Abstract
Mitochondrial network architecture plays a critical role in cellular physiology. Indeed, alterations in the shape of mitochondria upon exposure to cellular stress can cause the dysfunction of these organelles. In this scenario, mitochondrial dynamics proteins and the phospholipid composition of the mitochondrial membrane are key for fine-tuning the modulation of mitochondrial architecture. In addition, several factors including post-translational modifications such as the phosphorylation, acetylation, SUMOylation, and o-GlcNAcylation of mitochondrial dynamics proteins contribute to shaping the plasticity of this architecture. In this regard, several studies have evidenced that, upon metabolic stress, mitochondrial dynamics proteins are post-translationally modified, leading to the alteration of mitochondrial architecture. Interestingly, several proteins that sustain the mitochondrial lipid composition also modulate mitochondrial morphology and organelle communication. In this context, pharmacological studies have revealed that the modulation of mitochondrial shape and function emerges as a potential therapeutic strategy for metabolic diseases. Here, we review the factors that modulate mitochondrial architecture.
1. Mitochondrial Network Architecture
Mitochondrial architecture is determined by several components, which include the following: mitochondrial distribution in the cytosol, supported by interaction with the cytoskeleton; events of fission and fusion, mediated by mitochondrial dynamics proteins; mitochondrial network contact with other organelles (e.g., endoplasmic reticulum (ER), lipid droplets (LDs), lysosomes, and plasma membrane); and the lipid composition of mitochondrial membranes [1,2,3,4,5,6,7,8].
In addition, the ultrastructure of mitochondria may be modified by mitochondrial ion transport fluxes such as Ca2+ and K+ [9,10]. An influx of these ions can alter the morphology of these organelles, leading to swelling or a donut-like shape [4,11].
Cytoskeleton: Recent work evidenced that cytoskeleton elements differentially modulate the mobility and shape of mitochondria [7,12]. Nocodazole, a drug that disrupts microtubule polymerization, reduces mitochondrial network cellular coverage, disrupts mitochondrial alignment with microtubules, and decreases mitochondrial mobility. In contrast, F-actin or intermediate filaments maintain mitochondria confined to the microtubule network, and their disruption alters mitochondrial shape [7,12]. Of note, it has been reported that ARP2/3 and Formin-dependent actin cycling (actin assembly and disassembly) occur through the mitochondrial network [1]. Furthermore, actin polymerization promotes mitochondrial fission, whereas F-actin disruption causes mitochondria fusion [1]. In this regard, F-actin is assembled into the outer mitochondrial membrane (OMM), thereby contributing to mitochondrial fission [1,13].
Mitochondrial dynamics: Continual shifting of mitochondrial network architecture is supported by mitochondrial dynamics proteins. In the last 20 years, several proteins that coordinate mitochondrial fission and fusion events have been defined [14,15,16,17,18]. Moreover, recent studies have indicated that mitochondrial fission and fusion occur in the same site on the mitochondria [2], and also that ER wrapping around mitochondria is required to sustain the dynamics of these organelles [6]. A recent report describes two distinct types of mitochondrial fission. Peripheral fission sustains mitochondrial degradation mediated by mitophagy while midzone mitochondrial fission is required to maintain mitochondrial biogenesis and dynamics [19] (see Section 2).
Mitochondrial contacts with other organelles. A growing number of studies have demonstrated the interaction of mitochondria with different organelles. This is an emergent field of research that will permit understanding of how some metabolic and cell signaling mechanisms originate.
Mitochondria–endoplasmic reticulum contacts. Several investigations have reported that mitochondria–endoplasmic reticulum interactions are required to modulate the calcium signal. Close contact between mitochondria and endoplasmic reticulum plays a critical role in mitochondrial calcium uptake. For instance, the complex VDAC1-GRP75-PI3R mediates mitochondria–endoplasmic reticulum tethering and is required to sustain mitochondrial calcium homeostasis [20]. Moreover, mitochondria–endoplasmic reticulum interaction is involved in apoptosis and autophagy activation [21,22,23]. Overnutrition induced via a high fat diet in mice leads to alteration in calcium handling in the liver [24,25]. These studies have demonstrated that mitochondrial–endoplasmic reticulum contacts are disrupted upon ingestion of a high fat diet. On the other hand, the mechanism of phosphatidylethanolamine (PE) synthesis and shuttling requires mitochondrial–endoplasmic reticulum contacts. The enzymes required for phosphatidic acid (PA) conversion to PE are localized in mitochondrial–endoplasmic reticulum contact. Several proteins contribute to mitochondria–endoplasmic reticulum tethering and the maintenance of calcium transfer and lipid synthesis (revised by Wenzel et al., 2022) [26].
Mitochondria–lipid droplets contacts. Contacts between mitochondria and lipid droplets (LDs) are mediated by several proteins including PLIN1, PLIN5, MFN2, and MIGA2 [27] (see Section 4). The highly dynamic interactions between both organelles allow fatty acid migration from LD to mitochondria, where it is oxidized to produce ATP or heat. Furthermore, they are also needed for LD expansion, which stores fatty acids and lipid intermediates to avoid cellular lipotoxicity [28,29].
Mitochondria–peroxisome contacts. Mitochondrial–peroxisome interaction plays a relevant role in fatty acid metabolism and in cellular redox homeostasis; a coordinate function of these organelles is required to sustain mitochondrial activity. Studies have shown that peroxisomal protein (PEX5 or PEX16) ablation in mice livers led to mitochondrial dysfunction and fragmentation [30,31]. Moreover, PEX16 knockout mice showed that peroxisomes are required to sustain mitochondrial homeostasis upon metabolic stress induced by a high fat diet [31]. Two proteins involved in mitochondria–peroxisome tethering have been identified, enoyl-CoA-δ isomerase 2 (ECI2) and the lipid transport protein VPS13D [32,33]. Interestingly, knockdown of VPS13D expression promotes mitochondrial fragmentation, revealing the role of this protein in mitochondrial architecture [34]. Also interesting, a recent study of yeast demonstrated that PEX4 and Fzo1 (the yeast orthologue of MFN1 and MFN2 proteins) are involved in mitochondria–peroxisome tethering [35]. Further investigations are required to elucidate the role of this protein in mammalian cells.
Mitochondria–endosome contacts. A recent report has documented that VDAC2 tethers RAS-PI3K-positive early endosomes with mitochondria [36]. This study also shows that this interaction promotes endosome acidification and maturation [36]. Depletion of the mitochondrial fusion protein MFN1 promotes the association of endosomes and mitochondria through a process that requires Rab5C [37]. In addition, it has been described that endosomes are in close contact with mitochondria in axons of retinal ganglion cells [38]. Rab7a is localized in endosomes, and ribonucleoprotein particles and mitochondria are in contact in axons. In this way, these contacts are required for local translation of mRNA that encodes to mitochondrial proteins. Moreover, Rab7a mutations lead to alteration of mitochondrial morphology, decrease mitochondrial membrane potential, and modify the mitochondrial retrograde and anterograde transport in axons [38].
Mitochondria–lysosome contacts. Lysosome–mitochondrial tethering is mediated by Rab7, is localized in lysosomes, and is modulated by TBC1D15, a Rab7 GTPase activating protein, and Fis1, localized in mitochondria [39]. This study also documented that lysosomes localize in mitochondrial fission sites, suggesting that they modulate mitochondrial dynamics [39]. The functional role of mitochondrial–lysosome tethering on calcium homeostasis has recently been reported [40]. The cation channel TRPML1 localized in lysosomes and late endosomes mediates the direct calcium transfer into mitochondria. Moreover, TRPML1 loss-of-function impairs calcium transfer, and it results in alterations of tethering dynamics [40].
Mitochondrial architecture is altered by environmental stimuli. It has been widely described in cell models that metabolic stress, induced by OXPHOS inhibitors, nutrient starvation, endoplasmic reticulum (ER) stressors, protein and RNA synthesis suppressors, and UV irradiation, induce transient mitochondrial elongation upon acute treatment [41,42,43,44]. Moreover, mitochondrial elongation improves the OXPHOS activity under these stimuli [41,45]. It has been demonstrated that the relationship between mitochondrial morphology and OXPHOS is complex. Given that, maximal mitochondrial respiration is carried out upon mitochondrial uncoupling agents (such as CCCP) treatment and these compounds promote mitochondrial depolarization and fragmentation. A simple relation between mitochondrial elongation and increased OXPHOS activity may not occur under all conditions. Thus, genetic ablation of the mitochondrial fusion protein Mitofusin 1, in which the mitochondrial network is fragmented, showed similar OXPHOS activity in glucose- or galactose-supplemented culture media [46]. In keeping with this, it has been reported that hepatic MFN1 ablation increases mitochondrial mass and OXPHOS activity [47]. Recent evidence has shed new light on the link between the morphology and function of mitochondria. Ngo et al. demonstrated that mitochondrial fragmentation enhances long-chain fatty acid oxidation. Moreover, mitochondrial fragmentation decreases the inhibition of malonyl-CoA-dependent carnitine palmitoyltransferase I. These data reveal that mitochondrial fragmentation upon lipid overload activates mitochondrial β-oxidation [48].
Mitochondrial shape alterations have been reported in conditions such as obesity, diabetes, ischemic–reperfusion, senescence, and cancer [24,49,50,51,52,53,54]. Under metabolic stress induced by nutrient overload, mitochondrial network architecture is fragmented, and this alteration is associated with a metabolic dysfunction that results in a decrease in OXPHOS activity, apoptosis, or mitophagy [55]. These alterations have been evidenced in several cell models, including β-pancreatic cells, skeletal muscle cells, cardiomyocytes, and adipocytes [56,57,58,59,60]. Similarly, disruption of the mitochondrial network architecture and mitochondrial dysfunction have been reported based on in vivo mouse models of diabetes, cardiomyopathy, non-alcoholic fatty liver disease, and obesity [51,61,62,63]. Emergent evidence indicates that mitophagy is impaired in metabolic diseases. The perturbation of mitochondrial homeostasis that causes a decrease in mitochondrial membrane potential induces mitochondrial fragmentation and activates lysosome-dependent mitochondrial degradation [55,64,65]. Recently, Han and colleagues demonstrated the presence of distinct populations of mitochondria in non-small cell lung cancer tumors. They reported a peri-droplet mitochondrial network throughout the cytoplasm in oxidative lung adenocarcinoma (LUAD) cells that was accompanied by an increase in fatty acid oxidation. On the other hand, cells with higher glycolytic activity (lung squamous cell carcinoma) show perinuclear mitochondria. These exciting results support the importance of mitochondrial architecture in sustaining tumor metabolism [54]. Thus, modulation of mitochondrial architecture appears to play a crucial role in cellular responses leading to metabolic diseases.
The discovery of proteins that modulate mitochondrial architecture and the mechanism of action of the same has allowed intense research efforts regarding the link between metabolism and mitochondrial function. These efforts have revealed that changes in the expression or post-translational modification of mitochondrial dynamics proteins are associated with metabolic diseases. As a result of these investigations, new fields of drug discovery aiming to modulate mitochondrial dynamics and thereby redirect cellular metabolism have emerged.
Given the continuous and rapid changes in mitochondrial architecture, sophisticated microscopy approaches are required to elucidate the biological factors that regulate mitochondrial morphology in vivo. Recent studies using super-resolution microscopy have revealed the cellular mechanisms by which mitochondrial network architecture is modulated [2,5,6,19]. In this regard, live-cell imaging has shown that ER–mitochondrial constriction sites prime and coordinate the mitochondrial fission and fusion protein machinery [2]. Remarkably, recent evidence using proximal proteomic and live-cell microscopy demonstrates that ABHD16A, a hydrolase enzyme that mediates the conversion of phosphatidylserine (PS) to lysophosphatidylserine (lysoPS), is localized at ER–mitochondria contacts and is required for mitochondrial fission and fusion [8].
The use of intravital multiphoton microscopy-based technology, which allows collection of cell images in live animals, has revealed that the mitochondrial network is disrupted upon pathological conditions in vivo. This work has demonstrated the transient disruption of the mitochondrial network upon brain ischemia. The mitochondria of dendrites at pyramidal neurons have an elongated shape, as revealed by intravital microscopy and electron microscopy. In contrast, upon transient brain ischemia, mitochondria undergo fragmentation and have a globular morphology, and “mitochondria-on-a-string” (MOAS) structures appear. Ischemic recovery rescues mitochondrial elongation and decreases globular mitochondria, and MOAS morphology is not observed in dendrites [66]. Taken together, these observations indicate that a transient and reversible alteration of mitochondrial morphology occurs under ischemia in vivo. Interestingly, MOAS morphology has also been described in neurons in cellular and mouse models of Alzheimer’s disease, aging, and hypoxia. It has been proposed that energetic collapse upon ischemic injury leads to unfinished mitochondrial fission [67,68]. Intravital microscopy has also been used to study live-cell mitochondrial dynamics and mitophagy in the livers of mice after acute ethanol treatment [68]. This report demonstrated that this treatment disrupts mitochondrial membrane potential; however, only a subpopulation of mitochondria undergoes mitophagy. In addition, ethanol activates selective mitophagy instead of lipophagy in the livers of this model [69]. In this context, a novel state-of-the-art methodological approach will unravel the function and modification of the mitochondrial network under physiological and pathological conditions (Figure 1 and Table 1).
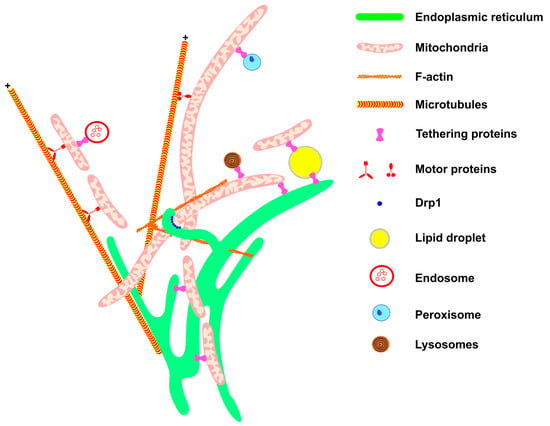
Figure 1.
Mitochondrial network architecture. Mitochondria morphology is sustained and affected by interactions with other cellular structures. Communication with the ER (green) provides mitochondrial phospholipids and sustains calcium homeostasis. Interaction with the components of the cytoskeleton (F-actin filaments and microtubules, in orange) moves the mitochondria to specific regions of the cell, such as the appropriate location for cell division, or the periphery of the cell in the case of neurons. Motor protein kinesins and dyneins help to move mitochondria along the cytoskeletons of microtubules (for review, see McElroy et al., 2023) [70]. The endoplasmic reticulum wraps the mitochondria, thereby helping to recruit DRP1 (blue) on the mitochondrial membrane. Through communication with LDs (yellow), mitochondria are provided with fatty acids for β-oxidation and in turn provide energy for LD expansion. The contact between mitochondria and endosome (red) is important for mitochondria quality control through lipid and ion transference. Peroxisome (blue) and mitochondria communication allows the complementation of fatty acid oxidation and anti-oxidant system activation (for review, see Chen et al., 2020) [71]. Mitochondria–lysosome interaction (red and black) facilitates the direct transference of calcium from lysosomes to mitochondria. All these interactions are mediated by specific proteins that connect or tether the membranes.

Table 1.
Cellular components that modulate mitochondrial architecture.
2. Mitochondrial Dynamics Proteins
The function of mitochondrial dynamics proteins is modulated by protein expression, proteolytic processing, subcellular localization, and post-translational modifications. Additionally, some accessory mitochondrial proteins and mitochondrial lipid composition help to modulate mitochondrial shape. Thus, the fine-tuned coordination of these factors regulates the architecture and function of the mitochondrial network. Mitochondrial dynamics proteins have been widely studied, and their function has been characterized in several tissues under a range of physiological and pathological conditions. In this section, we will describe the major and recent advances in the field of mitochondrial dynamics.
2.1. Mitochondrial Fission Proteins
Mitochondrial fission is characterized by the division of a mitochondrion into two smaller mitochondria. This process is key for mitochondrial distribution during mitosis, mitochondrial clearance, ATP distribution (e.g., distal axons), and metabolism [48,64,77,78]. The main determinant protein for fission is the dynamin-related protein 1 (DRP1), which, together with adaptor proteins (FIS1 and MFF), the ER, and the cytoskeleton, promotes mitochondrial constriction and fragmentation [6]. It has been demonstrated that β-cells and brown adipocytes present predominantly mitochondrial fission under physiological stimulus or nutrient exposition; moreover, mitochondrial fission is required for dendritic spine plasticity and neuronal synapses [65,78,79,80,81,82].
2.1.1. DRP1
Dynamin-related protein 1, also called dynamin-like protein, is a GTPase that translocates between the cytosol and the mitochondrial outer membrane, and it is regulated by phosphorylation and cytoskeleton interaction [83]. DRP1 promotes mitochondrial fission by forming a ring at mitochondria–ER contacts where membrane constriction occurs [6,14]. DRP1 also participates in peroxisome fission and mitochondrial calcium handling. The coordinated action of DRP1, peroxisomal membrane protein 11 (PEX11), and MFF promotes the constriction and division of peroxisomes [84,85]. In addition, DRP1 participates in the regulation of mitochondrial calcium levels. DRP1 knockout (KO) hippocampal neurons expressing a mutant amyloid precursor protein (APP) show mitochondrial calcium overload and increased APP-dependent neurotoxicity. These data suggest that DRP1 exerts a protective function in Alzheimer’s disease [86,87].
Although mitochondrial fission is associated with energy expenditure in brown adipocytes or excess nutrients and low ATP demand in β-cells [88], the increase in fission is usually associated with mitochondrial dysfunction or cancer invasion. For instance, ERK1/2-dependent DRP1 phosphorylation promoted by SIRT6 has been associated with increased mitochondrial fission and ovarian cancer invasion [89]. In colorectal, breast, and lung cancer and acute lymphoblastic leukemia, there is an increase in fission, and DRP1 phosphorylation is also associated with chemoresistance [90,91].
The increase in mitochondrial fission has been related to neurodegenerative diseases such as Parkinson’s disease (PD) and Alzheimer’s disease (AD). Regarding AD, the levels of DRP1 are frequently increased in postmortem tissues while mitochondrial fusion proteins Mitofusin 1 (MFN1) and Mitofusin 2 (MFN2) are reduced in these tissues. One might also conclude that AD may be associated with decreased mitochondrial fusion [92].
DRP1-associated dysfunctions are related to autophagy deficiency [93] or an increase in Radical Oxygen Species (ROS) levels [94]. The untreated diabetic condition, mimicked by prolonged exposure to high glucose levels, causes mitochondrial fission and an increase in ROS production [94]. Recently, a very interesting study showed that spontaneous fission can occur in two mitochondrial sites: peripheral or midzone. The occurrence of peripheral fission is less frequent in a healthy mitochondrion and is associated with the loss of mitochondrial membrane potential and high ROS and calcium levels. The product of peripheral fission is directed to mitophagy, while daughter mitochondria from midzone fission undergo mitochondrial dynamics [19].
2.1.2. MFF
Mitochondrial Fission Factor (MFF) was described as an accessory receptor for mitochondrial fission, and it has an additional role in peroxisome fission [18]. The analysis of several human tissues demonstrates that MFF is highly expressed in the heart, liver, brain, muscle, and stomach [18]. MFF is the predominant DRP1 receptor in mammals, and its deficiency is associated with developmental delay, peripheral neuropathy, and optic atrophy. MFF-associated mitochondrial fission occurs independently of FIS1. It was demonstrated that mitochondrial fission is compromised by DRP1 and MFF knockdown, but not by FIS1 deficiency [95].
Liu and Chan (2015) characterized the interaction between DRP1 and MFF and concluded that MFF interacts with only complex DRP1 forms (tetramer or higher order) and that these forms are necessary for efficient fission [96]. On the other hand, MiD49 and 51, other DRP1 receptors, can interact with monomeric forms of DRP1 [96].
