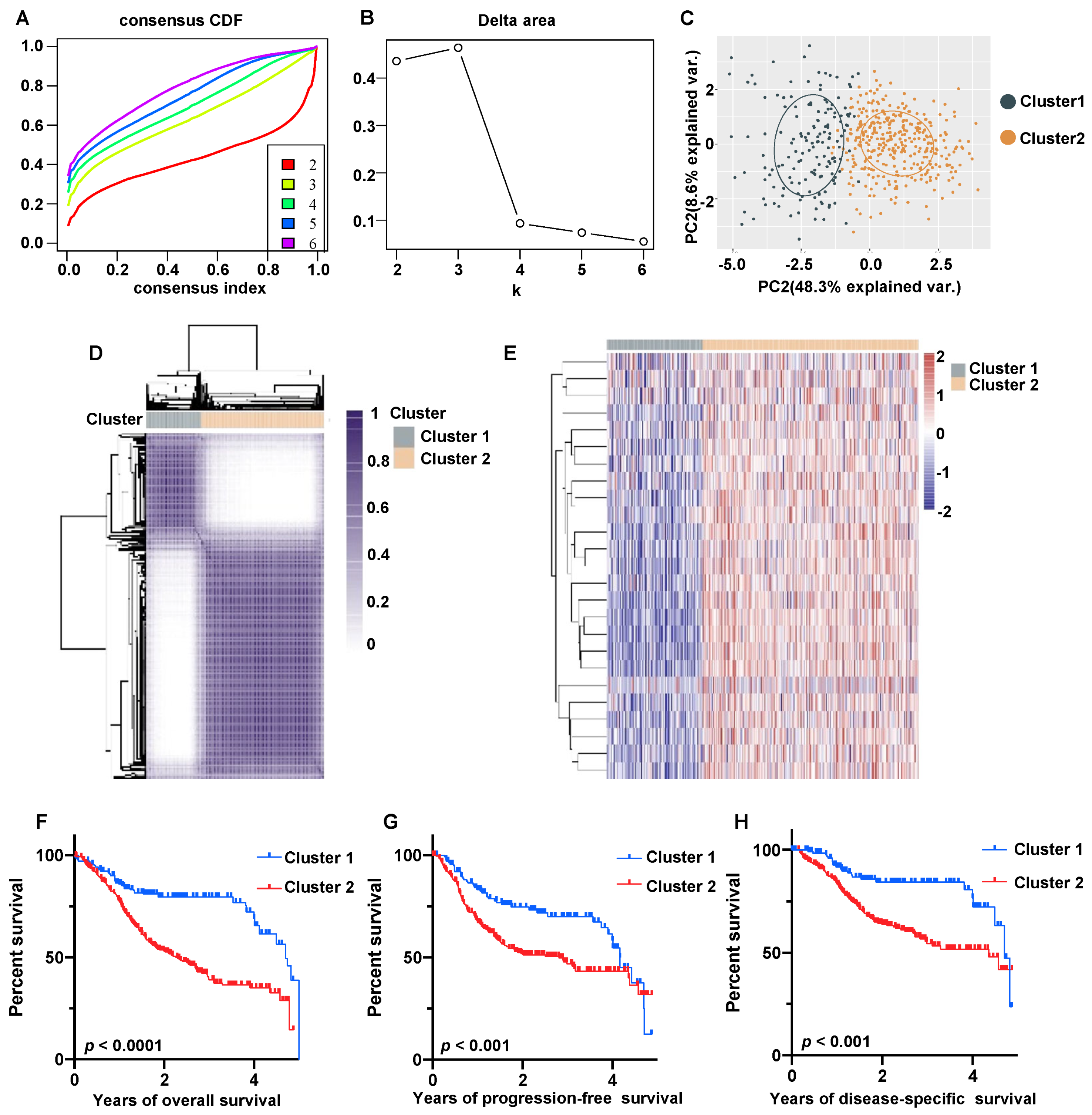
2.7. Construction of Molecular Subgroups Using Consensus Clustering
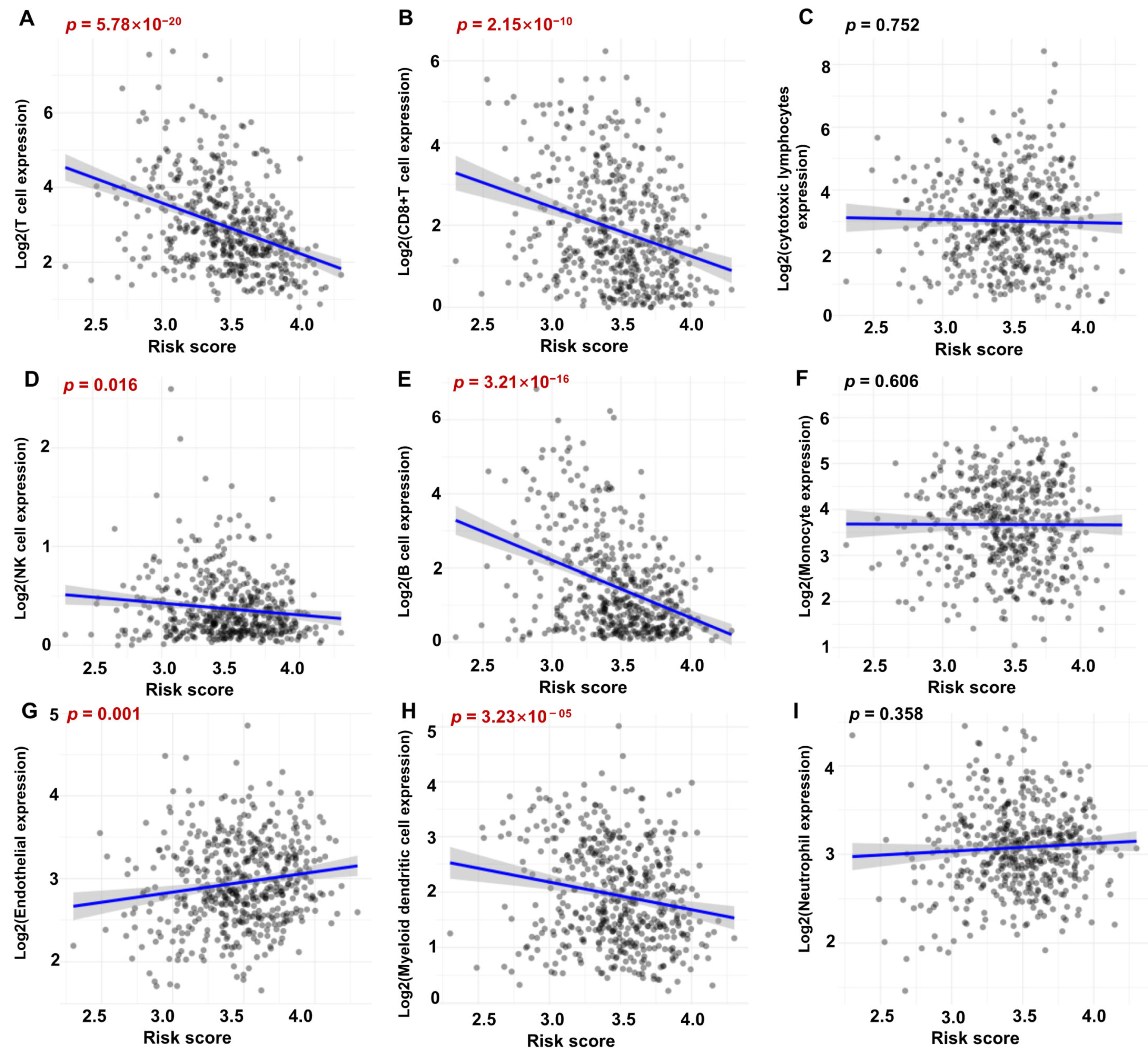
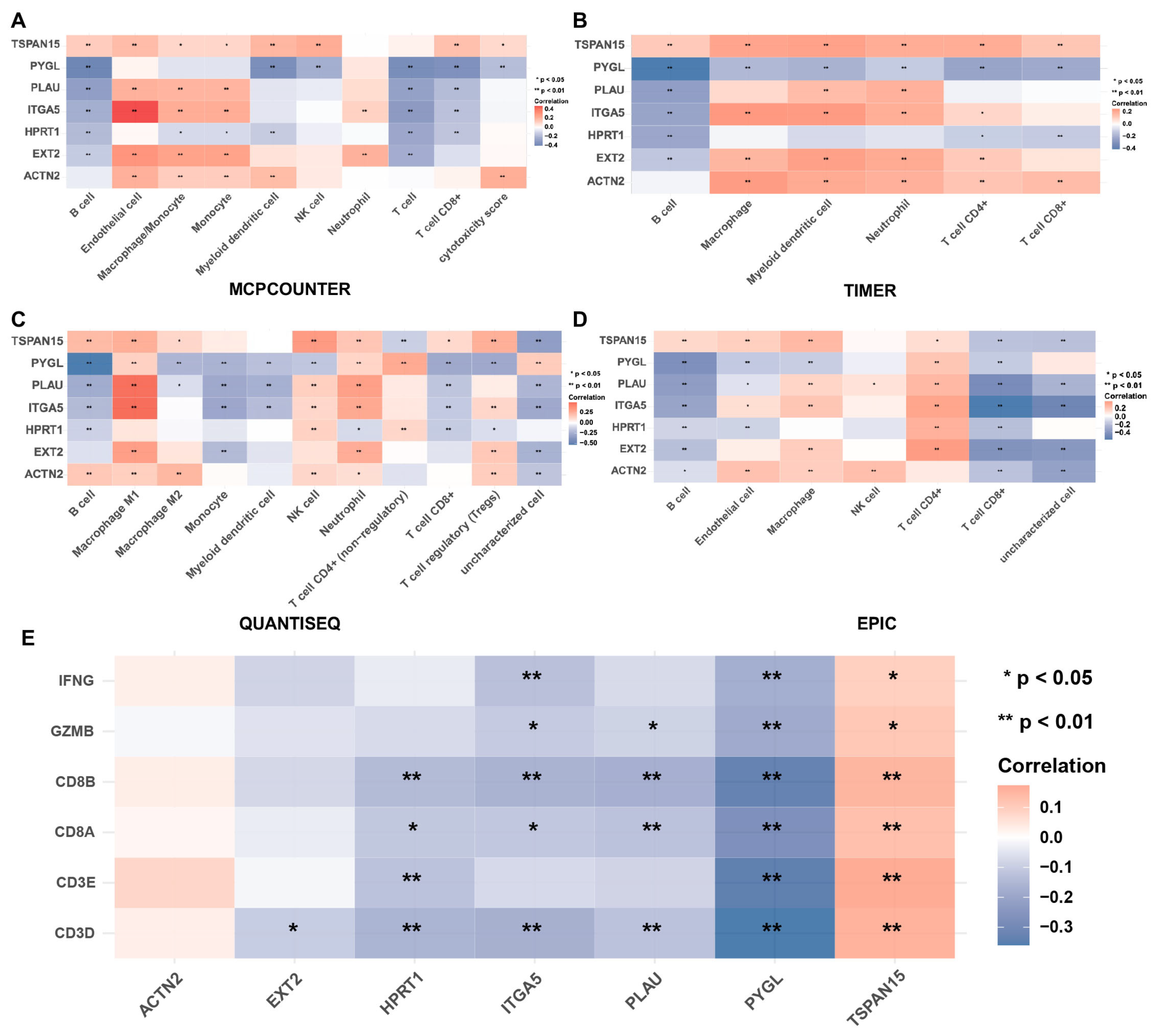
2.8. Immune Infiltration Analysis
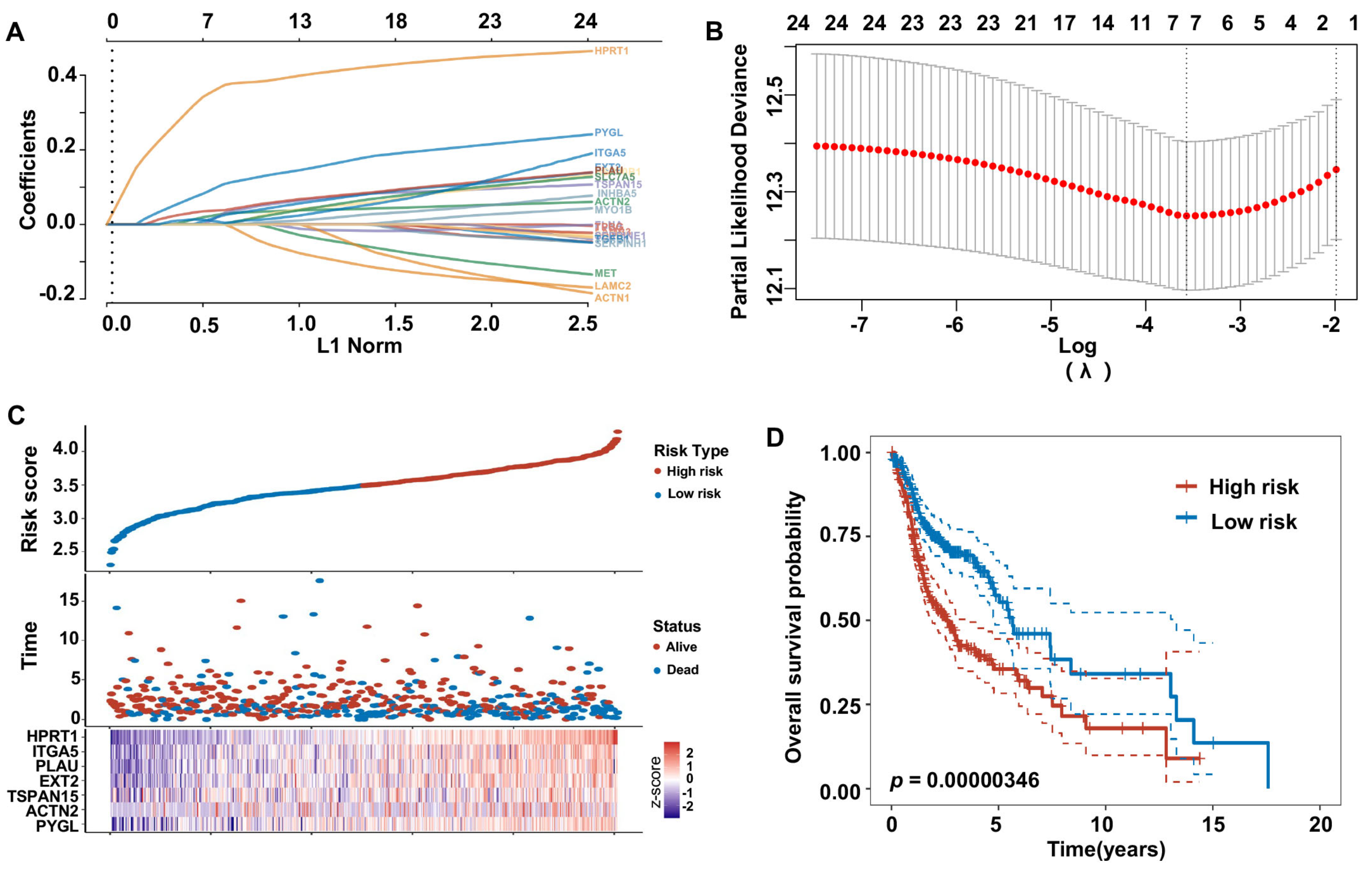
2.9. Construction of Risk Model and Nomograms Based on Prognostic HNSCC ERGs
2.10. Immunohistochemistry (IHC) Staining
3. Results
3.1. Identification of HNSCC Exosome-Related Genes
3.2. Functional Enrichment Analysis and Determination of Prognostic HNSCC ERGs
3.3. Expression Profiles of 25 Prognostic HNSCC ERGs
3.4. Molecular Subtype of HNSCC Based on 25 Prognostic ERGs
3.5. HNSCC ERG-High Subtype Was Associated with Immunosuppressive TME
3.6. Construction of 25 Prognostic HNSCC ERGs Risk Model
3.7. Correlation between Immune Score and Risky ERGs in HNSCC
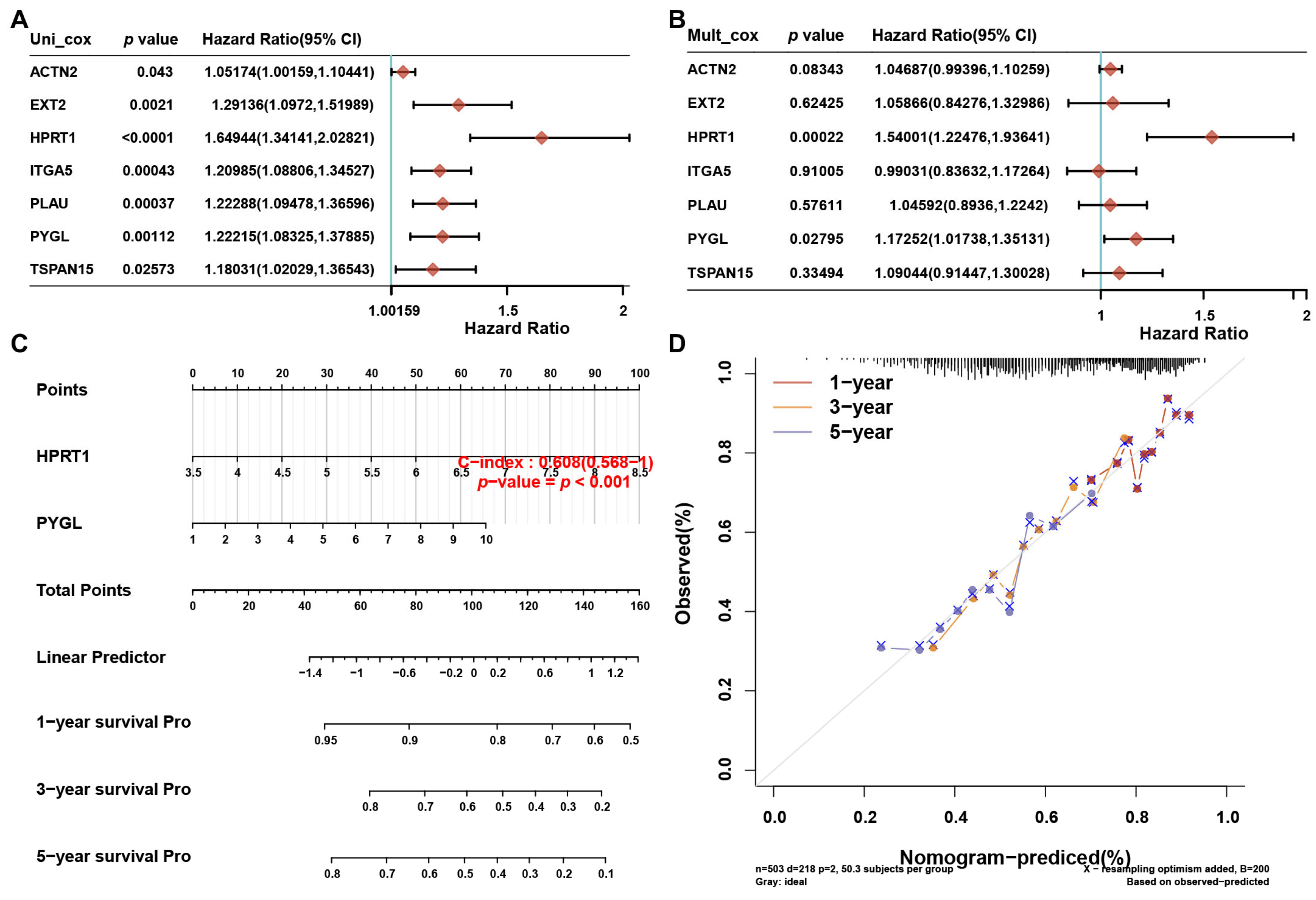
3.8. A prognostic Nomogram Based on the 7 Risky ERGs
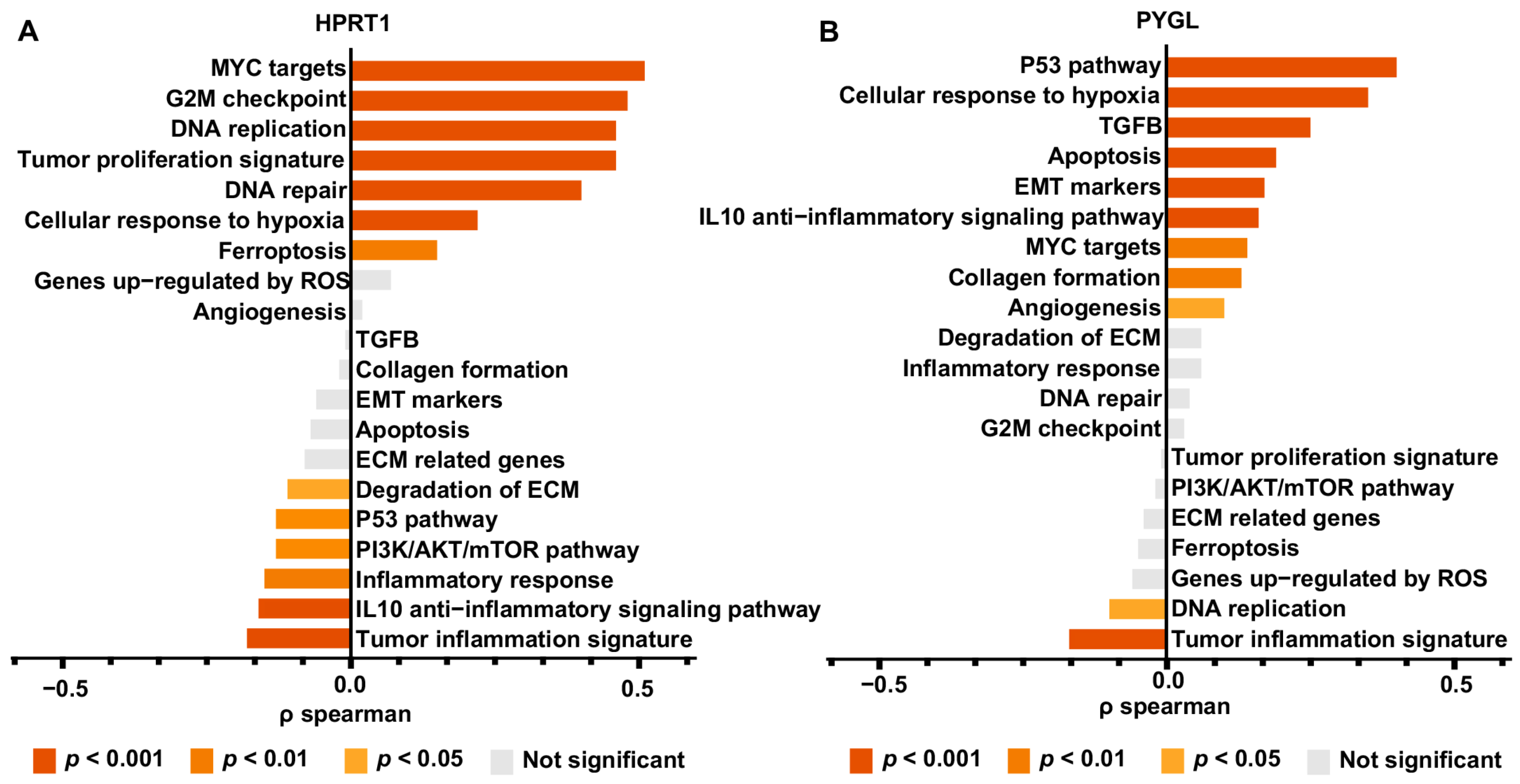
3.9. Genetic Function Analysis of HPRT1 and PYGL
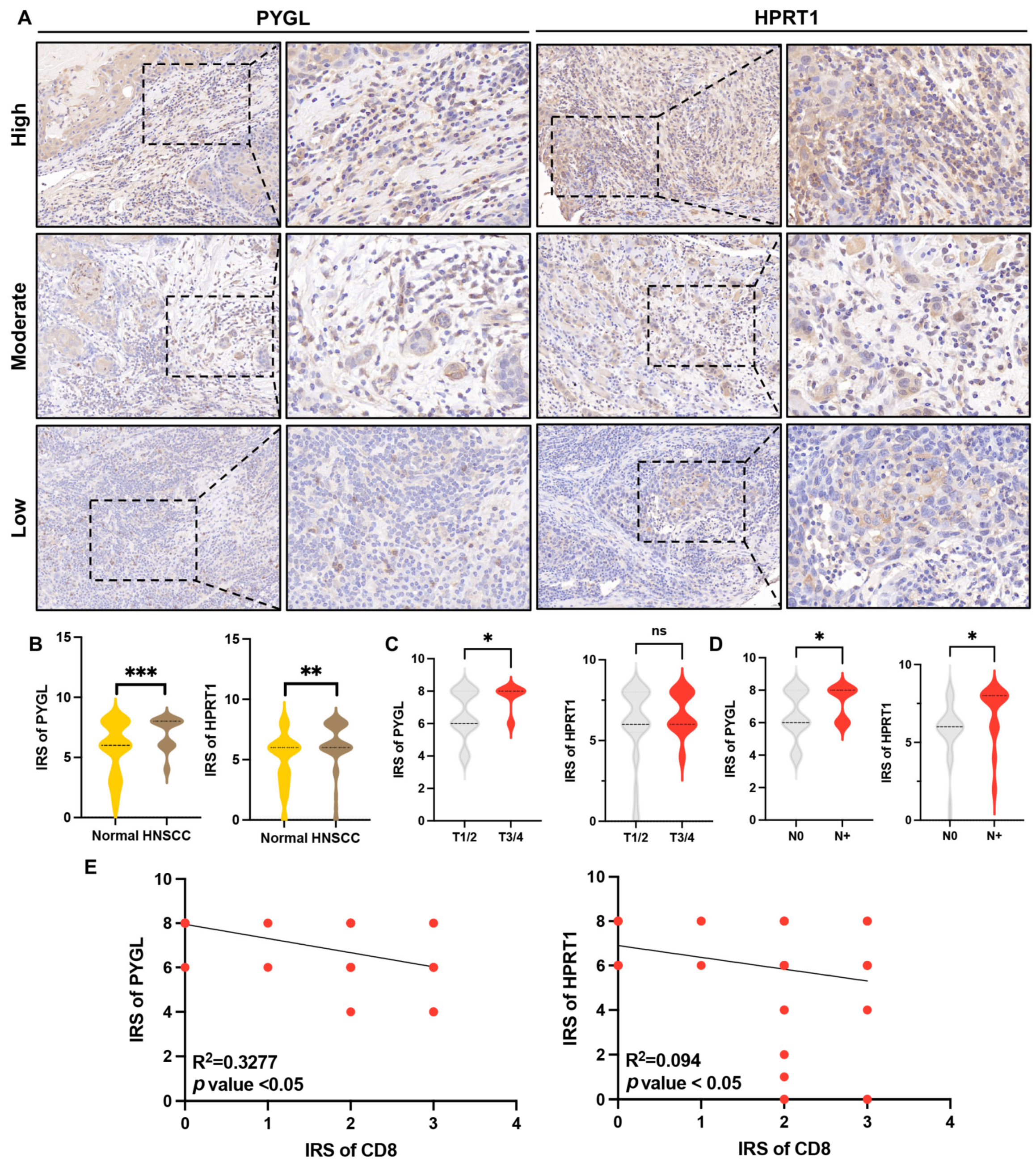