2.1.3. FIS1
Mitochondrial fission protein 1 (FIS1) was first identified as essential for mitochondrial fission in yeast. Based on that observation, for many years it was considered a receptor/adaptor of DRP1 in humans [97]. However, growing evidence shows that the role of FIS1 can be cell-type dependent and that it is not essential for DRP1-mediated fission [95,98]. The depletion of FIS1 in some cells leads to mitochondrial elongation [99], while in others it does not alter the morphology of these organelles [95].
Experiments with yeast deficient in fission machinery demonstrate that reconstitution with MFF/DRP1 restores fission capacity, whereas FIS1/DRP1 does not have this effect [100].
Studies by Yu and colleagues (2019) raised the possibility that FIS1-induced fragmentation is a result of fusion inhibition rather than fission activation [98]. In DRP1 KO HEK293 cells, FIS1 overexpression causes mitochondria fragmentation through MFN1, MFN2, and OPA1 interaction and inhibition of their GTPase activity [98]. Studies focusing on identifying new DRP1 inhibitors indicate that residues 60–66 of FIS1 show high homology to residues 49–55 of DRP1 and that the use of a peptide inhibitor for these regions decreases mitochondrial fragmentation and oxidative stress and inhibits apoptosis [101].
Recently, structural studies showed that the arm in the N-terminus (residues 1–9) of FIS1 is decisive for the IN and OUT conformation of this protein and is necessary for its interaction with DRP1. The absence of these residues in the arm increases the interaction with fusion-related proteins. These observations thus suggest that, depending on cellular conditions, FIS1 inhibits mitochondrial fusion by interacting with fusion proteins through its arm [102].
Also, the discrepant results regarding the effects of DRP1 and FIS1 can be explained by Kleele’s results (2021), which revealed several aspects of fission [19]. Assays of cellular localization showed that MFF forms bright foci at the constriction site of mitochondrial midzone fission while FIS1 is distributed throughout the OMM in non-dividing mitochondria. In mitochondria undergoing peripheral fission, FIS1 is enriched and, in the case of FIS1 depletion, peripheral fission rates are decreased [19]. Additionally, the role of FIS1 in mitochondrial peripheral fission can be protective, since cardiomyocytes lacking FIS1 undergo major apoptosis [19].
FIS1 depletion associated with mitochondrial elongation promotes mitochondrial DNA (mtDNA) damage and a senescence-associated phenotype. These observations also may be associated with the selective FIS1 function in mitophagy [103,104]. High expression of FIS1 has been reported in patient samples of oral melanoma, and the mitochondrial fission induced by FIS1 in lung cancer stem cells leads to autophagy and cancer cell survival [105].
2.1.4. MiD49 and MiD51
Mitochondrial elongation factors 1 and 2 (MIEF 1/2) or mitochondrial dynamics proteins of 49 kDa (MiD49) and 51 kDa (MiD51) were found in a screening to identify new uncharacterized proteins [106]. MiD49/51 is a strong DRP1 receptor that does not localize at peroxisomes but is present at mitochondria–ER contact sites [107,108]. The first implication of the involvement of MiD49/51 in fission machinery was provided by observations made by Palmer and colleagues. In this regard, MiD49/51 overexpression for short time periods was found to cause mitochondrial fission. This finding was reinforced by other studies that show that double knockdown also leads to mitochondrial elongation [109,110]. Moreover, MiD49/51 promotes fission independently of FIS1 or MFF [110]. However, other studies point to a fission inhibition role of MiD49/51. Zhao and co-workers described MiD51 as a fusion promoter because elevated levels of this protein cause mitochondrial elongation, while its depletion promotes mitochondrial fragmentation [111]. These authors proposed that MiD51 interacts with DRP1 and sequesters it, thus inhibiting fission [111]. Furthermore, the same group characterized MiD49 and observed similar effects causing mitochondrial fusion [112]. Palmer and colleagues attributed the association of MiD49/51 overexpression and mitochondrial fusion to extended overexpression [107].
Experiments with DRP1 mutants revealed that DRP1 oligomerization is required for interaction with MFF but not MiD49/51, thereby suggesting that the latter recruits an immature or inactive form of DRP1, thereby decreasing the availability of active DRP1 oligomers to interact with MFF [96]. Moreover, Lóson and colleagues showed that DRP1 phosphorylation at S637 is necessary for complex formation with MiD49/51 [110].
It is interesting that although MiD49/51 does not participate directly in peroxisome fission, by binding and sequestering DRP1, it also inhibits peroxisome fragmentation [107]. This result indicates that MiD49/51 is a stronger DRP1 recruiter than MFF or FIS1 [107].
In this case, MiD49/51 could facilitate mitochondrial fusion because of its self-association that approximates two adjacent mitochondria, facilitating MFN1/2-mediated tethering of two mitochondrial outer membranes [107]. This scenario is reasonable considering that fusion and fission machinery are in the same mitochondria–ER contact sites [2]. Indeed, mitochondrial elongation caused by MiD49/51 overexpression requires the action of fusion mediators MFN1 and MFN2 [107].
Atkins and colleagues have proposed that the discrepancy in connection with the functional role of MiD49/51 proteins in mitochondria dynamics (whether MiD49/51 mediate fusion or fission) can be attributed to cell type and the levels of MiD49/51 expression [113].
They also speculated that MiD49/51 can be used as a therapeutic target in diseases involving the dysregulation of mitochondrial dynamics, such as hyperproliferative conditions like cancer and pulmonary arterial hypertension (PAH) [113]. In PAH, for example, fission exceeds fusion, and the levels of MiDs are elevated, and, consequently, there is protection from apoptosis [114]. The silencing of MiD49/51 promotes mitochondrial elongation and a decrease in proliferation [114]. On the other hand, the inhibition of MiDs in acute ischemia–reperfusion injury decreases fission and protects the heart against cell death [115].
Studies of tissues from patients with ovarian cancer show that high expression of MiD51 is associated with poor prognosis. In contrast, in pancreatic cancer, MiD49 is considered a tumor suppressor [116,117].
2.2. Mitochondrial Fission Protein Phosphorylation
2.2.1. DRP1 Phosphorylation
Two main phosphorylation sites have been described for human DRP1, namely S637 and S616. Several kinases are related to the phosphorylated forms of DRP1 S616. CDK1/cyclin B1 is the main kinase described, and this phosphorylation is very important because it occurs in the mitotic phase and is required for the equal distribution of mitochondria for daughter cells [118]. CDK5 also phosphorylates S616, but the consequence of this modification is still controversial. While some authors observed fission inhibition in post-mitotic neurons upon CDK5 phosphorylation at a DRP1 S616 equivalent residue in rats [119], the results of other studies indicate that phosphorylation by CDK5 at the equivalent serine residue in mice induces mitochondrial fission, which is associated with neuronal death upon NMDA-induced injury [120].
In a model of high glucose stimulation, mitochondrial fission is increased and associated with DRP1 S616 phosphorylation by ERK1/2 in vitro [121]. In human pancreatic cancer, DRP1 S616 phosphorylation by ERK2 and mitochondrial fragmentation is related to tumor growth [122]. This phosphorylation is associated with a series of negative consequences. For instance, Yuan and colleagues (2021) showed that hypoxia leads to ERK1 activation, DRP1 S616 phosphorylation, mitochondria fragmentation, and amyloid-β (Aβ) production [123].
Also, MAPK1 phosphorylated DRP1 at S616 and induced mitochondrial fragmentation and dysfunction in striatal cells of a Huntington’s disease model [124]. Studies with rat models of hypertension-induced encephalopathy showed that PKCδ phosphorylates DRP1 at S579 (equivalent to human S616) promoted mitochondrial fragmentation and neuronal cell death [125].
Finally, PTEN-induced kinase 1 (PINK1) also phosphorylated DRP1 at S616, which is necessary for autophagy independent of Parkin and is relevant for Parkinson’s disease (PD) [126].
Phosphorylation at T595 has been described in a model of PD. In this case, a mutated leucine-rich repeat kinase 2 (LRKK2) protein, which is frequently found in PD, phosphorylated DRP1 at T595, increased GTPase activity and mitochondrial fission, and led to exacerbated autophagy [127].
S637 phosphorylation is commonly associated with an inhibitory post-translational modification of DRP1 because this residue is located at the DRP1 GTPase effector domain (GED), and its GTPase activity would be inhibited after phosphorylation. However, the effect of S637 phosphorylation is rather controversial because some studies showed DRP1 inhibition [128] while others related S637 phosphorylation to fission activation [90,129].
This residue can be phosphorylated by protein kinase A (PKA) and B (AKT), leading to fission inhibition [130,131]. This notion was reinforced by studies using mitochondria depolarizing agents, which promoted an increase in calcium levels and activation of the phosphatase calcineurin. Calcineurin thus dephosphorylates DRP1 S637 and promotes DRP1 translocation to mitochondria, allowing fission to take place [132]. On the other hand, the dephosphorylation of S637 by PGAM5 inhibits fission [133]. Recently, Valera and colleagues (2021) demonstrated crosstalk among the DRP1 phosphorylation sites in mice. They showed that the phosphorylation of murine S600 (equivalent to S637 in humans) occurs before that of S579 (equivalent to S616 in humans) and that it is enough to induce an increase in p-S579 levels. In mice, they attribute the discrepancy in previous results to the effect of S600 phosphorylation on DRP1 isoforms and also to the requirement of the downstream phosphorylation of S579. In the absence of S579 phosphorylation, mitochondrial elongation can be observed after S600 phosphorylation [134].
Phosphorylation at S637 also occurs after A-kinase anchoring protein (AKAP)1 activation in rats with streptozotocin (STZ)-induced diabetes. This activation is accompanied by mitochondrial fission, podocyte damage with loss of mitochondrial membrane potential, a decrease in ATP synthesis, and increased ROS levels, and thus podocyte apoptosis [135]. A similar response to hyperglycemic stress has been reported in podocytes and endothelial cells, but this time, the phosphorylation of DRP1 at S600 (equivalent to S637) and mitochondrial fission were mediated by Rho-associated coiled-coil containing protein kinase 1 (ROCK1) [136].
In primary mouse chondrocytes, TANK-binding kinase 1 (TBK1) phosphorylated DRP1 at S637, thus attenuating TNF-α-induced apoptosis [137]. These authors suggested that this phosphorylation by TBK1 is necessary for autophagy. TBK1 also phosphorylated S412 at the DRP1 middle domain (MD) and S684 in the GED upon innate RNA sensing. These phosphorylations also inhibit DRP1 activity and are important for antiviral immunity [138].
2.2.2. MFF Phosphorylation
MFF was identified in a screening for AMPK substrates. MFF contains two possible sites for AMPK phosphorylation. Although S172 was found to be a direct substrate of AMPK, the phosphorylation signal of MFF is lost only when S155 is concomitantly mutated with S172. AMPK is activated in response to mitochondrial stress, and it triggers mitophagy. Therefore, MFF phosphorylation can induce mitochondrial fragmentation to allow mitophagy to take place. This process occurs, for example, when there is an electron transport chain dysfunction, with a decrease in ATP levels. AMPK is activated, and it phosphorylates MFF, culminating in mitochondrial fragmentation [139].
As occurs for DRP1, MFF is also specifically phosphorylated in mitosis. The protein kinase PKD phosphorylates S155, S172, and S275 induce mitochondrial fission in mitosis but not during interphase [140].
2.2.3. FIS1 Phosphorylation
FIS1 Y38 phosphorylation by Met tyrosine kinase, a receptor for hepatocyte growth factor (HGF), promotes mitochondrial fission and facilitates hepatocellular carcinoma (HCC) tumor growth [141]. This modification increases DRP1/FIS1 interaction. In addition, phosphorylation at Y34 occurs, and it also increases the affinity for DRP1 and induces mitochondrial fragmentation. This phosphorylation is mediated by DNA-PKcs in acute kidney injury and oxidative stress in renal endothelial cells after ischemia/reperfusion injury [142] (Figure 2).
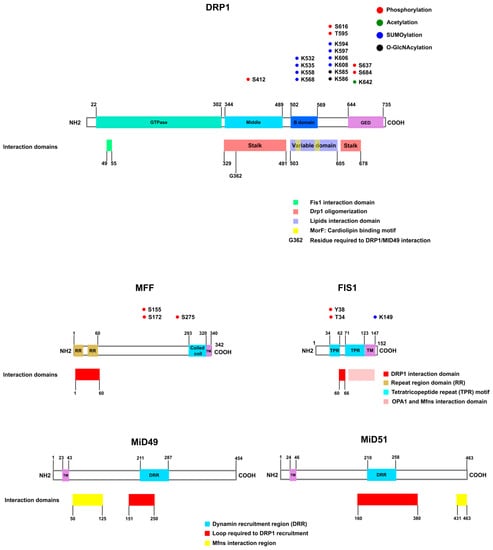
Figure 2.
Mitochondrial fission proteins. The main players in mitochondrial fission are DRP1 and its mitochondrial adaptors. DRP1 is a dynamin-like GTPase that comprises the dynamin type G domain at the N-terminus (green), a middle domain (light blue), a B domain (blue), and a GTPase effector domain (GED) (magenta) at the C-terminus. Several post-translational modifications occur at DRP1, such as phosphorylation (represented in red), acetylation (green), SUMOylation (blue), and O-GlcNAcylation (black). The interaction regions in DRP1, where DRP1 interacts with its adaptors or itself, described to date are shown. FIS1 interacts with the dynamin-type G domain on DRP1. DRP1 oligomerization occurs via intermolecular stalk interaction. The stalk of DRP1 comprises the middle domain and the GED. The variable domain (also called insert B) contains the lipid interaction residues and the cardiolipin interaction region. A glycine residue in the stalk domain is required for DRP1 interaction with MiD49. MFF presents two domain repeats (R1 and R2), a coiled-coil domain (CC), and the transmembrane segment (TM). Interaction with DRP1 occurs via RR1 and RR2. FIS1 is formed by two tetratricopeptide repeat (TPR) motifs and a transmembrane domain. FIS1 interacts with DRP1 via residues located at the TPR motif. Also, the region in FIS1 that interacts with Mitofusins and OPA1 is shown. The regions of interaction with DRP1 are presented in FIS1 and MFF. MiD49/51 contains a transmembrane domain at the N-terminus and a Dynamin Recruitment Region (DRR). The region of interaction between MiD49 or MiD51 and Mitofusins is also shown [95,96,98,101,143,144,145,146,147].
2.3. Mitochondrial Fusion Proteins
Mitochondrial fusion is regulated mainly by GTPase dynamin-related proteins MFN1 and MFN2 located at the OMM, and autosomal dominant optic atrophy 1 (OPA1) at the inner mitochondrial membrane (IMM). Mitochondrial fusion requires both MFN1 and OPA1, while MFN2 is not essential for this function [148].
Mitofusins are mitochondrial outer transmembrane GTPases that participate in mitochondrial fusion and in the maintenance of contacts between membranes. The large N-terminus domains of Mitofusins are located at the cytosol, and they contain the GTPase domain, which is followed by heptad repeat (HR) 1; two transmembrane domains; and then a C-terminus, coiled-coil HR2, which, according to classical models, is also exposed towards the cytosol [15,149].
Interestingly, bioinformatic analysis and biochemical assays have found only one transmembrane domain, so the HR2 domain would be exposed to the intermembrane space (IMS). Moreover, it has been proposed that HR2–HR2 domain interaction in the IMS would permit Mitofusin oligomerization, and redox-sensitive cysteine Cys 684 and Cys 700 are key to promoting mitochondrial fusion [150].
Mice lacking MFN1 or MFN2 are embryonically lethal. This observation points to these molecules playing a key role in embryonic development [151].
Human Mitofusins are 62% identical and 77% similar to one another, and their expression levels vary depending on the tissue. Moreover, several cells express both proteins, thereby indicating that they do not have the same biological function. Both MFN1 and MFN2 depletion lead to mitochondrial fragmentation in mouse embryonic fibroblast (MEF) cells, although in a different way and intensity [151]. While MFN1 shows higher GTPase activity and a greater effect on mitochondrial fusion than MFN2, both proteins can form homo- or heterodimers to mediate fusion [151].
After GTP binding, a rearrangement of residues around the nucleotide-binding pocket occurs, losing intramolecular HR1–HR2 interactions and exposing the HR2 to the cytosol. This allows the dimerization between both HR2 domains from two opposite Mitofusins. After dimerization and GTP hydrolysis, a conformational change induces the close state, dragging the opposite membranes close, allowing fusion [152,153,154,155,156].
This proposed model does not fit with Mattie and colleagues’ conclusions, whereby trans-dimerization occurs in the oxidative environment of the IMS that allows the formation of hydrogen bridges between two HR2s from two adjacent Mitofusins [150] (Figure 3).
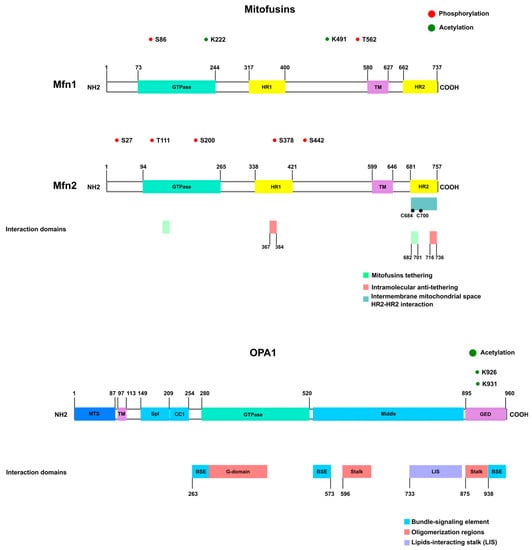
Figure 3.
Mitochondrial fusion proteins. Mitofusins are dynamin-like GTPases comprising the GTPase domain at the N-terminus, followed by HR1, the transmembrane region (TM), and HR2 at the C-terminus. The phosphorylated and acetylated residues are shown in red and green, respectively. The HR domains are essential for the anti-tethering and pro-tethering states. Intramolecular interaction between HR1 and HR2 is associated with the anti-tethering conformation (red). Upon nucleotide binding, a conformational change exposes the HR2 domain to the cytosol, allowing interaction with the HR2 from adjacent mitochondria and tethering (represented in green). The GTPase domain also participates in the interaction for fusion [152,153,154,155,156]. An alternative model proposed that HR2–HR2 domain interaction occurs in the intermembrane mitochondrial space [150]. Isoform 1 of OPA (the long form) presents the transmembrane region at the N-terminus, followed by a coiled coil (CC), G domain (GTPase), middle domain, and GED. OPA1 oligomerization involves the G domain, middle domain, and GED domain [152,156,157].
Several lines of evidence point to overlapping functional roles for MFN1 and MFN2, as well as exclusive functions. We will explore the specific functions of each protein below.
2.3.1. Mitofusin 1
MFN1 plays a key role in OMM fusion that is related to its GTPase activity. Surprisingly, in contrast to OPA1 and MFN2, no human pathologies related to MFN1 mutations have been identified to date. This observation thus suggests that MFN1 plays an important role during gestation and that the loss-of-function is incompatible with human life [158].
The metabolic effect of MFN1 KO or knockdown depends on the cell type. For example, the fragmentation observed in some tissues, such as the liver and myocardium, from MFN1 KO-diabetic mice is associated with an increase in mitochondrial respiration related to lipid metabolism. In the liver, MFN1 KO can have a protective role against insulin resistance because lipids are used as the main energy source. These mice also show an increase in Complex I activity [47]. However, in skeletal muscle or neurons, the reduction or ablation of MFN1 expression leads to a decrease in the mitochondrial oxidative phosphorylation system (OXPHOS), while in brown adipose tissue (BAT), MFN1 KO has no effect on energy expenditure (for review, see [159]).