3.10. Overexpression of HPRT1 and PYGL Were Correlated with Tumor Progression and CD8+ T Cell Infiltration in HNSCC Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andersen, M.H. Tumor microenvironment antigens. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Van den Bossche, V.; Zaryouh, H.; Vara-Messler, M.; Vignau, J.; Machiels, J.-P.; Wouters, A.; Schmitz, S.; Corbet, C. Microenvironment-driven intratumoral heterogeneity in head and neck cancers: Clinical challenges and opportunities for precision medicine. Drug Resist. Updat. 2022, 60, 100806. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Ruppin, E. Multiomics Prediction of Response Rates to Therapies to Inhibit Programmed Cell Death 1 and Programmed Cell Death 1 Ligand 1. JAMA Oncol. 2019, 5, 1614–1618. [Google Scholar] [CrossRef]
- Clancy, J.W.; D’Souza-Schorey, C. Tumor-Derived Extracellular Vesicles: Multifunctional Entities in the Tumor Microenvironment. Annu. Rev. Pathol. 2022, 18, 205–229. [Google Scholar] [CrossRef]
- Saleem, S.N.; Abdel-Mageed, A.B. Tumor-derived exosomes in oncogenic reprogramming and cancer progression. Cell. Mol. Life Sci. 2015, 72, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.; Tian, Z.; Du, Z.; Wu, K.; Xu, G.; Dai, M.; Wang, Y.; Xiao, M. M1-like tumor-associated macrophages cascade a mesenchymal/stem-like phenotype of oral squamous cell carcinoma via the IL6/Stat3/THBS1 feedback loop. J. Exp. Clin. Cancer Res. 2022, 41, 10. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, J.; Chen, W.; Chen, W. M1-like tumor-associated macrophages activated by exosome-transferred THBS1 promote malignant migration in oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 143. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Soder, R.; Abhyankar, S.; Abdelhakim, H.; Braun, M.W.; Trinidad, C.V.; Pathak, H.B.; Pessetto, Z.; Deighan, C.; Ganguly, S.; et al. WJMSC-derived small extracellular vesicle enhance T cell suppression through PD-L1. J. Extracell. Vesicles 2021, 10, e12067. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Donninger, H.; Eaton, J.; Yaddanapudi, K. Regulatory Role of Immune Cell-Derived Extracellular Vesicles in Cancer: The Message Is in the Envelope. Front. Immunol. 2020, 11, 1525. [Google Scholar] [CrossRef] [PubMed]
- Mentkowski, K.I.; Snitzer, J.D.; Rusnak, S.; Lang, J.K. Therapeutic Potential of Engineered Extracellular Vesicles. AAPS J. 2018, 20, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, K.; Fu, W.; Li, T.; Zhao, J.; Lei, C.; Hu, S. The roles of small extracellular vesicles in cancer and immune regulation and translational potential in cancer therapy. J. Exp. Clin. Cancer Res. 2022, 41, 286. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.H.; Li, Y.Y.; Wu, T.L.; Chang, J.W.; Chou, W.C.; Hsieh, L.L.; Chen, J.R.; Yeh, K.Y. Culture supernatants of different colon cancer cell lines induce specific phenotype switching and functional alteration of THP-1 cells. Cell. Immunol. 2014, 290, 107–115. [Google Scholar] [CrossRef]
- Anand, S.; Samuel, M.; Ang, C.S.; Keerthikumar, S.; Mathivanan, S. Label-Based and Label-Free Strategies for Protein Quantitation. Methods Mol. Biol. 2017, 1549, 31–43. [Google Scholar]
- Safran, M.; Rosen, N.; Twik, M.; Barshir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite; Springer Nature: Singapore, 2021; pp. 27–56. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019, 35, 436–445. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.A.-O.; Gentles, A.A.-O.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A.-O. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Racle, J.; Gfeller, D. EPIC: A Tool to Estimate the Proportions of Different Cell Types from Bulk Gene Expression Data. Methods Mol. Biol. 2020, 2120, 233–248. [Google Scholar] [PubMed]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Tu, G.; Peng, W.; Cai, Q.; Zhao, Z.; Peng, X.; He, B.; Zhang, P.; Shi, S.; Wang, X. A Novel Model Based on Genomic Instability-Associated Long Non-Coding RNAs for Predicting Prognosis and Response to Immunotherapy in Patients with Lung Adenocarcinoma. Front. Genet. 2021, 12, 720013. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Zhang, M.; He, S.; Lu, K.; Chen, Y.; Xing, G.; Lu, Y.; Liu, P.; Li, Y.; Wang, S.; et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat. Commun. 2014, 5, 3733. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Zhang, Z.; Zhang, J.; Pan, X.; Zhu, X.; Wang, X.; Li, Z.; Ruan, M.; Li, H.; Chen, W.; et al. CD73 in small extracellular vesicles derived from HNSCC defines tumour-associated immunosuppression mediated by macrophages in the microenvironment. J. Extracell. Vesicles 2022, 11, e12218. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Hsieh, C.H.; Lin, P.H.; Chen, Y.T.; Hsu, D.S.; Tai, S.K.; Chu, P.Y.; Yang, M.H. Snail-regulated exosomal microRNA-21 suppresses NLRP3 inflammasome activity to enhance cisplatin resistance. J. Immunother. Cancer 2022, 10, e004832. [Google Scholar] [CrossRef]
- Xie, C.; Ji, N.; Tang, Z.; Li, J.; Chen, Q. The role of extracellular vesicles from different origin in the microenvironment of head and neck cancers. Mol. Cancer 2019, 18, 83. [Google Scholar] [CrossRef] [Green Version]