The shift from oxidative to glycolytic metabolism and mitochondrial fission has been described in several types of cancer (lung, ovarian, colorectal, and pancreatic, among others). However, fusion and oxidative metabolism were also found in pancreatic cancer, suggesting that changes in mitochondrial dynamics favor or support cancer development (for review, see [159]). In hepatocellular carcinoma (HCC), MFN1 downregulation is related to metastasis and poor prognosis. MFN1 depletion triggers the epithelial–mesenchymal transition, and the inhibition of MFN1 GTPase activity via FUNDC2 interaction is associated with metabolic reprogramming and tumor growth [160,161]. In addition to causing metabolic changes, MFN1 can affect cell death. In this regard, MFN1 KO MEF cells are reported to be resistant to apoptotic stimuli [160]. This resistance was attributed to the inefficient accumulation of BAX on the OMM with incorrect curvature caused by hyper-fragmentation.
Both MFN1 and MFN2 interact with BAX and BAK. However, only the former is related to apoptosis. After apoptotic stimuli, BAK dissociates from MFN2 and associates with MFN1, facilitating BAK oligomerization and then cytochrome c release and cell death [162,163,164].
2.3.2. Mitofusin 2
While MFN1 is the main player in OMM fusion, MFN2 plays a more prominent role in metabolism regulation. MFN2 stimulates mitochondrial respiration, glucose oxidation, and mitochondrial membrane potential, and its effects are not related to GTPase or fusion activity. MFN2 appears to regulate the expression of subunits of all OXPHOS complexes. In this regard, MFN2 loss-of-function reduces the subunits from OXPHOS Complexes I, II, III, and V, and the overexpression of this Mitofusin increases the expression of subunits related to Complexes I, IV, and V [165].
MFN2 expression was induced in mouse muscle during myogenesis, and its depletion led to mitochondrial fragmentation, reduced glucose oxidation and cell respiration, loss of mitochondrial membrane potential, and proton leak [3].
MFN2 is also present at the ER, and it interacts with MFN1 or MFN2 located on the OMM [166]. MFN2 location at the ER is essential for MFN2 function related to mitochondrial metabolism. This interaction is important to maintain contacts between these two organelles [166,167,168] and for Ca2+ and PS transfer from the ER to mitochondria, thus increasing mitochondrial metabolism and phosphatidylethanolamine (PE) synthesis [73,169].
Furthermore, MFN2 also plays a key role in maintaining mitochondrial contacts with other membranous structures, such as LDs and melanosomes (for review, see [170]). Although the effect of MFN2 at these contacts is still being studied, MFN2 knockdown frequently reduces the contacts between mitochondria and other membranes.
MFN2, but not MFN1, plays a crucial role in the ER stress response. MFN2 loss of function in MEF cells leads to an increase in ER chaperone levels. Also, ER stress caused by thapsigargin or tunicamycin induces an increase in MFN2 expression [171]. The depletion of MFN2 in MEF cells causes basal activation of the PERK pathway, and, despite exacerbation of the unfolding protein response (UPR), these cells are not able to activate apoptosis and/or autophagy [172].
Mutations in MFN2 cause the autosomal dominant neuropathy named Charcot–Marie–Tooth type 2A (CMT2), characterized by chronic axonal neuropathy [173,174]. So far, more than 100 different MFN2 mutations have been described in CMT2 patients [173,174,175,176,177]; however, the molecular defects are not completely understood.
MFN2 mutations have already been associated with reduced axonal mitochondrial activity [178,179], but mitochondrial morphology changes are not so clear and are dependent on the specific mutation and the cell type studied. Electron micrographs of the sural nerve from biopsies of two patients showed small, electron-dense, and swollen mitochondrial axons [176]. On other hand, cells from patients with different MFN2 mutations did not show any mitochondrial changes in morphology [180], with the exception of fibroblasts derived from patients with R364W mutations that showed changes in mitochondria–ER contacts. R364W cells present an increase in the length of mitochondria–ER contacts and also an increase in the distance between organelles. The cholesterol metabolism and lipid droplet formation also increased in these cells [180]. Studies with MFN1 and MFN2 double KO cells showed that the overexpression of MFN2 mutants induces several different phenotypes confirming that endogenous MFN1 or non-mutated MFN2 can compensate for the defects induced by MFN2 mutations [181]. Moreover, a study with a small MFN2 agonist showed effects only when endogenous MFN1 was present [179]. As CMT2A patients are heterozygous, endogenous MFN1 and non-mutated MFN2 can compensate for defects in mitochondria morphology and respiration.
Most mutations detected in CMT2A are missense located at the GTPase or coiled-coil domains (Figure 3). R94 was found mutated in several studies (R94Q or R94W), as were R280H and R104W or R364W (between GTPase and HR1) and T105M [177].
Rocha and colleagues have proposed that MFN2 activity is mediated by the interaction between Methionine 376 and Histidine 380 with Aspartate 725 and Leucine 727 and the phosphorylation at Serine 378 [179]. They showed that a MFN2 agonist mimics the peptide–peptide interface of MFN2, allosterically activating it, and promoting mitochondrial fusion. The agonist does not show activity above the mutants but increases the activity of functional MFN2 rescuing the defects associated to CMT2A [179].
Mitochondrial dysfunction is associated with insulin resistance, and muscles from obese or type 2 diabetic patients show a reduced expression of MFN2 [182].
MFN1 and MFN2 act together to maintain glucose homeostasis by preventing TFAM loss and consequently preserving the mtDNA content in β-cells. Neither MFN1 nor MFN2 is individually essential to preserve β-cell function, but together they coordinately promote the regulation of glucose levels and glucose-stimulated insulin secretion [183].
In BAT from mice on a high-fat diet, the deletion of MFN2, but not MFN1, reversed mitochondrial dysfunction, leading to an increase in insulin sensitivity and resistance to obesity [184]. In contrast, this deletion in skeletal muscle and liver promotes mitochondrial fragmentation, a decrease in oxidative phosphorylation, glucose intolerance, and enhanced hepatic gluconeogenesis [185,186]. In mouse skeletal muscle, MFN2 protected mitochondria from damage. The reduction of MFN2 in that tissue led to a decrease in autophagy and caused the accumulation of dysfunctional mitochondria, a phenotype related to aging [187].
MFN2 has a protective role in liver diseases such as non-alcoholic steatohepatitis (NASH). Liver-specific ablation of MFN2 in mice results in inflammation, fibrosis, and liver cancer. These effects are related to the role of MFN2 in mitochondria–ER contacts and are associated with a reduction of PS transfer and thus decreased phospholipid synthesis, and consequently ER stress [73].
MFN2 KO in neurons leads to a reduction in neuritic growth, and this is reversed by the expression of an ER-targeted MFN2 [169]. MFN2 is involved in mitochondrial quality control by participating in Parkin recruitment and then autophagy [188].
2.3.3. OPA1
Autosomal dominant optic atrophy (OPA1) is the mammalian ortholog of yeast Mgm1p, and it is associated with mitochondrial fusion [189]. The name refers to the observation that mutations in this gene cause inherited optic neuropathy [190,191].
OPA1 is essential for mitochondrial fusion. However, independent of this function, it also has effects on cristae formation and mtDNA maintenance. Indeed, in the case of energetic stress, cells harboring OPA1 fusion-deficient mutants are able to sustain ATP levels and retain cristae structure [17,192,193].
Delettre and colleagues reported eight mRNA isoforms for OPA1. All OPA1 variants contain a cleavage site, S1, for mitochondrial processing peptidase, and some of them have an additional cleavage site, S2 [194].
After translation, OPA1 splicing variants are processed and can produce several proteins (at least five distinct bands in SDS-PAGE from 80–100 kDa are identified). OPA1 is translated with a matrix-targeting signal (which is cleaved by the mitochondrial processing peptidase (MPP) in the mitochondrial matrix) followed by the transmembrane domain (Figure 3). After MPP cleavage, the mature OPA1 isoform (also named L-isoform) in the matrix can be further processed, giving rise to short variants. This processing can occur through cleavage sites S1 (Exon 5) and S2 (Exon 5b) [195,196].
Song and colleagues conducted assays using MEF OPA1 null cells expressing individual OPA1 splicing variants. They observed that cells expressing OPA1 isoforms 3, 5, 6, or 8 showed predominantly fragmented mitochondria, and none of them expressed the long OPA1 protein form (L-OPA1) [196].
The expression of the long form of OPA1(L-OPA1) is always accompanied by a short form (S-OPA1). In complementation assays, both short and long forms were found to be necessary for efficient mitochondrial fusion, thereby indicating that S1 cleavage is essential for OPA1 activity related to fusion. Indeed, assays using proteoliposome (OPA1 + phosphatidylcholine) and liposome (cardiolipin and phosphatidylcholine) to simulate the IMM and OMM showed heterotypic tethering between cardiolipin and L-OPA1 before GTP hydrolysis and fusion. This tethering can be facilitated by S-OPA1. Also, this assay revealed that cristae maintenance is due to trans-OPA1 tethering when low levels of cardiolipin are present [197].
The origin of short isoforms was initially associated with mitochondrial membrane potential loss and mitochondrial fragmentation [195]. However, it is now known that, in addition to inducing S1 cleavage, CCCP, a mitochondrial membrane potential uncoupler, causes the degradation of L-OPA1 forms. Moreover, CCCP does not induce S2 cleavage [196]. Despite morphological differences, the cristae structure, amount of mtDNA, and energy efficiency can be regulated for any OPA1 isoform, regardless of length, independently of protein products [198].
The regulation of OPA1 is highly complex, and the generation of small isoforms depends on the activity of at least three proteases, namely m-AAA; a matrix metalloprotease named OMA1 (overlapping with m-AAA protease); the inner membrane rhomboid PARL (presenilin-associated rhomboid-like) protease; and the inner membrane protease, named i-AAA protease YME1L (ATP-dependent protease yeast mtDNA escape 1-like). YME1L cleaves OPA1 at the S2 site while OMA1 uses S1 [195,196,199].
OPA1 cleavage by OMA1 is increased under different types of cellular/mitochondrial stress and is regulated by autocatalytic proteolysis, allowing the reversibility of fusion inhibition [200]. The ablation of OMA1 in mice leads to hepatic steatosis, decreased energy expenditure, and impaired thermogenic regulation associated with transcriptional changes in genes involved in lipid and glucose metabolism [199].
YME1L, on the other hand, cleaves OPA1 under increased OXPHOS activity, and it is degraded in ATP-depleted cells. YME1L deficiency is associated with heart failure, impaired eye development, and spinal cord degeneration. OMA1 and YMEL1 can be reciprocally degraded depending on mitochondrial conditions (ATP levels, membrane potential state, OXPHOS activity, etc.) [46,201,202,203,204].
PARL is related to the antiapoptotic function of OPA1, such that PARL ablation in mice leads to muscle loss, progressive cachexia, and death at only 8 weeks of age, and these effects are associated with extensive lymphocyte apoptosis. Indeed, PARL cleaves OPA1, thereby generating soluble short OPA1 forms that are located in the IMS. OPA1 plays an anti-apoptotic role because it maintains the cristae structure by preventing cytochrome c release via the presence at IMS of the oligomers formed after PARL cleavage [192,205]. PARL also has a key role in the respiratory chain because it regulates coenzyme Q biosynthesis and Complex III activity, and its deficiency causes alterations in mitochondrial structure, neuronal necrosis, and a phenotype similar to that of Leigh syndrome [206].
Since OPA1 was first linked to dominant optic atrophy, a disease that affects retinal ganglion cells and the optic nerve, several other diseases related to the nervous system, namely spastic paraplegia, multiple sclerosis-like syndrome, parkinsonism, and dementia, have been associated with OPA1 gene mutations (for review, see [207]).
Mutations in the OPA1 gene are found in 32–90% of patients with autosomal dominant optic atrophy (ADOA) [208,209,210,211]. More than 150 mutations have already been identified [211], and most of them cause premature truncations of OPA1 and are located at the GTPase or GED domains [208,212]. V903G, R290Q, R904D, N158S, and E907G are among the most frequent mutations [190,211]. Also, there are mutations in splicing sites resulting in changes in OPA1 isoform expression. A change in a splicing site can lead to exon skipping or amino acid residue substitution (D438Y or D438H) that can affect protein dimerization [211].
Studies in patient-derived cells have documented aberrant cristae structure and mitochondrial fragmentation (G300E or R290Q mutations, for example). On the other hand, OPA1 58 delta (with a frame-shift deletion of 58 residues in the GED domain) showed normal filamentous mitochondria, although OPA1 protein expression was almost half compared to control cells [212]. Cartes-Saavedra and colleagues (2023) showed that GED is not necessary for OPA1 oligomer formation and fusion, but it is necessary for GTPase activity [213].
A mutation found in lymphoblastoid cell lines derived from Asian ADOA-affected patients led to P400A change; decrease in mtDNA; and defects in respiratory capacity, ATP synthesis, and mitochondrial membrane potential. These conditions caused increased oxidative damage and apoptosis [214].
OPA1 deficiency is also related to inflammation and myopathies. OPA1 ablation in the skeletal muscle of mice results in mitochondrial fragmentation, reduced respiration, ER stress, and the secretion of systemic inflammatory markers and FGF21 [215,216,217]. OPA1 KO mice also show improved glucose tolerance and are resistant to age- and diet-induced insulin resistance [216,217]. The OPA1 role in browning is not well understood because mice with selective KO of OPA1 in brown adipose tissue present increased levels of FGF21 and enhanced browning of white adipose tissue [218]. On the other hand, OPA1 overexpression has been associated with white adipose tissue expandability and the improvement of glucose tolerance and insulin sensitivity [219].
OPA1 is also important for cancer development, although its function or final effect is not clear. In breast cancer, OPA1 upregulation is associated with poor prognosis. In addition, in hepatocellular carcinoma (HCC) and lung adenocarcinoma, OPA1 expression determines the response to chemotherapeutics, and OPA1 knockdown sensitizes cells to the treatment. OPA1 involvement in tumor development may be related to its role in angiogenesis [220,221,222].
2.4. Mitochondrial Fusion Protein Phosphorylation
Mitofusin 1 and Mitofusin 2 Phosphorylation
The mechanisms that regulate Mitofusin activity are poorly understood. To date, only two MFN1 residues have been described as phosphorylated, affecting the activity or organization of the protein. MFN1 is phosphorylated at T562 by ERK1/2 after apoptotic stimuli, leading to MFN1 oligomerization inhibition and mitochondrial fragmentation [162].
MFN1 is also phosphorylated at S86 by PKC IIB, leading to a decrease in MFN1 GTPase activity and, consequently, the inhibition of fusion [223].
For MFN2, on the other hand, several target residues and kinases have been documented. However, these phosphorylations are associated with MFN2 degradation and not the regulation of MFN2 activity [188,224,225].
JNK1 and PINK1 phosphorylate MFN2 at residues S27 and S442, respectively. These phosphorylations permit MFN2 ubiquitination by Parkin and subsequently mitophagy activation [188].
MFN2 is also phosphorylated at S442 by AMPK upon energy stress, leading to mitochondrial fission, autophagy, and an increase in the number of contacts between the ER and mitochondria [226]. As mentioned before, AMPK also phosphorylates MFF. Hu and colleagues suggested that, during acute stress, AMPK phosphorylates MFN2, inducing autophagy and survival. However, under prolonged stress, the association of AMPK with MFF is increased, thereby reinforcing mitochondrial fission and autophagy [226].
Recently, MFN2 was identified as a substrate of mTORC [227]. MFN2 S200 is phosphorylated, and it leads to an increase in GTPase activity and respiration. This phosphorylation is particularly interesting in the context of cancer because cancer cells usually show dysregulated fusion/fission processes. The authors propose that MFN2 S200 phosphorylation in cancer tissues increases MFN2/Pyruvate kinase isoform 2 (PKM2) interaction, thereby favoring mitochondrial fusion and the switch between OXPHOS and glycolysis. This interaction would be a mechanism with which to protect mitochondria from over-fragmentation. Table 2.
Table 2.
Phosphorylation sites of mitochondrial dynamics proteins.
3. Other Post-Translational Modifications of Mitochondrial Dynamics Proteins
In addition to phosphorylation, several other post-translational modifications have been reported in mitochondrial dynamics proteins. Additional DRP1 post-translational modifications include acetylation [229], o-GlcNAcylation [230,231], and SUMOylation [232,233]. FIS1 is SUMOylated [234] and acetylated [235]. MFN1 and MFN2 are SUMOylated [236], and MFN1 acetylation has also been reported [42,237]. OPA1 is acetylated [238] (Figure 2 and Figure 3).
3.1. Mitochondrial Dynamics Protein Acetylation
Protein acetylation is a reversible introduction of an acyl group at the ε-sidechain of a lysine residue in a target protein. This process neutralizes the lysine charge and favors alterations in protein conformation [239]. A wide family of acetyltransferases and deacetylases has been reported [240]. Surprisingly, total cellular proteome studies reveal that 20% of mitochondrial proteins are acetylated, among which 80% are mitochondrial metabolic enzymes [241]. Additional research evidences an inverse relation between proteome acetylation and mitochondrial metabolism [242]. Thus, acetylation–deacetylation post-translational modifications play a critical role in sustaining the cellular metabolism balance.
Overnutrition induced by a high-fat diet (HFD) results in an increase in fatty acid oxidation and the accumulation of acetylCoA. In addition, excessive oxidative stress can decrease the NAD+ pool required to activate deacetylases. Thus, under metabolic stress, protein acetylation is enhanced. Several studies suggest that increased acetylation of mitochondrial dynamics proteins promote mitochondrial fragmentation and decreased mitochondrial function.
In this regard, HFD-induced obesity increases DRP1 acetylation at K642 and promotes cardiac dysfunction in mice. Moreover, DRP1 K642R, a deacetylation mimic mutation, suppresses DRP1 oligomerization and mitochondrial fission in cardiomyocytes. These findings suggest that DRP1 acetylation impairs the function and dynamics of mitochondria through exacerbated fission in these organelles. In line with this, OPA1 acetylation and mitochondrial fragmentation have been reported in pathological heart hypertrophy models, including db/db mice, pressure overload transverse aortic constriction (TAC), and angiotensin chronic infusion [229].
SIRT3, a mitochondrial deacetylase, decreases OPA1 acetylation and promotes its GTPase activity, thereby activating mitochondrial fusion in the heart [238]. Additionally, SIRT3-dependent OPA1 deacetylation promotes the differentiation and OXPHOS activation of human cardiomyocytes derived from human induced pluripotent stem cells (hiPSC) [243].
MFN1 acetylation has been explored in MEF cells and skeletal muscle. Under nutrient starvation, MFN1 is associated with HDAC6 deacetylase, and it increases mitochondrial elongation. Moreover, HDAC6 ablation suppresses mitochondrial elongation in MEF cells and the tibialis anterior muscle of HDAC6 KO mice upon fasting. Also, a mutation in the potential acetylation lysine residue K222 promotes mitochondrial elongation [237]. Future studies should seek to validate MFN1 acetylation at K222 using mass spectrometry.
On the other hand, studies suggest that the acetylation of mitochondrial dynamics proteins serves as a label for mitochondrial quality control. Under acute mitochondrial stress induced by antimycin, rapid and transitory mitochondrial network elongation improves metabolism and decreases oxidative stress [11]. These effects on mitochondrial morphology and function are associated with increased acetylation of MFN1, which allows the activation of mitochondrial quality control [42]. MFN1 acetylation at K491 facilitates interaction with MARCH5, a ubiquitin ligase. Under acute mitochondrial stress, the mitochondrial network is hyperfused, and the quality control of the mitochondrial network is activated. MARCH5 ubiquitinates acetylated MFN1 and promotes its turnover under conditions of mitochondrial stress [42].