- Paskeh, M.D.A.; Entezari, M.; Mirzaei, S.; Zabolian, A.; Saleki, H.; Naghdi, M.J.; Sabet, S.; Khoshbakht, M.A.; Hashemi, M.; Hushmandi, K.; et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J. Hematol. Oncol. 2022, 15, 83. [Google Scholar] [CrossRef]
- Han, Q.F.; Li, W.J.; Hu, K.S.; Gao, J.; Zhai, W.L.; Yang, J.H.; Zhang, S.J. Exosome biogenesis: Machinery, regulation, and therapeutic implications in cancer. Mol. Cancer 2022, 21, 207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, E.; Zhuang, H.; Xie, L.; Feng, X.; Liu, J.; Yu, Y. Construction of a novel gene-based model for prognosis prediction of clear cell renal cell carcinoma. Cancer Cell Int. 2020, 20, 27. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Jiao, Z.; Li, Y.; Su, X.; Yao, F.; Peng, J.; Chen, W.; Yang, A. HPRT1 Promotes Chemoresistance in Oral Squamous Cell Carcinoma via Activating MMP1/PI3K/Akt Signaling Pathway. Cancers 2022, 14, 855. [Google Scholar] [CrossRef]
- Wu, H.; Zhao, X.; Zhu, T.; Rong, D.; Wang, Y.; Leng, D.; Wu, D. A Glycosyltransferase-Related Signature for Predicting Overall Survival in Head and Neck Squamous Cell Carcinoma. Front. Genet. 2022, 13, 856671. [Google Scholar] [CrossRef] [PubMed]
- Moy, J.D.; Moskovitz, J.M.; Ferris, R.L. Biological mechanisms of immune escape and implications for immunotherapy in head and neck squamous cell carcinoma. Eur. J. Cancer 2017, 76, 152–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notarangelo, G.; Spinelli, J.B.; Perez, E.M.; Baker, G.J.; Kurmi, K.; Elia, I.; Stopka, S.A.; Baquer, G.; Lin, J.R.; Golby, A.J.; et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8+ T cell function. Science 2022, 377, 1519–1529. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Cluster 1 | Cluster 2 | p-Value | |
|---|---|---|---|---|
| Age | Mean (SD) | 61 (11.6) | 61.1 (12) | |
| Median [MIN, MAX] | 61 [24, 87] | 61 [19, 90] | 0.969 | |
| Gender | Female | 37 | 97 | |
| Male | 119 | 251 | 0.386 | |
| Smoking | Non-smoking | 41 | 72 | |
| Smoking | 112 | 269 | 0.202 | |
| pT stage | T1 | 15 | 19 | |
| T2 | 49 | 97 | ||
| T3 | 40 | 93 | 0.125 | |
| T4 | 8 | 17 | ||
| T4a | 38 | 114 | ||
| TX | 6 | 5 | ||
| T4b | 3 | |||
| pN stage | N0 | 70 | 172 | |
| N1 | 21 | 60 | ||
| N2 | 3 | 16 | ||
| N2a | 9 | 9 | ||
| N2b | 31 | 46 | 0.035 | |
| N2c | 11 | 30 | ||
| N3 | 1 | 6 | ||
| NX | 10 | 9 | ||
| pM stage | M0 | 146 | 333 | |
| M1 | 1 | 4 | ||