In line with these observations, a study using MEF cells and hiPSC demonstrated that acetylation facilitates FIS1 ubiquitination and degradation. Cellular reprogramming to pluripotency requires mitochondrial fission and acetyl carboxylase 1 (ACC1)-dependent de novo lipid synthesis. ACC1 expression decreases intracellular acetylCoA, whereby FIS1 is deacetylated and mitochondrial fission is activated. Hence, crosstalk between lipid metabolism and mitochondrial fission mediated by ACC1 allows human fibroblast reprograming [235] (Table 3).
Table 3.
Acetylation of mitochondrial dynamics proteins.
These results give rise to the question: are there specific acetylation sites in mitochondrial dynamics proteins that cause mitochondrial fission and mitochondrial metabolism suppression, and are others associated with mitochondrial turnover?
3.2. SUMOylation of Mitochondrial Dynamics Proteins
SUMOylation is an enzymatic and reversible conjugation of small ubiquitin-like modifier (SUMO) proteins at lysine residues of target proteins. Mediated by SUMO 1 or SUMO 2/3, the reaction can be reversed by several isoforms of SUMO-specific peptidases (SENPs).
Recent work reports that FIS1 is SUMOylated at K149 by SUMO2/3, and this modification suppresses FIS1 localization at the OMM. Moreover, the authors demonstrated that FIS1 deSUMOylation at K149 by SENP3 allows mitophagy to take place in deferoxamine-treated cells [234]. Consistent with this finding, it has been reported that neuronal ischemic stress promotes PERK-dependent SENP3 lysosomal degradation. Under this condition, DRP1 remains SUMOylated, and the mitochondrial network is elongated. In contrast, reperfusion restores SENP3 levels, promotes DRP1 deSUMOylation and mitochondrial fragmentation, and triggers apoptosis [233].
Additional work has revealed a close relationship between DRP1 S616 phosphorylation and SUMOylation. DUSP6 is a redox-sensitive phosphatase that dephosphorylates DRP1 at S616 and promotes mitochondrial elongation. Under oxidative stress or ischemic/reperfusion, DUSP6 is deSUMOylated by SENP1, ubiquitinated, and degraded. Thus, DRP1 phosphorylation at S616 is sustained under stress conditions, and the mitochondrial network is fragmented [244]. This mechanism supports the notion that deSUMOylation by SENP3 protease might modulate mitochondrial architecture and metabolism.
In contrast, several studies describe that SUMOylation promotes mitochondrial fission and apoptosis. DRP1 is SUMOylated by SUMO1 during staurosporin-activated apoptosis. This modification helps stabilize mitochondria–ER contacts to promote the activation of apoptosis. Furthermore, DRP1 SUMOylation is required for mitochondrial calcium uptake and cristae remodeling [245].
In addition, DRP1 SUMOylation appears to play a key role in tumor growth and metastasis. Cancer progression has been associated with an increase in mitochondrial fission and a decrease in mitochondrial fusion protein expression. In breast cancer cells, SYNJ2BP-COX16 (a readthrough of synaptojanin 2 binding protein and COX16) SUMOylation regulates mitochondrial morphology through DRP1 SUMOylation and DRP1 phosphorylation at S616. Hence, mitochondrial network fragmentation occurs, thereby contributing to cancer cell proliferation and metastasis [246].
Furthermore, a recent report demonstrates that mitochondrial fusion proteins MFN1 and MFN2 are SUMOylated by SUMO 1 under metabolic stress induced by CCCP treatments. The authors propose that the SUMOylation of these Mitofusins helps to select and separate depolarized mitochondria from the mitochondrial network. Moreover, this step may be necessary to recruit mitophagy machinery at the mitochondrial membrane [236].
Emerging evidence points to an association between SUMOylation and acetylation post-translational modifications. In this regard, in fasting conditions, SENP1 deSUMOylase is translocated to mitochondria, and it removes SIRT3 SUMOylation at K288 and promotes SIRT3 deacetylase activity [247]. Moreover, a SIRT1 K223R (mouse homolog K288 residue) mutation that suppresses SIRT1 acetylation improves metabolism. Thus, SIRT1 K223R transgenic mice show less weight gain than control mice under HFD-induced obesity [247].
In this regard, a report describes a metabolic link between protein SUMOylation, acetylation, and modulation of the metabolic reprogramming required for the development of memory lymphocytes T (TM). TM cells show reduced levels of acetylated proteome compared to effector lymphocytes T (TE). Moreover, AMPK activates the SENP1-SIRT3 axis in TM cells. Thus, YME1L1 deacetylation by SIRT3 reduces its protease activity, increases L-OPA1 levels, and promotes mitochondrial fusion and OXPHOS activation in TM cells [248] (Table 4).
Table 4.
SUMOylation of mitochondrial dynamics proteins.
3.3. O-GlcNAcylation of Mitochondrial Dynamics Proteins
Nutrient overload increases oxidative stress and mitochondrial dysfunction and impairs OXPHOS activity [56,250,251]. Several studies have linked o-GlcNAcylation to decreased lipid and glucose metabolism. Recent work revealed that conditional inducible overexpression of GLUT-4 in the heart of diabetic mice promotes an increase in o-GlcNAcylation, mitochondrial dysfunction, and diastolic dysfunction [252]. Knockout mice for O-GlcNAc transferase (OGT), an enzyme that transfers GlcNAc from UDP-GlcNAc to protein threonine or serine residues, with HFD-induced obesity show an increase in lipolysis in visceral fat [253]. Moreover, a decrease in perlipin1 (Plin1) o-GlcNAcylation by OGT promotes its phosphorylation and lipolysis [254].
DRP1 o-GlcNAcylation has been detected in the heart of streptozotocin-induced diabetes in mice [230]. o-GlcNAcylation of DRP1 at T585 and T586 in cardiomyocytes decreases DRP1 S637 phosphorylation and increases mitochondrial fragmentation in cardiomyocytes [230]. In addition, the ablation of O-GlcNAcase (OGA), a glycosidase that removes o-GlcNAcylation, increases mitochondrial fragmentation, decreases the NDUFB8 complex I subunit, and promotes DRP1 o-GlcNAcylation [231]. A study suggests that OPA1 is also O-GlcNAcylated upon high glucose treatments. This modification is associated with dysfunction of both mitochondrial fission and OXPHOS activity. Further investigation should be directed to demonstrate the direct O-GlcNAcylation of OPA1 protein [255].
Taken together, these studies suggest that O-GlcNAcylation of mitochondrial dynamics leads to mitochondrial dysfunction. Hence, this post-translational modification could be a potential target for drug design.
4. Participation of Lipids in Mitochondrial Morphology
4.1. Mitochondrial Phospholipids
In addition to the mitochondrial dynamics proteins mentioned above, a specific phospholipid composition of the OMM and IMM is required for the transient rupture of the membrane bilayer through fusion and fission processes. In this regard, a proper phospholipid composition is important not for only maintaining membrane fluidity and curvature but also for the correct membrane binding of protein machineries that allow mitochondrial biogenesis and function [256,257,258].
Mitochondria cannot produce phospholipids such as phosphatidylcholine (PC), phosphatidylinositol (PI), or phosphatidylserine (PS), which are derived from other cellular organelles. However, they can produce cardiolipin (CL), phosphatidylethanolamine (PE), and phosphatidylglycerol (PG) using the lipid precursors phosphatidic acid (PA) and PS, which are imported from the ER [72].
4.2. Mitochondrial Phospholipid Composition
The phospholipid compositions of the OMM and IMM differ significantly. Most CL is localized in the IMM and accounts for 15–20% of the phospholipids present in this membrane [259], while PC is the most abundant phospholipid in both membranes [260]. This phospholipid composition generates unique mitochondrial membrane properties.
Whereas PC, PI, and PS are cylindrical-shaped phospholipids that promote bilayer formation, PE, CL, and PA are conical-shaped, and they impose negative curvature stress on the membrane, allowing the formation of non-bilayer structures that facilitate fission and fusion events and cristae organization [261].
Mitochondrial membrane fusion and fission can be modulated by non-bilayer phospholipid concentrations. It has been reported that cardiomyocytes from Barth Syndrome patients, in whom CL level is decreased, show enlarged mitochondria and abnormal cristae morphology [262]. In this regard, CL is needed for IMM fusion, via its interaction with OPA1 [197], and for mitochondrial fission, through protein–lipid interaction with the B-insert domain of DRP1, which enhances its oligomerization and assembly (Figure 2) [144,263].
At the IMM, PE plays crucial roles in maintaining the tubular morphology and respiratory function of mitochondria. Studies demonstrate that when mitochondrial PE content is reduced by ~20%, mitochondrial oxygen consumption and ATP production decrease, consistent with defects in OXPHOS Complexes I and IV [264]. In addition, cells lacking phosphatidylserine decarboxylase (PSD), the enzyme that catalyzes the formation of PE from PS in mitochondria, present mitochondrial fragmentation and a swollen morphology [265,266].
On the other hand, PA, a minor component of the mitochondrial membranes, is a negative regulator of mitochondrial fission through its interaction with DRP1. Mitochondrial phospholipase D (MitoPLD), an enzyme that catalyzes the formation of PA from phospholipids such as PC or CL, mediates the binding of DRP1 to the PA head group at the OMM. The activity of DRP1 is thus inhibited, thereby impairing mitochondrial fission [267]. In this regard, recent studies have demonstrated that several proteins involved in maintaining the lipid composition of mitochondrial membranes modulate mitochondrial morphology (Table 5).
Mitochondrial dysfunction induced by lipid dysregulation, either by alteration of synthesis, turnover, or transport, has been associated with the genesis and progression of several diseases. Detailed information about diseases triggered by defective mitochondrial membrane phospholipids can be found in the recent review by Dong et al. [268]. This section will focus on mitochondrial lipid transfer proteins (LPTs) and scramblases involved in the maintenance of mitochondrial PL levels.
Table 5.
Proteins that sustain mitochondrial membrane lipid composition.
Table 5.
Proteins that sustain mitochondrial membrane lipid composition.
| Protein | Function | Localization | Protein Loss-of-Function | Ref. |
|---|---|---|---|---|
| Mitochondrial Phenotype | ||||
| PSD | Conversion PS to PE | Mitochondria | Mitochondrial fragmentation | [264,265] |
| MitoPLD | Conversion PA to PC and CL | Mitochondria | Mitochondrial fragmentation | [269] |
| StarD7 | Lipid transfer activity, PC distribution into mitochondria | Mitochondria | Mitochondrial fragmentation, mitochondrial cristae alterations | [270,271,272] |
| ORP5 | Transfer PS from ER to mitochondria | MCSs | Mitochondrial cristae alterations | [273,274] |
| ORP8 | Transfer PS from ER to mitochondria | MCSs | Mitochondrial cristae alterations | [273,274] |
| MFN2 | Tethering | MCSs, mitochondria | Mitochondrial fragmentation | [166] |
| PTPIP51 | PE to MCSs | MCSs, mitochondria | - | [275] |
| MIGA2 | Tethering, transfer PS from ER to mitochondria | LD–mitochondria contact, MCSs | Mitochondrial fragmentation | [74,276] |
| PLIN1 | Scaffold | LD–mitochondria contact | Mitochondrial cristae alterations | [277] |
| PTPMT1 | Phosphatase | Mitochondria | Mitochondrial fragmentation | [278] |
| VPS13A | Tethering | MCSs | Mitochondrial fragmentation | [279] |
| VPS13C | Tethering | MCSs | Mitochondrial fragmentation | [280] |
| VPS13D | Tethering | MCSs | Mitochondrial fragmentation | [34] |
| VAP-A | Tethering | MCSs | Mitochondrial fragmentation | [281] |
| PRELID1 | Lipid transfer activity, supply PA for CL biosynthetic | Mitochondria | Mitochondrial fragmentation | [282] |
| TRIAP1 | Lipid transfer activity, supply PA to the CL biosynthetic pathway | Mitochondria | Mitochondrial fragmentation | [282] |
| TAFFAZIN | Acyl transferase | Mitochondria | Reduced cristae density | [283] |
| PLSCR3 | CL transference between OMM and IMM | Mitochondria | Mitochondrial cristae alterations | [284] |
| ABHD16A | PS to Lyso-PS | Endoplasmic reticulum, MCSs | Mitochondrial fragmentation | [8] |
4.3. Lipid Transfer Proteins
LTPs play a major role in regulating the lipid composition of mitochondrial membranes by promoting non-vesicular lipid transport. The function of LTPs at mitochondrial contact sites has recently been addressed. LTPs contain a lipid-binding/transfer domain that include the Steroidogenic Acute Regulatory Protein-related Lipid Transfer (START); the oxysterol-binding protein (OSBP)-related proteins; or the proteins of relevant evolutionary and lymphoid interest (PRELI)-like domains, which act as transporters, tethers or lipid sensors at contact sites between mitochondrial membranes and the ER or LD membranes [285] (Figure 4).
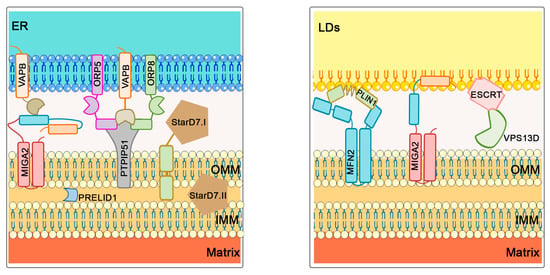
Figure 4.
Phospholipid transport at ER–mitochondria and LD–mitochondria contact sites. MIGA2 binds mitochondria directly to LDs, and mitochondria to the ER by interacting with the ER protein VAP-A/B. ORP5 and ORP58 mediate the transfer of PS from the ER to mitochondria and interact with VAPB and PTPIP51 proteins, while StarD7 regulates PC distribution. Across the mitochondrial membranes, PRELID1 is responsible for PA transfer across the membranes, allowing CL biosynthesis in the IMM. In addition to MIGA2, the PLIN1 protein is also involved in the formation of LD–mitochondria contact sites by interacting with MFN2. VPS13D protein is known to interact with ESCRT on the surface of LDs, thereby also allowing FA transport.
The lipid transfer protein StarD7 regulates the distribution of PC within mitochondrial membranes and the ER. It is synthesized as a precursor form, StarD7-I, which contains a mitochondrial targeting sequence (MTS) and is cleaved by the mitochondrial peptidases MPP and PARL to produce the mature form StarD7-II [286]. StarD7 deficiency modifies mitochondrial network morphology and mitochondrial cristae structure, thereby causing mitochondrial dysfunction [270,271,272].
Two members of the OSBP family, the lipid transfer proteins ORP5 and ORP8, transfer PS from the ER to mitochondria and are required for sustaining mitochondrial morphology and respiration [273,274]. In addition, MFN2, which promotes mitochondria–ER tethering [287], also facilitates PS transfer from the ER to the mitochondria [73].
The protein tyrosine phosphatase-interacting protein 51 (PTPIP51) is reported to regulate CL levels by transferring its precursor PA via its TPR domain at mitochondrial–ER contact sites, and this transfer is independent of its interaction via its FFAT-like motif with the ER-VAMP associated protein (VAP) B [275]. Furthermore, PTPIP51 interacts with ORP5/8, thereby pointing to a functional link to lipid transfer [273].
MIGA2 is the only LD–mitochondria-binding protein that directly binds to both organelles via its amphipathic fragment. It is a transmembrane protein localized in LDs whose FFAT motif binds to the ER protein VAP-A/B, which promotes the formation of mitochondria–ER contacts and links reactions of de novo lipogenesis in mitochondria to triglyceride production in the ER [288]. MIGA2 also plays a role in mitochondrial morphology as its depletion leads to mitochondrial fragmentation [74,269]. Additionally, recent studies show evidence that MIGA2 binds and transports PS at mitochondria–ER contacts [74,276].
The perilipin 1 (PLIN1) protein is also involved in the formation of LD–mitochondria contact sites in BAT via its interaction with MFN2. In this model, depletion or knockout of MFN2 results in fewer LD–mitochondria membrane contact sites (MCS), altered lipid metabolism, and reduced FA oxidation by mitochondria [289].
Vacuolar protein sorting 13 (VPS13) A is involved in ER binding to mitochondria and the regulation of lipid transfer [279]. Loss-of-function mutations in VPS13A are associated with Chorea Acanthocytosis, a Huntington-like syndrome associated with abnormalities in red blood cells [290,291]. VPS13C depletion is linked to mitochondrial fragmentation, impaired respiration, and increased mitophagy, and it is associated with Parkinson’s disease [280]. An FFAT motif known to interact with VPS13A and VPS13C is present in both proteins [292], and it can be strengthened and reduced via phosphorylation of the FFAT motif [293,294].
VPS13D has a ubiquitin-binding domain required for mitochondria–ER contacts and mitochondrial clearance, and its loss results in enlarged mitochondria [295]. VPS13D is known to interact with ESCRT on the surface of LDs, transferring FA and contributing to membrane remodeling in that region [296]. Loss-of-function mutations in VPS13D cause autosomal recessive ataxia with abnormal mitochondrial morphology and reduced energy generation [297].
After being taken up into the mitochondria, phospholipids need to be distributed between the two mitochondrial membranes. The PRELID1/TRIAP1 protein complex transfers PA across the membranes, providing PA to the CL biosynthetic enzymes in the IMM [282].
4.4. Phospholipid Scramblases
The asymmetric distribution of phospholipids across the membrane bilayer necessary to preserve mitochondrial morphology depends on scramblases. These proteins facilitate lipid translocation from one membrane leaflet to another. Several scrambling proteins, such as PLSCRs, TMEM16 family members, and Xkr, facilitate the mixing of membrane lipids. In mitochondrial membranes, Phospholipid Scramblase 3 (PLSCR3) is responsible for CL transfer between the OMM and IMM [298,299]. It plays an important role in regulating mitochondrial morphology and respiratory function, as cells expressing a mutant PLS3 (F258V) have fewer and larger mitochondria with densely packed cristae as compared to control cells and have reduced oxygen consumption and intracellular ATP [284].
In addition to the transfer proteins mentioned above, mitochondrial morphology is regulated by the increased level of ER hydrolase ABHD16A, which mediates the hydrolysis of PS to lysophosphatidylserine (Lyso-PS) [300]. ABHD16A is required for ER-associated mitochondrial membrane constriction, and its depletion affects the recruitment of fission and fusion machinery, not only due to changes in Lyso-PS levels but also due to its ability to depalmitoylate mitochondrial transmembrane proteins [301]. Metabolic dysregulation of lyso-PS levels leads to the human genetic neurological disorder PHARC (polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataracts).
5. Pharmacological Approaches to Modulate Mitochondrial Architecture
Mitochondrial dynamics mediated by several fission and fusion events might be a strategic target with which to modify cellular metabolism. Finding new drugs to modulate mitochondrial architecture is an emergent field. In recent years, there have been reports of some compounds that modulate mitochondrial shape and metabolism.
Imeglimin is an antidiabetic drug structurally related to metformin that improves insulin sensitivity and mitochondrial function and decreases hepatic steatosis of high-fat, high-sucrose diet-treated mice [302,303,304]. This compound increases insulin release in obese db/db diabetic mice [305]. Moreover, this compound also modifies the mitochondrial morphology in pancreatic β-cells of db/db mice [305]. Studies in hepatic HepG2 cell lines show that like metformin, Imeglimin activates similar cellular signals, including AMPK activation and a decrease in mitochondrial respiration [306]. Interestingly, Imeglimin, as opposed to metformin, promotes the expression of Complex I and Complex III mitochondrial subunits [306]. Further investigations are required to elucidate the molecular mechanism of action of Imeglimin in mitochondria.
Mdiv-1 is a quinazoline-derived allosteric DRP1 inhibitor [307]. This compound promotes mitochondrial fusion, and it has been suggested that it protects from mitochondrial dysfunction caused by metabolic diseases and ischemic stroke [308,309,310,311]. Despite these reports, this compound shows Complex I off-target inhibition, and it impairs mitochondrial and cytosolic calcium homeostasis [312]. Also, several studies describe toxic cellular effects in some cells [313]. A recent report describes a novel DRP1 inhibitor, DRP1i27, that interacts with DRP1 and decreases its GTPase activity. This compound promotes mitochondrial elongation and protects from ischemic and reperfusion injury [314].
P110 is a seven-amino acid peptide that decreases the DRP1/FIS1 interaction and increases mitochondrial elongation [101,315]. In addition, this compound reduces oxidative stress and preserves mitochondrial respiration in LPS-treated cardiomyocytes [316]. In vivo studies demonstrated that p110 treatment for 10 or 24 days improves the motor and locomotion function of amyotrophic lateral sclerosis (ALS) mice. These mice carry the G39A SOD1 mutation that promotes oxidative stress, muscle atrophy, mitochondrial dysfunction, and cristae alterations. Overall these parameters were improved after P110 treatment [317].
Recent investigations, using a screening of molecules that can interact with the switch I-adjacent grove (SWAG) on DRP1 have led to the discovery of a novel DRP1 inhibitor, SC9. This molecule increases mitochondrial elongation under a cellular stress condition, such as LPS stimulus [315].
A recent study screened the mitochondrial morphology effect of 10,275 compounds using a high-content live-cell imaging approach [318]. Using this approach, PFK15 (6-phosphofructo-2-kinase (PFKFB3) inhibitor) was identified as a mitochondrial fission inhibitor. Based on the structure of this pharmacophore, a novel mitochondrial fission inhibitor, named MIDI, was synthesized. This compound increases mitochondrial elongation in cells treated with different stressors, such as hydrogen peroxide or mitochondrial function disruptors. Furthermore, MIDI promotes mitochondrial elongation of MFN1 KO cells. Mechanistically, this compound covalently interacts with cysteine C367 in the stalk domain on DRP1 [318].
Furthermore, antioxidant molecules can increase mitochondrial elongation. SS-31 (elamipretide) is a mitochondrial-target tetrapeptide with antioxidant capacity, and it improves mitochondrial elongation and mitochondrial fusion and promotes mitophagy [319,320]. SS-31 has been used in several models of cardiovascular disease. In addition, it is being tested in several clinical trials focused on heart failure, mitochondrial myopathy, renal disease, and aging [321,322,323,324].
The repurposing of FDA-approved drugs has opened new perspectives for drug indications. Leflunomide, a drug used for the treatment of rheumatoid arthritis, increases mitochondrial fusion by activating MFN1 and MFN2 expression [325]. Studies propose that a reversion of fragmented mitochondria in cancer might be a potential therapy. In this regard, leflunomide promotes mitochondrial elongation and increases the survival of pancreatic cancer patients [326].
A recent approach demonstrates that direct modulation of mitochondrial fusion using the TAT-367-384Gly fusogenic peptide creates a potential drug to treat mitochondrial disease. This peptide decreases HR1–HR2 interaction of MFN1 or MFN2 and results in the exposure of the MFN1/MFN2 HR2 domain to the cytoplasm. The open conformation of the HR2 domain might interact with another HR2 domain in other mitochondria and promote mitochondrial fusion. This peptide reverses the mitochondrial defect observed in CMT2A pathology [156]. Moreover, pharmacophore studies focused on HR1–HR2 MFN2 domain interaction and HR1 S378 phosphorylation revealed that a small chimeric molecule (Chimera B-A/l) promotes MFN2-dependent mitochondrial fusion [179]. Based on this molecule, MiM111 was developed. This compound improves mitochondrial transport and promotes axonal regeneration in models of CMT2A pathology [327,328,329].
A recent report describes a small molecule MASM7, which promotes MFN1 and/or MFN2 tethering through HR2 domain interaction. MASM7 increases mitochondrial fusion and enhances mitochondrial respiration, mitochondrial potential, and ATP production [330]. On the other hand, another molecule, MFI8, suppresses Mitofusin tethering, promotes mitochondrial fragmentation, and decreases mitochondrial function [330]. Remarkably, the design of these molecules has been based on the classical Mitofusin topology. Studies that describe Mitofusins as single-spanning membrane proteins may be also used to design novel pharmacological approximations, through the cysteine (Cys 684 and Cys 700) targeting of Mitofusins or the HR2–HR2 interaction region localized in IMS [150]. These studies may open new chemical approaches to modulate mitochondrial architecture. Direct activation or inhibition of the MFN1/2 tethering mechanism emerges as a promising strategy to directly modulate mitochondrial function. Table 6.
Table 6.
Agents that target mitochondria and mitochondrial diseases.
Author Contributions
Conceptualization, J.P.M., F.L.B., M.L.R., D.G. and A.Z.; writing—original draft preparation J.P.M., F.L.B., M.L.R., D.G. and A.Z.; writing—review and editing, J.P.M., F.L.B., M.L.R., D.G. and A.Z.; visualization, J.P.M., F.L.B., M.L.R. and D.G. All authors have read and agreed to the published version of the manuscript.
Funding
J.P.M. CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) is a project of Instituto de Salud Carlos III; F.L.B. was funded by Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP, through Fellowship 2018/05350-3); L.R. was funded by CONICET through PhD fellowship (N° Res 4878, 2015). A.Z. was funded by MICINN (PID2019-106209RB-I00), the Generalitat de Catalunya (Grant 2017SGR1015), the Fundación BBVA, the Fundació Marató de TV3 (20132330), EFSD, AFM Téléthon and “La Caixa” Foundation, Health Research Grant 2021 (LCF/PR/HR21/52410007). A.Z. is a recipient of an ICREA “Academia” Award (Generalitat de Catalunya).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank T. Yates for editorial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L.F. Dynamic Actin Cycling through Mitochondrial Subpopulations Locally Regulates the Fission-Fusion Balance within Mitochondrial Networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and Fusion Machineries Converge at ER Contact Sites to Regulate Mitochondrial Morphology. J. Cell Biol. 2020, 219, e201911122. [Google Scholar] [CrossRef]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 Determines Mitochondrial Network Architecture and Mitochondrial Metabolism. A Novel Regulatory Mechanism Altered in Obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Flinck, M.; Pardo, L.A. The Interplay between Dysregulated Ion Transport and Mitochondrial Architecture as a Dangerous Liaison in Cancer. Int. J. Mol. Sci. 2021, 22, 5209. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Webster, B.M.; Mastronarde, D.N.; Verhey, K.J.; Voeltz, G.K. ER Sliding Dynamics and ER-Mitochondrial Contacts Occur on Acetylated Microtubules. J. Cell Biol. 2010, 190, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Fernández Casafuz, A.B.; De Rossi, M.C.; Bruno, L. Mitochondrial Cellular Organization and Shape Fluctuations Are Differentially Modulated by Cytoskeletal Networks. Sci. Rep. 2023, 13, 4065. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Voeltz, G.K. An ER Phospholipid Hydrolase Drives ER-Associated Mitochondrial Constriction for Fission and Fusion. eLife 2022, 11, e84279. [Google Scholar] [CrossRef]
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 Controls Cristae Junction and Spatially Anchors Mitochondrial Ca2+ Uniporter Complex. Nat. Commun. 2019, 10, 3732. [Google Scholar] [CrossRef]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-Sensitive Potassium Channel in Mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Liu, X.; Hajnóczky, G. Altered Fusion Dynamics Underlie Unique Morphological Changes in Mitochondria during Hypoxia-Reoxygenation Stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Coscia, S.M.; Simpson, C.L.; Ortega, F.E.; Wait, E.C.; Heddleston, J.M.; Nirschl, J.J.; Obara, C.J.; Guedes-Dias, P.; Boecker, C.A.; et al. Actin Cables and Comet Tails Organize Mitochondrial Networks in Mitosis. Nature 2021, 591, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A Human Dynamin-Related Protein Controls the Distribution of Mitochondria. J. Cell Biol. 1998, 143, 351–358. [Google Scholar] [CrossRef]
- Santel, A.; Fuller, M.T. Control of Mitochondrial Morphology by a Human Mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.-C. HFis1, a Novel Component of the Mammalian Mitochondrial Fission Machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The Human Dynamin-Related Protein OPA1 Is Anchored to the Mitochondrial Inner Membrane Facing the Inter-Membrane Space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef]
- Gandre-Babbe, S.; van der Bliek, A.M. The Novel Tail-Anchored Membrane Protein Mff Controls Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct Fission Signatures Predict Mitochondrial Degradation or Biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Xu, L.; Qiu, Y.; Wang, X.; Shang, W.; Bai, J.; Shi, K.; Liu, H.; Liu, J.-P.; Wang, L.; Tong, C. ER-Mitochondrial Contact Protein Miga Regulates Autophagy through Atg14 and Uvrag. Cell Rep. 2022, 41, 111583. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic Enrichment of Hepatic Endoplasmic Reticulum-Mitochondria Contact Leads to Mitochondrial Dysfunction in Obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef]
- Beaulant, A.; Dia, M.; Pillot, B.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Bendridi, N.; Chanon, S.; Vieille-Marchiset, A.; Da Silva, C.C.; et al. Endoplasmic Reticulum-Mitochondria Miscommunication Is an Early and Causal Trigger of Hepatic Insulin Resistance and Steatosis. J. Hepatol. 2022, 77, 710–722. [Google Scholar] [CrossRef]
- Wenzel, E.M.; Elfmark, L.A.; Stenmark, H.; Raiborg, C. ER as Master Regulator of Membrane Trafficking and Organelle Function. J. Cell Biol. 2022, 221, e202205135. [Google Scholar] [CrossRef]
- Thiam, A.R.; Farese, R.V.; Walther, T.C. The Biophysics and Cell Biology of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics That Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6. [Google Scholar] [CrossRef] [PubMed]
- Talari, N.K.; Mattam, U.; Meher, N.K.; Paripati, A.K.; Mahadev, K.; Krishnamoorthy, T.; Sepuri, N.B.V. Lipid-Droplet Associated Mitochondria Promote Fatty-Acid Oxidation through a Distinct Bioenergetic Pattern in Male Wistar Rats. Nat. Commun. 2023, 14, 766. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Chi, L.; Lee, D.; Leung, S.; Hu, G.; Wen, B.; Delgado-Olguin, P.; Vissa, M.; Li, R.; Brumell, J.H.; Kim, P.K.; et al. Loss of Functional Peroxisomes Leads to Increased Mitochondrial Biogenesis and Reduced Autophagy That Preserve Mitochondrial Function. Cell. Mol. Life Sci. 2023, 80, 183. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, X.; Issop, L.; Culty, M.; Papadopoulos, V. ACBD2/ECI2-Mediated Peroxisome-Mitochondria Interactions in Leydig Cell Steroid Biosynthesis. Mol. Endocrinol. 2016, 30, 763–782. [Google Scholar] [CrossRef] [PubMed]
- Guillén-Samander, A.; Leonzino, M.; Hanna, M.G.; Tang, N.; Shen, H.; De Camilli, P. VPS13D Bridges the ER to Mitochondria and Peroxisomes via Miro. J. Cell Biol. 2021, 220, e202010004. [Google Scholar] [CrossRef]
- Baldwin, H.A.; Wang, C.; Kanfer, G.; Shah, H.V.; Velayos-Baeza, A.; Dulovic-Mahlow, M.; Brüggemann, N.; Anding, A.; Baehrecke, E.H.; Maric, D.; et al. VPS13D Promotes Peroxisome Biogenesis. J. Cell Biol. 2021, 220, e202001188. [Google Scholar] [CrossRef] [PubMed]
- Shai, N.; Yifrach, E.; Van Roermund, C.W.T.; Cohen, N.; Bibi, C.; IJlst, L.; Cavellini, L.; Meurisse, J.; Schuster, R.; Zada, L.; et al. Systematic Mapping of Contact Sites Reveals Tethers and a Function for the Peroxisome-Mitochondria Contact. Nat. Commun. 2018, 9, 1761. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.O.; Fujioka, Y.; Kashiwagi, S.; Yoshida, A.; Fujioka, M.; Sasajima, H.; Nanbo, A.; Amano, M.; Ohba, Y. Interaction between PI3K and the VDAC2 Channel Tethers Ras-PI3K-Positive Endosomes to Mitochondria and Promotes Endosome Maturation. Cell Rep. 2023, 42, 112229. [Google Scholar] [CrossRef]
- Irazoki, A.; Gordaliza-Alaguero, I.; Frank, E.; Giakoumakis, N.N.; Seco, J.; Palacín, M.; Gumà, A.; Sylow, L.; Sebastián, D.; Zorzano, A. Disruption of Mitochondrial Dynamics Triggers Muscle Inflammation through Interorganellar Contacts and Mitochondrial DNA Mislocation. Nat. Commun. 2023, 14, 108. [Google Scholar] [CrossRef]
- Cioni, J.-M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as MRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72.e15. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-Lysosome Contacts Regulate Mitochondrial Fission via RAB7 GTP Hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-Lysosome Contacts Regulate Mitochondrial Ca2+ Dynamics via Lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-Y.; Nguyen, O.T.K.; Kang, H.; Cho, H. MARCH5-Mediated Quality Control on Acetylated Mfn1 Facilitates Mitochondrial Homeostasis and Cell Survival. Cell Death Dis. 2014, 5, e1172. [Google Scholar] [CrossRef] [PubMed]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 Is Required for Stress-Induced Mitochondrial Hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular Network Formation Protects Mitochondria from Autophagosomal Degradation during Nutrient Starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic Cleavage of Opa1 Stimulates Mitochondrial Inner Membrane Fusion and Couples Fusion to Oxidative Phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Joffraud, M.; Boutant, M.; Ratajczak, J.; Gao, A.W.; Maclachlan, C.; Hernandez-Alvarez, M.I.; Raymond, F.; Metairon, S.; Descombes, P.; et al. Mfn1 Deficiency in the Liver Protects against Diet-Induced Insulin Resistance and Enhances the Hypoglycemic Effect of Metformin. Diabetes 2016, 65, 3552–3560. [Google Scholar] [CrossRef]
- Ngo, J.; Choi, D.W.; Stanley, I.A.; Stiles, L.; Molina, A.J.A.; Chen, P.-H.; Lako, A.; Sung, I.C.H.; Goswami, R.; Kim, M.-Y.; et al. Mitochondrial Morphology Controls Fatty Acid Utilization by Changing CPT1 Sensitivity to Malonyl-CoA. EMBO J. 2023, 42, e111901. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.M.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria Are Required for Pro-Ageing Features of the Senescent Phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial Energetics in the Heart in Obesity-Related Diabetes: Direct Evidence for Increased Uncoupled Respiration and Activation of Uncoupling Proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef]
- Rajab, B.S.; Kassab, S.; Stonall, C.D.; Daghistani, H.; Gibbons, S.; Mamas, M.; Smith, D.; Mironov, A.; AlBalawi, Z.; Zhang, Y.H.; et al. Differential Remodelling of Mitochondrial Subpopulations and Mitochondrial Dysfunction Are a Feature of Early Stage Diabetes. Sci. Rep. 2022, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Chen, D.; Riehle, C.; Soto, J.; Theobald, H.A.; Hu, X.X.; Ganesan, B.; Weimer, B.C.; Abel, E.D. Tissue-Specific Remodeling of the Mitochondrial Proteome in Type 1 Diabetic Akita Mice. Diabetes 2009, 58, 1986–1997. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Tanguy, S.; Foriel, S.; Grichine, A.; Sanchez, C.; Pernet-Gallay, K.; Kaambre, T.; Kuznetsov, A.V.; Usson, Y.; Boucher, F.; et al. The Impact of Cardiac Ischemia/Reperfusion on the Mitochondria-Cytoskeleton Interactions. Biochim. Biophys. Acta 2016, 1862, 1159–1171. [Google Scholar] [CrossRef]
- Han, M.; Bushong, E.A.; Segawa, M.; Tiard, A.; Wong, A.; Brady, M.R.; Momcilovic, M.; Wolf, D.M.; Zhang, R.; Petcherski, A.; et al. Spatial Mapping of Mitochondrial Networks and Bioenergetics in Lung Cancer. Nature 2023, 615, 712–719. [Google Scholar] [CrossRef]
- Nisr, R.B.; Shah, D.S.; Ganley, I.G.; Hundal, H.S. Proinflammatory NFkB Signalling Promotes Mitochondrial Dysfunction in Skeletal Muscle in Response to Cellular Fuel Overloading. Cell. Mol. Life Sci. 2019, 76, 4887–4904. [Google Scholar] [CrossRef]
- Molina, A.J.A.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects Beta-Cells from Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Stiles, L.; Shirihai, O.S. Mitochondrial Dynamics and Morphology in Beta-Cells. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 725–738. [Google Scholar] [CrossRef]
- Nisr, R.B.; Shah, D.S.; Hundal, H.S. Mono- and Polyunsaturated Fatty Acids Counter Palmitate-Induced Mitochondrial Dysfunction in Rat Skeletal Muscle Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2020, 54, 975–993. [Google Scholar] [CrossRef]
- Parra, V.; Verdejo, H.E.; Iglewski, M.; Del Campo, A.; Troncoso, R.; Jones, D.; Zhu, Y.; Kuzmicic, J.; Pennanen, C.; Lopez-Crisosto, C.; et al. Insulin Stimulates Mitochondrial Fusion and Function in Cardiomyocytes via the Akt-MTOR-NFκB-Opa-1 Signaling Pathway. Diabetes 2014, 63, 75–88. [Google Scholar] [CrossRef]
- Zhou, Z.; Torres, M.; Sha, H.; Halbrook, C.J.; Van den Bergh, F.; Reinert, R.B.; Yamada, T.; Wang, S.; Luo, Y.; Hunter, A.H.; et al. Endoplasmic Reticulum-Associated Degradation Regulates Mitochondrial Dynamics in Brown Adipocytes. Science 2020, 368, 54–60. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease With Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef] [PubMed]