| MX | 9 | 11 | 0.337 | |
| pTNM stage | I | 8 | 17 | |
| II | 23 | 58 | ||
| III | 33 | 58 | 0.621 | |
| IVA | 86 | 205 | ||
| IVB | 4 | 9 | ||
| IVC | 2 | 1 | ||
| Grade | G1 | 24 | 38 | |
| G2 | 78 | 223 | ||
| G3 | 40 | 79 | ||
| G4 | 2 | |||
| GX | 9 | 8 | 0.023 | |
| New tumor event type | Metastasis | 10 | 9 | |
| Primary | 3 | 6 | ||
| Recurrence | 11 | 28 | 0.188 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, Y.; Du, Z.; Xu, G.; Tian, Z.; Xiao, M.; Wang, Y. Identification of Exosome-Related Genes Associated with Prognosis and Immune Infiltration Features in Head-Neck Squamous Cell Carcinoma. Biomolecules 2023, 13, 958. https://doi.org/10.3390/biom13060958
You Y, Du Z, Xu G, Tian Z, Xiao M, Wang Y. Identification of Exosome-Related Genes Associated with Prognosis and Immune Infiltration Features in Head-Neck Squamous Cell Carcinoma. Biomolecules. 2023; 13(6):958. https://doi.org/10.3390/biom13060958
Chicago/Turabian StyleYou, Yuanhe, Zhong Du, Guisong Xu, Zhuowei Tian, Meng Xiao, and Yanan Wang. 2023. "Identification of Exosome-Related Genes Associated with Prognosis and Immune Infiltration Features in Head-Neck Squamous Cell Carcinoma" Biomolecules 13, no. 6: 958. https://doi.org/10.3390/biom13060958