- Shami, G.J.; Cheng, D.; Verhaegh, P.; Koek, G.; Wisse, E.; Braet, F. Three-Dimensional Ultrastructure of Giant Mitochondria in Human Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2021, 11, 3319. [Google Scholar] [CrossRef]
- Hammerschmidt, P.; Ostkotte, D.; Nolte, H.; Gerl, M.J.; Jais, A.; Brunner, H.L.; Sprenger, H.-G.; Awazawa, M.; Nicholls, H.T.; Turpin-Nolan, S.M.; et al. CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell 2019, 177, 1536–1552.e23. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and Selective Fusion Govern Mitochondrial Segregation and Elimination by Autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Wikstrom, J.D.; Mahdaviani, K.; Liesa, M.; Sereda, S.B.; Si, Y.; Las, G.; Twig, G.; Petrovic, N.; Zingaretti, C.; Graham, A.; et al. Hormone-Induced Mitochondrial Fission Is Utilized by Brown Adipocytes as an Amplification Pathway for Energy Expenditure. EMBO J. 2014, 33, 418–436. [Google Scholar] [CrossRef]
- Kirov, S.A.; Fomitcheva, I.V.; Sword, J. Rapid Neuronal Ultrastructure Disruption and Recovery during Spreading Depolarization-Induced Cytotoxic Edema. Cereb. Cortex 2020, 30, 5517–5531. [Google Scholar] [CrossRef]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered Brain Energetics Induces Mitochondrial Fission Arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef]
- Morozov, Y.M.; Datta, D.; Paspalas, C.D.; Arnsten, A.F.T. Ultrastructural Evidence for Impaired Mitochondrial Fission in the Aged Rhesus Monkey Dorsolateral Prefrontal Cortex. Neurobiol. Aging 2017, 51, 9–18. [Google Scholar] [CrossRef]
- Samuvel, D.J.; Li, L.; Krishnasamy, Y.; Gooz, M.; Takemoto, K.; Woster, P.M.; Lemasters, J.J.; Zhong, Z. Mitochondrial Depolarization after Acute Ethanol Treatment Drives Mitophagy in Living Mice. Autophagy 2022, 18, 2671–2685. [Google Scholar] [CrossRef]
- McElroy, T.; Zeidan, R.S.; Rathor, L.; Han, S.M.; Xiao, R. The Role of Mitochondria in the Recovery of Neurons after Injury. Neural Regen. Res. 2023, 18, 317–318. [Google Scholar] [CrossRef]
- Chen, C.; Li, J.; Qin, X.; Wang, W. Peroxisomal Membrane Contact Sites in Mammalian Cells. Front. Cell Dev. Biol. 2020, 8, 512. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Adlakha, J.; Wan, N.; Guinn, E.; Giska, F.; Gupta, K.; Melia, T.J.; Reinisch, K.M. Mitoguardin-2-Mediated Lipid Transfer Preserves Mitochondrial Morphology and Lipid Droplet Formation. J. Cell Biol. 2022, 221, e202207022. [Google Scholar] [CrossRef]
- Barra, J.; Crosbourne, I.; Wang, L.; Nelson, I.; Goldwag, J.; Jourd’heuil, F.; Jourd’heuil, D.; Barroso, M. DMT1-mediated Endosome-mitochondria Interactions Regulates Iron Homeostasis and Mitochondrial Metabolism. FASEB J. 2022, 36, fasebj.2022.36.S1.R5276. [Google Scholar] [CrossRef]
- Wang, X.; Schwarz, T.L. The Mechanism of Ca2+ -Dependent Regulation of Kinesin-Mediated Mitochondrial Motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef]
- Kanfer, G.; Kornmann, B. Dynamics of the Mitochondrial Network during Mitosis. Biochem. Soc. Trans. 2016, 44, 510–516. [Google Scholar] [CrossRef]
- Shields, L.Y.; Kim, H.; Zhu, L.; Haddad, D.; Berthet, A.; Pathak, D.; Lam, M.; Ponnusamy, R.; Diaz-Ramirez, L.G.; Gill, T.M.; et al. Dynamin-Related Protein 1 Is Required for Normal Mitochondrial Bioenergetic and Synaptic Function in CA1 Hippocampal Neurons. Cell Death Dis. 2015, 6, e1725. [Google Scholar] [CrossRef]
- Li, H.; Chen, Y.; Jones, A.F.; Sanger, R.H.; Collis, L.P.; Flannery, R.; McNay, E.C.; Yu, T.; Schwarzenbacher, R.; Bossy, B.; et al. Bcl-x L Induces Drp1-Dependent Synapse Formation in Cultured Hippocampal Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okamoto, K.-I.; Hayashi, Y.; Sheng, M. The Importance of Dendritic Mitochondria in the Morphogenesis and Plasticity of Spines and Synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.-I.; et al. Mitochondrial Fission Factor Drp1 Is Essential for Embryonic Development and Synapse Formation in Mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Lee, J.S.; Moon, J.H.; Lee, S.-A.; Park, Y.J.; Lee, D.-S.; Chung, S.S.; Park, K.S. SENP2 Regulates Mitochondrial Function and Insulin Secretion in Pancreatic β Cells. Exp. Mol. Med. 2022, 54, 72–80. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-Related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Thiemann, M.; Grabenbauer, M.; Yoon, Y.; McNiven, M.A.; Schrader, M. Dynamin-like Protein 1 Is Involved in Peroxisomal Fission. J. Biol. Chem. 2003, 278, 8597–8605. [Google Scholar] [CrossRef]
- Li, X.; Gould, S.J. The Dynamin-like GTPase DLP1 Is Essential for Peroxisome Division and Is Recruited to Peroxisomes in Part by PEX11. J. Biol. Chem. 2003, 278, 17012–17020. [Google Scholar] [CrossRef] [PubMed]
- Favaro, G.; Romanello, V.; Varanita, T.; Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-Mediated Mitochondrial Shape Controls Calcium Homeostasis and Muscle Mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef]
- Shields, L.Y.; Li, H.; Nguyen, K.; Kim, H.; Doric, Z.; Garcia, J.H.; Gill, T.M.; Haddad, D.; Vossel, K.; Calvert, M.; et al. Mitochondrial Fission Is a Critical Modulator of Mutant APP-Induced Neural Toxicity. J. Biol. Chem. 2021, 296, 100469. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef]
- Bandopadhyay, S.; Prasad, P.; Ray, U.; Das Ghosh, D.; Roy, S.S. SIRT6 Promotes Mitochondrial Fission and Subsequent Cellular Invasion in Ovarian Cancer. FEBS Open Bio 2022, 12, 1657–1676. [Google Scholar] [CrossRef]
- Chang, Y.-J.; Chen, K.-W.; Chen, L. Mitochondrial ROS1 Increases Mitochondrial Fission and Respiration in Oral Squamous Cancer Carcinoma. Cancers 2020, 12, 2845. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Jia, R. The Role of Mitochondrial Dynamics in Human Cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Beta with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer’s Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Höke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent Mitophagy Requires D Rp1 and Maintains the Integrity of Mammalian Heart and Brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased Production of Reactive Oxygen Species in Hyperglycemic Conditions Requires Dynamic Change of Mitochondrial Morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff Is an Essential Factor for Mitochondrial Recruitment of Drp1 during Mitochondrial Fission in Mammalian Cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef]
- Liu, R.; Chan, D.C. The Mitochondrial Fission Receptor Mff Selectively Recruits Oligomerized Drp1. Mol. Biol. Cell 2015, 26, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Mozdy, A.D.; McCaffery, J.M.; Shaw, J.M. Dnm1p GTPase-Mediated Mitochondrial Fission Is a Multi-Step Process Requiring the Novel Integral Membrane Component Fis1p. J. Cell Biol. 2000, 151, 367–380. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 Regulates Mitochondrial Dynamics through Inhibition of the Fusion Machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The Mitochondrial Protein HFis1 Regulates Mitochondrial Fission in Mammalian Cells through an Interaction with the Dynamin-Like Protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef]
- Koirala, S.; Guo, Q.; Kalia, R.; Bui, H.T.; Eckert, D.M.; Frost, A.; Shaw, J.M. Interchangeable Adaptors Regulate Mitochondrial Dynamin Assembly for Membrane Scission. Proc. Natl. Acad. Sci. USA 2013, 110, E1342–E1351. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 Inhibitor Diminishes Aberrant Mitochondrial Fission and Neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Egner, J.M.; Nolden, K.A.; Harwig, M.C.; Bonate, R.P.; De Anda, J.; Tessmer, M.H.; Noey, E.L.; Ihenacho, U.K.; Liu, Z.; Peterson, F.C.; et al. Structural Studies of Human Fission Protein FIS1 Reveal a Dynamic Region Important for GTPase DRP1 Recruitment and Mitochondrial Fission. J. Biol. Chem. 2022, 298, 102620. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jeong, S.-Y.; Lim, W.-C.; Kim, S.; Park, Y.-Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial Fission and Fusion Mediators, HFis1 and OPA1, Modulate Cellular Senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.-S.; Yoon, D.-S.; Lim, I.K.; Yoon, S.-H.; Chung, H.-Y.; Rojo, M.; Malka, F.; Jou, M.-J.; Martinou, J.-C.; Yoon, G. Formation of Elongated Giant Mitochondria in DFO-Induced Cellular Senescence: Involvement of Enhanced Fusion Process through Modulation of Fis1. J. Cell. Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Sun, Z.; Ye, T.; Li, J.; Zeng, B.; Zhao, Q.; Wang, J.; Xing, H.R. The Mitochondrial Fission Factor FIS1 Promotes Stemness of Human Lung Cancer Stem Cells via Mitophagy. FEBS Open Bio 2021, 11, 1997–2007. [Google Scholar] [CrossRef]
- Simpson, J.C.; Wellenreuther, R.; Poustka, A.; Pepperkok, R.; Wiemann, S. Systematic Subcellular Localization of Novel Proteins Identified by Large-Scale CDNA Sequencing. EMBO Rep. 2000, 1, 287–292. [Google Scholar] [CrossRef]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor Proteins MiD49 and MiD51 Can Act Independently of Mff and Fis1 in Drp1 Recruitment and Are Specific for Mitochondrial Fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef]
- Elgass, K.D.; Smith, E.A.; LeGros, M.A.; Larabell, C.A.; Ryan, M.T. Analysis of ER–Mitochondria Contacts Using Correlative Fluorescence Microscopy and Soft X-Ray Tomography of Mammalian Cells. J. Cell Sci. 2015, 128, 2795–2804. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, New Components of the Mitochondrial Fission Machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 Recruitment in Mitochondrial Fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, T.; Jin, S.; Wang, X.; Qu, M.; Uhlén, P.; Tomilin, N.; Shupliakov, O.; Lendahl, U.; Nistér, M. Human MIEF1 Recruits Drp1 to Mitochondrial Outer Membranes and Promotes Mitochondrial Fusion Rather than Fission. EMBO J. 2011, 30, 2762–2778. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yu, R.; Jin, S.-B.; Han, L.; Lendahl, U.; Zhao, J.; Nistér, M. The Mitochondrial Elongation Factors MIEF1 and MIEF2 Exert Partially Distinct Functions in Mitochondrial Dynamics. Exp. Cell Res. 2013, 319, 2893–2904. [Google Scholar] [CrossRef]
- Atkins, K.; Dasgupta, A.; Chen, K.-H.; Mewburn, J.; Archer, S.L. The Role of Drp1 Adaptor Proteins MiD49 and MiD51 in Mitochondrial Fission: Implications for Human Disease. Clin. Sci. 2016, 130, 1861–1874. [Google Scholar] [CrossRef]
- Chen, K.-H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation 2018, 138, 287–304. [Google Scholar] [CrossRef]
- Samangouei, P.; Crespo-Avilan, G.E.; Cabrera-Fuentes, H.; Hernández-Reséndiz, S.; Ismail, N.I.; Katwadi, K.B.; Boisvert, W.A.; Hausenloy, D.J. MiD49 and MiD51: New Mediators of Mitochondrial Fission and Novel Targets for Cardioprotection. Cond. Med. 2018, 1, 239–246. [Google Scholar]
- Bai, L.; Liang, J.; Li, L.; Li, E. Downregulation of MiD49 Contributes to Tumor Growth and Metastasis of Human Pancreatic Cancer. Oncol. Rep. 2020, 43, 1208–1220. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, X.; Shi, Y.; Cheng, L.; Song, T.; Wu, B.; Li, J.; Yang, H. MIEF2 Over-Expression Promotes Tumor Growth and Metastasis through Reprogramming of Glucose Metabolism in Ovarian Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 286. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-Related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.; Cho, H.M.; Kim, H.J.; Jeong, J.; Park, S.K.; Hwang, E.M.; Park, J.-Y.; Kim, W.R.; Kim, H.; Sun, W. CDK5-Dependent Inhibitory Phosphorylation of Drp1 during Neuronal Maturation. Exp. Mol. Med. 2014, 46, e105. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Huang, E.; Irrcher, I.; Rashidian, J.; Ishihara, N.; Lagace, D.C.; Slack, R.S.; Park, D.S. CDK5 Phosphorylates DRP1 and Drives Mitochondrial Defects in NMDA-Induced Neuronal Death. Hum. Mol. Genet. 2015, 24, 4573–4583. [Google Scholar] [CrossRef]
- Yu, T.; Jhun, B.S.; Yoon, Y. High-Glucose Stimulation Increases Reactive Oxygen Species Production Through the Calcium and Mitogen-Activated Protein Kinase-Mediated Activation of Mitochondrial Fission. Antioxid. Redox Signal. 2011, 14, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, J.; Ge, X.; Deng, J.; Xu, X.; Zhao, Y.; Wang, H. Activation of ERK–Drp1 Signaling Promotes Hypoxia-induced Aβ Accumulation by Upregulating Mitochondrial Fission and BACE1 Activity. FEBS Open Bio 2021, 11, 2740–2755. [Google Scholar] [CrossRef]
- Roe, A.J.; Qi, X. Drp1 Phosphorylation by MAPK1 Causes Mitochondrial Dysfunction in Cell Culture Model of Huntington’s Disease. Biochem. Biophys. Res. Commun. 2018, 496, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Disatnik, M.-H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant Mitochondrial Fission in Neurons Induced by Protein Kinase Cδ under Oxidative Stress Conditions in Vivo. Mol. Biol. Cell 2011, 22, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK 1 Phosphorylates Drp1 S616 to Regulate Mitophagy-independent Mitochondrial Dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-C.; Qi, X. Inhibition of Excessive Mitochondrial Fission Reduced Aberrant Autophagy and Neuronal Damage Caused by LRRK2 G2019S Mutation. Hum. Mol. Genet. 2013, 22, 4545–4561. [Google Scholar] [CrossRef]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-Dependent Protein Kinase Phosphorylation of Drp1 Regulates Its GTPase Activity and Mitochondrial Morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef]
- Han, X.-J.; Lu, Y.-F.; Li, S.-A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. CaM Kinase Iα–Induced Phosphorylation of Drp1 Regulates Mitochondrial Morphology. J. Cell Biol. 2008, 182, 573–585. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible Phosphorylation of Drp1 by Cyclic AMP-dependent Protein Kinase and Calcineurin Regulates Mitochondrial Fission and Cell Death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef]
- Cha, Y.; Kim, T.; Jeon, J.; Jang, Y.; Kim, P.B.; Lopes, C.; Leblanc, P.; Cohen, B.M.; Kim, K.-S. SIRT2 Regulates Mitochondrial Dynamics and Reprogramming via MEK1-ERK-DRP1 and AKT1-DRP1 Axes. Cell Rep. 2021, 37, 110155. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; de Brito, O.M.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by Calcineurin Regulates Translocation of Drp1 to Mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Ma, J.; Li, J.; Wang, D.; Wang, Z.; Wang, S. Mitochondrial Phosphatase PGAM5 Modulates Cellular Senescence by Regulating Mitochondrial Dynamics. Nat. Commun. 2020, 11, 2549. [Google Scholar] [CrossRef] [PubMed]
- Valera-Alberni, M.; Joffraud, M.; Miro-Blanch, J.; Capellades, J.; Junza, A.; Dayon, L.; Núñez Galindo, A.; Sanchez-Garcia, J.L.; Valsesia, A.; Cercillieux, A.; et al. Crosstalk between Drp1 Phosphorylation Sites during Mitochondrial Remodeling and Their Impact on Metabolic Adaptation. Cell Rep. 2021, 36, 109565. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, Y.; Yang, Q.; Hu, J.; Feng, J.; Liang, W.; Ding, G. AKAP1 Mediates High Glucose-Induced Mitochondrial Fission through the Phosphorylation of Drp1 in Podocytes. J. Cell. Physiol. 2020, 235, 7433–7448. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.J.; Schumacker, P.T.; Danesh, F.R. Mitochondrial Fission Triggered by Hyperglycemia Is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef]
- Hu, S.-L.; Mamun, A.A.; Shaw, J.; Li, S.-L.; Shi, Y.-F.; Jin, X.-M.; Yu, Y.-X.; Pang, C.-Z.; Li, Z.-Y.; Lu, J.-J.; et al. TBK1-Medicated DRP1 Phosphorylation Orchestrates Mitochondrial Dynamics and Autophagy Activation in Osteoarthritis. Acta Pharmacol. Sin. 2023, 44, 610–621. [Google Scholar] [CrossRef]
- Chen, S.; Liu, S.; Wang, J.; Wu, Q.; Wang, A.; Guan, H.; Zhang, Q.; Zhang, D.; Wang, X.; Song, H.; et al. TBK1-Mediated DRP1 Targeting Confers Nucleic Acid Sensing to Reprogram Mitochondrial Dynamics and Physiology. Mol. Cell 2020, 80, 810–827.e7. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-Activated Protein Kinase Mediates Mitochondrial Fission in Response to Energy Stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Pangou, E.; Bielska, O.; Guerber, L.; Schmucker, S.; Agote-Arán, A.; Ye, T.; Liao, Y.; Puig-Gamez, M.; Grandgirard, E.; Kleiss, C.; et al. A PKD-MFF Signaling Axis Couples Mitochondrial Fission to Mitotic Progression. Cell Rep. 2021, 35, 109129. [Google Scholar] [CrossRef]
- Yu, Y.; Peng, X.-D.; Qian, X.-J.; Zhang, K.-M.; Huang, X.; Chen, Y.-H.; Li, Y.-T.; Feng, G.-K.; Zhang, H.-L.; Xu, X.-L.; et al. Fis1 Phosphorylation by Met Promotes Mitochondrial Fission and Hepatocellular Carcinoma Metastasis. Signal Transduct. Target. Ther. 2021, 6, 401. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhu, H.; Li, R.; Mui, D.; Toan, S.; Chang, X.; Zhou, H. DNA-PKcs Interacts with and Phosphorylates Fis1 to Induce Mitochondrial Fragmentation in Tubular Cells during Acute Kidney Injury. Sci. Signal. 2022, 15, eabh1121. [Google Scholar] [CrossRef]
- Yu, R.; Liu, T.; Jin, S.-B.; Ankarcrona, M.; Lendahl, U.; Nistér, M.; Zhao, J. MIEF1/2 Orchestrate Mitochondrial Dynamics through Direct Engagement with Both the Fission and Fusion Machineries. BMC Biol. 2021, 19, 229. [Google Scholar] [CrossRef] [PubMed]
- Bustillo-Zabalbeitia, I.; Montessuit, S.; Raemy, E.; Basañez, G.; Terrones, O.; Martinou, J.-C. Specific Interaction with Cardiolipin Triggers Functional Activation of Dynamin-Related Protein 1. PLoS ONE 2014, 9, e102738. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhai, Y.; Chen, M.; Zhang, K.; Chen, Q.; Pang, X.; Sun, F. New Interfaces on MiD51 for Drp1 Recruitment and Regulation. PLoS ONE 2019, 14, e0211459. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Palmer, C.S.; Osellame, L.D.; Singh, A.P.; Elgass, K.; Stroud, D.A.; Sesaki, H.; Kvansakul, M.; Ryan, M.T. Structural and Functional Analysis of MiD51, a Dynamin Receptor Required for Mitochondrial Fission. J. Cell Biol. 2014, 204, 477–486. [Google Scholar] [CrossRef]
- Losón, O.C.; Liu, R.; Rome, M.E.; Meng, S.; Kaiser, J.T.; Shan, S.; Chan, D.C. The Mitochondrial Fission Receptor MiD51 Requires ADP as a Cofactor. Structure 2014, 22, 367–377. [Google Scholar] [CrossRef]
- Cipolat, S.; de Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 Requires Mitofusin 1 to Promote Mitochondrial Fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Membrane Topology and Mitochondrial Targeting of Mitofusins, Ubiquitous Mammalian Homologs of the Transmembrane GTPase Fzo. J. Cell Sci. 2002, 115, 1663–1674. [Google Scholar] [CrossRef]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A New Mitofusin Topology Places the Redox-Regulated C Terminus in the Mitochondrial Intermembrane Space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Li, Y.-J.; Cao, Y.-L.; Feng, J.-X.; Qi, Y.; Meng, S.; Yang, J.-F.; Zhong, Y.-T.; Kang, S.; Chen, X.; Lan, L.; et al. Structural Insights of Human Mitofusin-2 into Mitochondrial Fusion and CMT2A Onset. Nat. Commun. 2019, 10, 4914. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of Human Mitofusin 1 Provide Insight into Mitochondrial Tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural Basis for GTP Hydrolysis and Conformational Change of MFN1 in Mediating Membrane Fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef]
- Cao, Y.-L.; Meng, S.; Chen, Y.; Feng, J.-X.; Gu, D.-D.; Yu, B.; Li, Y.-J.; Yang, J.-Y.; Liao, S.; Chan, D.C.; et al. MFN1 Structures Reveal Nucleotide-Triggered Dimerization Critical for Mitochondrial Fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting Mitochondrial Fusion by Manipulating Mitofusin Conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial Dynamics in Health and Disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Jiang, L. Dysregulated Mitochondrial Dynamics and Metabolism in Obesity, Diabetes, and Cancer. Front. Endocrinol. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Han, S.; Zhang, Q.; Zhu, Y.; Zhang, H.; Wang, J.; Zhao, Y.; Zhao, J.; Su, L.; Li, L.; et al. FUNDC2 Promotes Liver Tumorigenesis by Inhibiting MFN1-Mediated Mitochondrial Fusion. Nat. Commun. 2022, 13, 3486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, T.-E.; Chen, M.; Xu, D.; Zhu, Y.; Hu, B.-Y.; Lin, Z.-F.; Pan, J.-J.; Wang, X.; Wu, C.; et al. MFN1-Dependent Alteration of Mitochondrial Dynamics Drives Hepatocellular Carcinoma Metastasis by Glucose Metabolic Reprogramming. Br. J. Cancer 2020, 122, 209–220. [Google Scholar] [CrossRef]
- Pyakurel, A.; Savoia, C.; Hess, D.; Scorrano, L. Extracellular Regulated Kinase Phosphorylates Mitofusin 1 to Control Mitochondrial Morphology and Apoptosis. Mol. Cell 2015, 58, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.-Y.; Youle, R.J. Role of Bax and Bak in Mitochondrial Morphogenesis. Nature 2006, 443, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Feng, L.; Dong, G.; Tao, Y.; Mei, L.; Xie, Z.-J.; Dong, Z. Bak Regulates Mitochondrial Morphology and Pathology during Apoptosis by Interacting with Mitofusins. Proc. Natl. Acad. Sci. USA 2007, 104, 11649–11654. [Google Scholar] [CrossRef] [PubMed]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacín, M.; Zorzano, A. The Charcot-Marie-Tooth Type 2A Gene Product, Mfn2, up-Regulates Fuel Oxidation through Expression of OXPHOS System. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 Ablation Increases Endoplasmic Reticulum–Mitochondria Coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical Reappraisal Confirms That Mitofusin 2 Is an Endoplasmic Reticulum-Mitochondria Tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [PubMed]
- Casellas-Díaz, S.; Larramona-Arcas, R.; Riqué-Pujol, G.; Tena-Morraja, P.; Müller-Sánchez, C.; Segarra-Mondejar, M.; Gavaldà-Navarro, A.; Villarroya, F.; Reina, M.; Martínez-Estrada, O.M.; et al. Mfn2 Localization in the ER Is Necessary for Its Bioenergetic Function and Neuritic Development. EMBO Rep. 2021, 22, e51954. [Google Scholar] [CrossRef]
- Gordaliza-Alaguero, I.; Cantó, C.; Zorzano, A. Metabolic Implications of Organelle–Mitochondria Communication. EMBO Rep. 2019, 20, e47928. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Papanicolaou, K.N.; Walsh, K. Loss of Mitofusin 2 Promotes Endoplasmic Reticulum Stress. J. Biol. Chem. 2012, 287, 20321–20332. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 Modulates the UPR and Mitochondrial Function via Repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the Mitochondrial GTPase Mitofusin 2 Cause Charcot-Marie-Tooth Neuropathy Type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Kijima, K.; Numakura, C.; Izumino, H.; Umetsu, K.; Nezu, A.; Shiiki, T.; Ogawa, M.; Ishizaki, Y.; Kitamura, T.; Shozawa, Y.; et al. Mitochondrial GTPase Mitofusin 2 Mutation in Charcot-Marie-Tooth Neuropathy Type 2A. Hum. Genet. 2005, 116, 23–27. [Google Scholar] [CrossRef]
- Lawson, V.H.; Graham, B.V.; Flanigan, K.M. Clinical and Electrophysiologic Features of CMT2A with Mutations in the Mitofusin 2 Gene. Neurology 2005, 65, 197–204. [Google Scholar] [CrossRef]
- Verhoeven, K. MFN2 Mutation Distribution and Genotype/Phenotype Correlation in Charcot-Marie-Tooth Type 2. Brain 2006, 129, 2093–2102. [Google Scholar] [CrossRef]
- Abati, E.; Manini, A.; Velardo, D.; Del Bo, R.; Napoli, L.; Rizzo, F.; Moggio, M.; Bresolin, N.; Bellone, E.; Bassi, M.T.; et al. Clinical and Genetic Features of a Cohort of Patients with MFN2-Related Neuropathy. Sci. Rep. 2022, 12, 6181. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H.; Schmidt, R.E.; Pestronk, A.; Milbrandt, J. Altered Axonal Mitochondrial Transport in the Pathogenesis of Charcot-Marie-Tooth Disease from Mitofusin 2 Mutations. J. Neurosci. 2007, 27, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 Agonists Reverse Mitochondrial Defects in Preclinical Models of Charcot-Marie-Tooth Disease Type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana–Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 Mutations in Charcot–Marie–Tooth Disease Alter Mitochondria-Associated ER Membrane Function but Do Not Impair Bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Complementation between Mouse Mfn1 and Mfn2 Protects Mitochondrial Fusion Defects Caused by CMT2A Disease Mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Palacín, M. Mitochondrial Dynamics as a Bridge between Mitochondrial Dysfunction and Insulin Resistance. Arch. Physiol. Biochem. 2009, 115, 1–12. [Google Scholar] [CrossRef]
- Sidarala, V.; Zhu, J.; Levi-D’Ancona, E.; Pearson, G.; Reck, E.; Walker, E.; Kaufman, B.; Soleimanpour, S. Mitofusin 1 and 2 Regulation of Mitochondrial DNA Content Is a Critical Determinant of Glucose Homeostasis. Nat. Commun. 2022, 13, 2340. [Google Scholar] [CrossRef]
- Mahdaviani, K.; Benador, I.Y.; Su, S.; Gharakhanian, R.A.; Stiles, L.; Trudeau, K.M.; Cardamone, M.; Enríquez-Zarralanga, V.; Ritou, E.; Aprahamian, T.; et al. Mfn2 Deletion in Brown Adipose Tissue Protects from Insulin Resistance and Impairs Thermogenesis. EMBO Rep. 2017, 18, 1123–1138. [Google Scholar] [CrossRef]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, Y.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the Charcot-Marie-Tooth Neuropathy Type 2A Gene, in Human Skeletal Muscle: Effects of Type 2 Diabetes, Obesity, Weight Loss, and the Regulatory Role of Tumor Necrosis Factor Alpha and Interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) Links Mitochondrial and Endoplasmic Reticulum Function with Insulin Signaling and Is Essential for Normal Glucose Homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef]
- Sebastián, D.; Zorzano, A. When MFN2 (Mitofusin 2) Met Autophagy: A New Age for Old Muscles. Autophagy 2016, 12, 2250–2251. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Wong, E.D.; Wagner, J.A.; Gorsich, S.W.; McCaffery, J.M.; Shaw, J.M.; Nunnari, J. The Dynamin-Related GTPase, Mgm1p, Is an Intermembrane Space Protein Required for Maintenance of Fusion Competent Mitochondria. J. Cell Biol. 2000, 151, 341–352. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, Is Mutated in Dominant Optic Atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, Encoding a Dynamin-Related GTPase, Is Mutated in Autosomal Dominant Optic Atrophy Linked to Chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; de Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; Strooper, B.D.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-Dependent Cristae Modulation Is Essential for Cellular Adaptation to Metabolic Demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef]
- Delettre, C.; Griffoin, J.M.; Kaplan, J.; Dollfus, H.; Lorenz, B.; Faivre, L.; Lenaers, G.; Belenguer, P.; Hamel, C.P. Mutation Spectrum and Splicing Variants in the OPA1 Gene. Hum. Genet. 2001, 109, 584–591. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of Mitochondrial Morphology through Proteolytic Cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 Processing Controls Mitochondrial Fusion and Is Regulated by MRNA Splicing, Membrane Potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular Basis of Selective Mitochondrial Fusion by Heterotypic Action between OPA1 and Cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef]
- Quirós, P.M.; Ramsay, A.J.; Sala, D.; Fernández-Vizarra, E.; Rodríguez, F.; Peinado, J.R.; Fernández-García, M.S.; Vega, J.A.; Enríquez, J.A.; Zorzano, A.; et al. Loss of Mitochondrial Protease OMA1 Alters Processing of the GTPase OPA1 and Causes Obesity and Defective Thermogenesis in Mice. EMBO J. 2012, 31, 2117–2133. [Google Scholar] [CrossRef]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-Induced OMA1 Activation and Autocatalytic Turnover Regulate OPA1-Dependent Mitochondrial Dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. YME1L Degradation Reduces Mitochondrial Proteolytic Capacity during Oxidative Stress. EMBO Rep. 2015, 16, 97–106. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef]
- Sprenger, H.-G.; Wani, G.; Hesseling, A.; König, T.; Patron, M.; MacVicar, T.; Ahola, S.; Wai, T.; Barth, E.; Rugarli, E.I.; et al. Loss of the Mitochondrial I-AAA Protease YME1L Leads to Ocular Dysfunction and Spinal Axonopathy. EMBO Mol. Med. 2019, 11, e9288. [Google Scholar] [CrossRef]
- Ohba, Y.; MacVicar, T.; Langer, T. Regulation of Mitochondrial Plasticity by the I-AAA Protease YME1L. Biol. Chem. 2020, 401, 877–890. [Google Scholar] [CrossRef]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial Rhomboid PARL Regulates Cytochrome c Release during Apoptosis via OPA1-Dependent Cristae Remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Radaelli, E.; Horré, K.; Arranz, A.M.; Gounko, N.V.; Agostinis, P.; Maia, T.M.; Impens, F.; Morais, V.A.; Lopez-Lluch, G.; et al. PARL Deficiency in Mouse Causes Complex III Defects, Coenzyme Q Depletion, and Leigh-like Syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Chao de la Barca, J.M.; Prunier-Mirebeau, D.; Amati-Bonneau, P.; Ferré, M.; Sarzi, E.; Bris, C.; Leruez, S.; Chevrollier, A.; Desquiret-Dumas, V.; Gueguen, N.; et al. OPA1-Related Disorders: Diversity of Clinical Expression, Modes of Inheritance and Pathophysiology. Neurobiol. Dis. 2016, 90, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Pesch, U.E.; Leo-Kottler, B.; Mayer, S.; Jurklies, B.; Kellner, U.; Apfelstedt-Sylla, E.; Zrenner, E.; Alexander, C.; Wissinger, B. OPA1 Mutations in Patients with Autosomal Dominant Optic Atrophy and Evidence for Semi-Dominant Inheritance. Hum. Mol. Genet. 2001, 10, 1359–1368. [Google Scholar] [CrossRef]
- Cohn, A.C.; Toomes, C.; Potter, C.; Towns, K.V.; Hewitt, A.W.; Inglehearn, C.F.; Craig, J.E.; Mackey, D.A. Autosomal Dominant Optic Atrophy: Penetrance and Expressivity in Patients with OPA1 Mutations. Am. J. Ophthalmol. 2007, 143, 656–662. [Google Scholar] [CrossRef]
- Almind, G.J.; Ek, J.; Rosenberg, T.; Eiberg, H.; Larsen, M.; Lucamp, L.; Brøndum-Nielsen, K.; Grønskov, K. Dominant Optic Atrophy in Denmark—Report of 15 Novel Mutations in OPA1, Using a Strategy with a Detection Rate of 90%. BMC Med. Genet. 2012, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Schimpf-Linzenbold, S.; Mazzola, P.; Kieninger, S.; Xiao, T.; Kellner, U.; Neuhann, T.; Kelbsch, C.; Tonagel, F.; Wilhelm, H.; et al. Mutation Spectrum of the OPA1 Gene in a Large Cohort of Patients with Suspected Dominant Optic Atrophy: Identification and Classification of 48 Novel Variants. PLoS ONE 2021, 16, e0253987. [Google Scholar] [CrossRef]
- Olichon, A.; Landes, T.; Arnauné-Pelloquin, L.; Emorine, L.J.; Mils, V.; Guichet, A.; Delettre, C.; Hamel, C.; Amati-Bonneau, P.; Bonneau, D.; et al. Effects of OPA1 Mutations on Mitochondrial Morphology and Apoptosis: Relevance to ADOA Pathogenesis. J. Cell. Physiol. 2007, 211, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Cartes-Saavedra, B.; Lagos, D.; Macuada, J.; Arancibia, D.; Burté, F.; Sjöberg-Herrera, M.K.; Andrés, M.E.; Horvath, R.; Yu-Wai-Man, P.; Hajnóczky, G.; et al. OPA1 Disease-Causing Mutants Have Domain-Specific Effects on Mitochondrial Ultrastructure and Fusion. Proc. Natl. Acad. Sci. USA 2023, 120, e2207471120. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Liang, X.; Lu, Y.; Zhu, L.; Fu, R.; Ji, Y.; Fan, W.; Chen, J.; Lin, B.; et al. A Novel ADOA-Associated OPA1 Mutation Alters the Mitochondrial Function, Membrane Potential, ROS Production and Apoptosis. Sci. Rep. 2017, 7, 5704. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 Drive Muscle Inflammation upon Opa1 Deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef]
- Pereira, R.O.; Tadinada, S.M.; Zasadny, F.M.; Oliveira, K.J.; Pires, K.M.P.; Olvera, A.; Jeffers, J.; Souvenir, R.; Mcglauflin, R.; Seei, A.; et al. OPA1 Deficiency Promotes Secretion of FGF21 from Muscle That Prevents Obesity and Insulin Resistance. EMBO J. 2017, 36, 2126–2145. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.O.; Marti, A.; Olvera, A.C.; Tadinada, S.M.; Bjorkman, S.H.; Weatherford, E.T.; Morgan, D.A.; Westphal, M.; Patel, P.H.; Kirby, A.K.; et al. OPA1 Deletion in Brown Adipose Tissue Improves Thermoregulation and Systemic Metabolism via FGF21. eLife 2021, 10, e66519. [Google Scholar] [CrossRef]
- Bean, C.; Audano, M.; Varanita, T.; Favaretto, F.; Medaglia, M.; Gerdol, M.; Pernas, L.; Stasi, F.; Giacomello, M.; Herkenne, S.; et al. The Mitochondrial Protein Opa1 Promotes Adipocyte Browning That Is Dependent on Urea Cycle Metabolites. Nat. Metab. 2021, 3, 1633–1647. [Google Scholar] [CrossRef]
- Herkenne, S.; Ek, O.; Zamberlan, M.; Pellattiero, A.; Chergova, M.; Chivite, I.; Novotná, E.; Rigoni, G.; Fonseca, T.B.; Samardzic, D.; et al. Developmental and Tumor Angiogenesis Requires the Mitochondria-Shaping Protein Opa1. Cell Metab. 2020, 31, 987–1003.e8. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, C.; Puszyk, W.M.; Ogunwobi, O.O.; Cao, M.; Wang, T.; Cabrera, R.; Nelson, D.R.; Liu, C. OPA1 Downregulation Is Involved in Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma. Lab. Investig. J. Tech. Methods Pathol. 2013, 93, 8–19. [Google Scholar] [CrossRef]
- Noguchi, M.; Kohno, S.; Pellattiero, A.; Machida, Y.; Shibata, K.; Shintani, N.; Kohno, T.; Gotoh, N.; Takahashi, C.; Hirao, A.; et al. Inhibition of the Mitochondria-Shaping Protein Opa1 Restores Sensitivity to Gefitinib in a Lung Adenocarcinomaresistant Cell Line. Cell Death Dis. 2023, 14, 241. [Google Scholar] [CrossRef]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.-H.; Dourado, P.M.M.; et al. A Selective Inhibitor of Mitofusin 1-ΒIIPKC Association Improves Heart Failure Outcome in Rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-Induced Phosphorylation and Proteasomal Degradation of Mitofusin 2 Facilitates Mitochondrial Fragmentation and Apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Chen, K.-H.; Lima, P.D.A.; Mewburn, J.; Wu, D.; Al-Qazazi, R.; Jones, O.; Tian, L.; Potus, F.; Bonnet, S.; et al. PINK1-Induced Phosphorylation of Mitofusin 2 at Serine 442 Causes Its Proteasomal Degradation and Promotes Cell Proliferation in Lung Cancer and Pulmonary Arterial Hypertension. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21771. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, H.; Zhang, L.; Lin, X.; Li, X.; Zhuang, H.; Fan, H.; Meng, T.; He, Z.; Huang, H.; et al. The AMPK-MFN2 Axis Regulates MAM Dynamics and Autophagy Induced by Energy Stresses. Autophagy 2021, 17, 1142–1156. [Google Scholar] [CrossRef]
- Li, T.; Han, J.; Jia, L.; Hu, X.; Chen, L.; Wang, Y. PKM2 Coordinates Glycolysis with Mitochondrial Fusion and Oxidative Phosphorylation. Protein Cell 2019, 10, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, J.; Jin, Z.; Zhang, Z. Structural and Evolutionary Characteristics of Dynamin-Related GTPase OPA1. PeerJ 2019, 7, e7285. [Google Scholar] [CrossRef]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 Activation Promotes DRP1-Dependent Mitochondrial Fission Necessary for Cell Reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, H.; Gutiérrez Cortés, N.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Gawlowski, T.; Suarez, J.; Scott, B.; Torres-Gonzalez, M.; Wang, H.; Schwappacher, R.; Han, X.; Yates, J.R.; Hoshijima, M.; Dillmann, W. Modulation of Dynamin-Related Protein 1 (DRP1) Function by Increased O-Linked-β-N-Acetylglucosamine Modification (O-GlcNAc) in Cardiac Myocytes. J. Biol. Chem. 2012, 287, 30024–30034. [Google Scholar] [CrossRef]
- Akinbiyi, E.O.; Abramowitz, L.K.; Bauer, B.L.; Stoll, M.S.K.; Hoppel, C.L.; Hsiao, C.-P.; Hanover, J.A.; Mears, J.A. Blocked O-GlcNAc Cycling Alters Mitochondrial Morphology, Function, and Mass. Sci. Rep. 2021, 11, 22106. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Iñiguez-Lluhí, J.A.; Stadler, J.; Chang, C.-R.; Arnoult, D.; Keller, P.J.; Hong, Y.; Blackstone, C.; Feldman, E.L. SUMOylation of the Mitochondrial Fission Protein Drp1 Occurs at Multiple Nonconsensus Sites within the B Domain and Is Linked to Its Activity Cycle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3917–3927. [Google Scholar] [CrossRef]
- Guo, C.; Hildick, K.L.; Luo, J.; Dearden, L.; Wilkinson, K.A.; Henley, J.M. SENP3-Mediated DeSUMOylation of Dynamin-Related Protein 1 Promotes Cell Death Following Ischaemia. EMBO J. 2013, 32, 1514–1528. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.; Wilkinson, K.A.; Harding, A.L.; Carmichael, R.E.; Robinson, D.; Colley, H.E.; Guo, C. The SUMO Protease SENP3 Regulates Mitochondrial Autophagy Mediated by Fis1. EMBO Rep. 2022, 23, e48754. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, T.; Wang, L.; Cai, Y.; Zhong, X.; He, X.; Hu, L.; Tian, S.; Wu, M.; Hui, L.; et al. Fatty Acid Synthesis Is Critical for Stem Cell Pluripotency via Promoting Mitochondrial Fission. EMBO J. 2017, 36, 1330–1347. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Juncker, M.; Reed, R.; Haas, A.; Guidry, J.; Matunis, M.; Yang, W.-C.; Schwartzenburg, J.; Desai, S. SUMOylation of Mitofusins: A Potential Mechanism for Perinuclear Mitochondrial Congression in Cells Treated with Mitochondrial Stressors. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166104. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kapur, M.; Li, M.; Choi, M.-C.; Choi, S.; Kim, H.-J.; Kim, I.; Lee, E.; Taylor, J.P.; Yao, T.-P. MFN1 Deacetylation Activates Adaptive Mitochondrial Fusion and Protects Metabolically Challenged Mitochondria. J. Cell Sci. 2014, 127, 4954–4963. [Google Scholar] [CrossRef]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 to Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef]
- Xiong, Y.; Guan, K.-L. Mechanistic Insights into the Regulation of Metabolic Enzymes by Acetylation. J. Cell Biol. 2012, 198, 155–164. [Google Scholar] [CrossRef]
- Li, P.; Ge, J.; Li, H. Lysine Acetyltransferases and Lysine Deacetylases as Targets for Cardiovascular Disease. Nat. Rev. Cardiol. 2020, 17, 96–115. [Google Scholar] [CrossRef]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Still, A.J.; Floyd, B.J.; Hebert, A.S.; Bingman, C.A.; Carson, J.J.; Gunderson, D.R.; Dolan, B.K.; Grimsrud, P.A.; Dittenhafer-Reed, K.E.; Stapleton, D.S.; et al. Quantification of Mitochondrial Acetylation Dynamics Highlights Prominent Sites of Metabolic Regulation. J. Biol. Chem. 2013, 288, 26209–26219. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, H.; Tan, B.; Yi, Q.; Sun, Y.; Xiang, H.; Chen, T.; Liu, H.; Xie, Q.; Wang, L.; et al. SIRT3 Promotes Metabolic Maturation of Human IPSC-Derived Cardiomyocytes via OPA1-Controlled Mitochondrial Dynamics. Free Radic. Biol. Med. 2023, 195, 270–282. [Google Scholar] [CrossRef]
- Ma, R.; Ma, L.; Weng, W.; Wang, Y.; Liu, H.; Guo, R.; Gao, Y.; Tu, J.; Xu, T.-L.; Cheng, J.; et al. DUSP6 SUMOylation Protects Cells from Oxidative Damage via Direct Regulation of Drp1 Dephosphorylation. Sci. Adv. 2020, 6, eaaz0361. [Google Scholar] [CrossRef]
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Mol. Cell 2015, 59, 941–955. [Google Scholar] [CrossRef]
- Wang, M.; Wei, R.; Li, G.; Bi, H.-L.; Jia, Z.; Zhang, M.; Pang, M.; Li, X.; Ma, L.; Tang, Y. SUMOylation of SYNJ2BP-COX16 Promotes Breast Cancer Progression through DRP1-Mediated Mitochondrial Fission. Cancer Lett. 2022, 547, 215871. [Google Scholar] [CrossRef]
- Wang, T.; Cao, Y.; Zheng, Q.; Tu, J.; Zhou, W.; He, J.; Zhong, J.; Chen, Y.; Wang, J.; Cai, R.; et al. SENP1-Sirt3 Signaling Controls Mitochondrial Protein Acetylation and Metabolism. Mol. Cell 2019, 75, 823–834.e5. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shangguan, X.; Zhou, W.; Cao, Y.; Zheng, Q.; Tu, J.; Hu, G.; Liang, Z.; Jiang, C.; Deng, L.; et al. Glucose Limitation Activates AMPK Coupled SENP1-Sirt3 Signalling in Mitochondria for T Cell Memory Development. Nat. Commun. 2021, 12, 4371. [Google Scholar] [CrossRef]
- Yamada, S.; Sato, A.; Ishihara, N.; Akiyama, H.; Sakakibara, S.-I. Drp1 SUMO/DeSUMOylation by Senp5 Isoforms Influences ER Tubulation and Mitochondrial Dynamics to Regulate Brain Development. iScience 2021, 24, 103484. [Google Scholar] [CrossRef]
- Hu, Y.; Suarez, J.; Fricovsky, E.; Wang, H.; Scott, B.T.; Trauger, S.A.; Han, W.; Hu, Y.; Oyeleye, M.O.; Dillmann, W.H. Increased Enzymatic O-GlcNAcylation of Mitochondrial Proteins Impairs Mitochondrial Function in Cardiac Myocytes Exposed to High Glucose. J. Biol. Chem. 2009, 284, 547–555. [Google Scholar] [CrossRef]
- Ma, J.; Banerjee, P.; Whelan, S.A.; Liu, T.; Wei, A.-C.; Ramirez-Correa, G.; McComb, M.E.; Costello, C.E.; O’Rourke, B.; Murphy, A.; et al. Comparative Proteomics Reveals Dysregulated Mitochondrial O-GlcNAcylation in Diabetic Hearts. J. Proteome Res. 2016, 15, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Wende, A.R.; Schell, J.C.; Ha, C.-M.; Pepin, M.E.; Khalimonchuk, O.; Schwertz, H.; Pereira, R.O.; Brahma, M.K.; Tuinei, J.; Contreras-Ferrat, A.; et al. Maintaining Myocardial Glucose Utilization in Diabetic Cardiomyopathy Accelerates Mitochondrial Dysfunction. Diabetes 2020, 69, 2094–2111. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-D.; Vera, N.B.; Yang, Y.; Zhang, B.; Ni, W.; Ziso-Qejvanaj, E.; Ding, S.; Zhang, K.; Yin, R.; Wang, S.; et al. Adipocyte OGT Governs Diet-Induced Hyperphagia and Obesity. Nat. Commun. 2018, 9, 5103. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fu, M.; Li, M.-D.; Zhang, K.; Zhang, B.; Wang, S.; Liu, Y.; Ni, W.; Ong, Q.; Mi, J.; et al. O-GlcNAc Transferase Inhibits Visceral Fat Lipolysis and Promotes Diet-Induced Obesity. Nat. Commun. 2020, 11, 181. [Google Scholar] [CrossRef]
- Makino, A.; Suarez, J.; Gawlowski, T.; Han, W.; Wang, H.; Scott, B.T.; Dillmann, W.H. Regulation of Mitochondrial Morphology and Function by O-GlcNAcylation in Neonatal Cardiac Myocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1296–R1302. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-Y.; Huang, P.; Jenkins, G.M.; Chan, D.C.; Schiller, J.; Frohman, M.A. A Common Lipid Links Mfn-Mediated Mitochondrial Fusion and SNARE-Regulated Exocytosis. Nat. Cell Biol. 2006, 8, 1255–1262. [Google Scholar] [CrossRef]
- Mårtensson, C.U.; Doan, K.N.; Becker, T. Effects of Lipids on Mitochondrial Functions. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 102–113. [Google Scholar] [CrossRef]
- Becker, T.; Horvath, S.E.; Böttinger, L.; Gebert, N.; Daum, G.; Pfanner, N. Role of Phosphatidylethanolamine in the Biogenesis of Mitochondrial Outer Membrane Proteins. J. Biol. Chem. 2013, 288, 16451–16459. [Google Scholar] [CrossRef]
- Hatch, G.M. Cell Biology of Cardiac Mitochondrial Phospholipids. Biochem. Cell Biol. Biochim. Biol. Cell. 2004, 82, 99–112. [Google Scholar] [CrossRef]
- Basu Ball, W.; Neff, J.K.; Gohil, V.M. The Role of Nonbilayer Phospholipids in Mitochondrial Structure and Function. FEBS Lett. 2018, 592, 1273–1290. [Google Scholar] [CrossRef]
- Holthuis, J.C.M.; Menon, A.K. Lipid Landscapes and Pipelines in Membrane Homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.N.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth Syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Francy, C.A.; Clinton, R.W.; Fröhlich, C.; Murphy, C.; Mears, J.A. Cryo-EM Studies of Drp1 Reveal Cardiolipin Interactions That Activate the Helical Oligomer. Sci. Rep. 2017, 7, 10744. [Google Scholar] [CrossRef] [PubMed]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine Deficiency in Mammalian Mitochondria Impairs Oxidative Phosphorylation and Alters Mitochondrial Morphology. J. Biol. Chem. 2013, 288, 4158–4173. [Google Scholar] [CrossRef]
- Steenbergen, R.; Nanowski, T.S.; Beigneux, A.; Kulinski, A.; Young, S.G.; Vance, J.E. Disruption of the Phosphatidylserine Decarboxylase Gene in Mice Causes Embryonic Lethality and Mitochondrial Defects. J. Biol. Chem. 2005, 280, 40032–40040. [Google Scholar] [CrossRef]
- Selathurai, A.; Kowalski, G.M.; Mason, S.A.; Callahan, D.L.; Foletta, V.C.; Della Gatta, P.A.; Lindsay, A.; Hamley, S.; Kaur, G.; Curtis, A.R.; et al. Phosphatidylserine Decarboxylase Is Critical for the Maintenance of Skeletal Muscle Mitochondrial Integrity and Muscle Mass. Mol. Metab. 2019, 27, 33–46. [Google Scholar] [CrossRef]
- Adachi, Y.; Itoh, K.; Yamada, T.; Cerveny, K.L.; Suzuki, T.L.; Macdonald, P.; Frohman, M.A.; Ramachandran, R.; Iijima, M.; Sesaki, H. Coincident Phosphatidic Acid Interaction Restrains Drp1 in Mitochondrial Division. Mol. Cell 2016, 63, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Ye, F.; Lin, J.; He, H.; Song, Z. The Metabolism and Function of Phospholipids in Mitochondria. Mitochondrial Communications 2023, 1, 2–12. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Bai, J.; Tian, X.; Zhao, X.; Liu, W.; Duan, X.; Shang, W.; Fan, H.-Y.; Tong, C. Mitoguardin Regulates Mitochondrial Fusion through MitoPLD and Is Required for Neuronal Homeostasis. Mol. Cell 2016, 61, 111–124. [Google Scholar] [CrossRef]
- Horibata, Y.; Ando, H.; Zhang, P.; Vergnes, L.; Aoyama, C.; Itoh, M.; Reue, K.; Sugimoto, H. StarD7 Protein Deficiency Adversely Affects the Phosphatidylcholine Composition, Respiratory Activity, and Cristae Structure of Mitochondria. J. Biol. Chem. 2016, 291, 24880–24891. [Google Scholar] [CrossRef]
- Flores-Martín, J.; Reyna, L.; Ridano, M.E.; Panzetta-Dutari, G.M.; Genti-Raimondi, S. Suppression of StarD7 Promotes Endoplasmic Reticulum Stress and Induces ROS Production. Free Radic. Biol. Med. 2016, 99, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.L.; Cruz Del Puerto, M.M.; Flores-Martín, J.; Racca, A.C.; Kourdova, L.T.; Miranda, A.L.; Panzetta-Dutari, G.M.; Genti-Raimondi, S. Role of the Lipid Transport Protein StarD7 in Mitochondrial Dynamics. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159029. [Google Scholar] [CrossRef] [PubMed]
- Galmes, R.; Houcine, A.; van Vliet, A.R.; Agostinis, P.; Jackson, C.L.; Giordano, F. ORP5/ORP8 Localize to Endoplasmic Reticulum-Mitochondria Contacts and Are Involved in Mitochondrial Function. EMBO Rep. 2016, 17, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Cardoso, V.F.; Rochin, L.; Arora, A.; Houcine, A.; Jääskeläinen, E.; Kivelä, A.M.; Sauvanet, C.; Le Bars, R.; Marien, E.; Dehairs, J.; et al. ORP5/8 and MIB/MICOS Link ER-Mitochondria and Intra-Mitochondrial Contacts for Non-Vesicular Transport of Phosphatidylserine. Cell Rep. 2022, 40, 111364. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.K.; Park, T.H.; Kim, H.Y.; Jang, H.; Lee, J.; Hwang, G.-S.; Ryu, S.E.; Park, S.H.; Song, H.K.; Ban, H.S.; et al. Phospholipid Transfer Function of PTPIP51 at Mitochondria-Associated ER Membranes. EMBO Rep. 2021, 22, e51323. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.; Jun, Y.; Lee, C. Structural Basis for Mitoguardin-2 Mediated Lipid Transport at ER-Mitochondrial Membrane Contact Sites. Nat. Commun. 2022, 13, 3702. [Google Scholar] [CrossRef]
- Liu, S.; Geng, B.; Zou, L.; Wei, S.; Wang, W.; Deng, J.; Xu, C.; Zhao, X.; Lyu, Y.; Su, X.; et al. Development of Hypertrophic Cardiomyopathy in Perilipin-1 Null Mice with Adipose Tissue Dysfunction. Cardiovasc. Res. 2015, 105, 20–30. [Google Scholar] [CrossRef]
- Shen, J.; Liu, X.; Yu, W.-M.; Liu, J.; Nibbelink, M.G.; Guo, C.; Finkel, T.; Qu, C.-K. A Critical Role of Mitochondrial Phosphatase Ptpmt1 in Embryogenesis Reveals a Mitochondrial Metabolic Stress-Induced Differentiation Checkpoint in Embryonic Stem Cells. Mol. Cell. Biol. 2011, 31, 4902–4916. [Google Scholar] [CrossRef]
- Yeshaw, W.M.; van der Zwaag, M.; Pinto, F.; Lahaye, L.L.; Faber, A.I.; Gómez-Sánchez, R.; Dolga, A.M.; Poland, C.; Monaco, A.P.; van IJzendoorn, S.C.; et al. Human VPS13A Is Associated with Multiple Organelles and Influences Mitochondrial Morphology and Lipid Droplet Motility. eLife 2019, 8, e43561. [Google Scholar] [CrossRef]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef]
- Subra, M.; Dezi, M.; Bigay, J.; Lacas-Gervais, S.; Di Cicco, A.; Araújo, A.R.D.; Abélanet, S.; Fleuriot, L.; Debayle, D.; Gautier, R.; et al. VAP-A Intrinsically Disordered Regions Enable Versatile Tethering at Membrane Contact Sites. Dev. Cell 2023, 58, 121–138.e9. [Google Scholar] [CrossRef]
- Potting, C.; Tatsuta, T.; König, T.; Haag, M.; Wai, T.; Aaltonen, M.J.; Langer, T. TRIAP1/PRELI Complexes Prevent Apoptosis by Mediating Intramitochondrial Transport of Phosphatidic Acid. Cell Metab. 2013, 18, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yazawa, E.; Keating, E.M.; Mazumdar, N.; Hauschild, A.; Ma, Q.; Wu, H.; Xu, Y.; Shi, X.; Strathdee, D.; et al. Genetic Modifiers Modulate Phenotypic Expression of Tafazzin Deficiency in a Mouse Model of Barth Syndrome. Hum. Mol. Genet. 2023, 32, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dai, Q.; Chen, J.; Durrant, D.; Freeman, A.; Liu, T.; Grossman, D.; Lee, R.M. Phospholipid Scramblase 3 Controls Mitochondrial Structure, Function, and Apoptotic Response. Mol. Cancer Res. 2003, 1, 892–902. [Google Scholar] [PubMed]
- Giordano, F. Non-Vesicular Lipid Trafficking at the Endoplasmic Reticulum-Mitochondria Interface. Biochem. Soc. Trans. 2018, 46, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Saita, S.; Tatsuta, T.; Lampe, P.A.; König, T.; Ohba, Y.; Langer, T. PARL Partitions the Lipid Transfer Protein STARD7 between the Cytosol and Mitochondria. EMBO J. 2018, 37, e97909. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Freyre, C.A.C.; Rauher, P.C.; Ejsing, C.S.; Klemm, R.W. MIGA2 Links Mitochondria, the ER, and Lipid Droplets and Promotes De Novo Lipogenesis in Adipocytes. Mol. Cell 2019, 76, 811–825.e14. [Google Scholar] [CrossRef]
- Boutant, M.; Kulkarni, S.S.; Joffraud, M.; Ratajczak, J.; Valera-Alberni, M.; Combe, R.; Zorzano, A.; Cantó, C. Mfn2 Is Critical for Brown Adipose Tissue Thermogenic Function. EMBO J. 2017, 36, 1543–1558. [Google Scholar] [CrossRef]
- Rampoldi, L.; Dobson-Stone, C.; Rubio, J.P.; Danek, A.; Chalmers, R.M.; Wood, N.W.; Verellen, C.; Ferrer, X.; Malandrini, A.; Fabrizi, G.M.; et al. A Conserved Sorting-Associated Protein Is Mutant in Chorea-Acanthocytosis. Nat. Genet. 2001, 28, 119–120. [Google Scholar] [CrossRef]
- Ueno, S.; Maruki, Y.; Nakamura, M.; Tomemori, Y.; Kamae, K.; Tanabe, H.; Yamashita, Y.; Matsuda, S.; Kaneko, S.; Sano, A. The Gene Encoding a Newly Discovered Protein, Chorein, Is Mutated in Chorea-Acanthocytosis. Nat. Genet. 2001, 28, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.E.; Levine, T.P. VAP, a Versatile Access Point for the Endoplasmic Reticulum: Review and Analysis of FFAT-like Motifs in the VAPome. Biochim. Biophys. Acta 2016, 1861, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Di Mattia, T.; Martinet, A.; Ikhlef, S.; McEwen, A.G.; Nominé, Y.; Wendling, C.; Poussin-Courmontagne, P.; Voilquin, L.; Eberling, P.; Ruffenach, F.; et al. FFAT Motif Phosphorylation Controls Formation and Lipid Transfer Function of Inter-Organelle Contacts. EMBO J. 2020, 39, e104369. [Google Scholar] [CrossRef] [PubMed]
- Kors, S.; Hacker, C.; Bolton, C.; Maier, R.; Reimann, L.; Kitchener, E.J.A.; Warscheid, B.; Costello, J.L.; Schrader, M. Regulating Peroxisome-ER Contacts via the ACBD5-VAPB Tether by FFAT Motif Phosphorylation and GSK3β. J. Cell Biol. 2022, 221, e202003143. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Wang, C.; Chang, T.-K.; Sliter, D.A.; Powers, C.M.; Hofmann, K.; Youle, R.J.; Baehrecke, E.H. Vps13D Encodes a Ubiquitin-Binding Protein That Is Required for the Regulation of Mitochondrial Size and Clearance. Curr. Biol. 2018, 28, 287–295.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fang, N.; Xiong, J.; Du, Y.; Cao, Y.; Ji, W.-K. An ESCRT-Dependent Step in Fatty Acid Transfer from Lipid Droplets to Mitochondria through VPS13D-TSG101 Interactions. Nat. Commun. 2021, 12, 1252. [Google Scholar] [CrossRef]
- Seong, E.; Insolera, R.; Dulovic, M.; Kamsteeg, E.-J.; Trinh, J.; Brüggemann, N.; Sandford, E.; Li, S.; Ozel, A.B.; Li, J.Z.; et al. Mutations in VPS13D Lead to a New Recessive Ataxia with Spasticity and Mitochondrial Defects. Ann. Neurol. 2018, 83, 1075–1088. [Google Scholar] [CrossRef]
- Wang, C.; Yuan, J.; Du, J. Resveratrol Alleviates Acute Lung Injury through Regulating PLSCR-3-Mediated Mitochondrial Dysfunction and Mitophagy in a Cecal Ligation and Puncture Model. Eur. J. Pharmacol. 2021, 913, 174643. [Google Scholar] [CrossRef]
- Van, Q.; Liu, J.; Lu, B.; Feingold, K.R.; Shi, Y.; Lee, R.M.; Hatch, G.M. Phospholipid Scramblase-3 Regulates Cardiolipin de Novo Biosynthesis and Its Resynthesis in Growing HeLa Cells. Biochem. J. 2007, 401, 103–109. [Google Scholar] [CrossRef]
- Singh, S.; Joshi, A.; Kamat, S.S. Mapping the Neuroanatomy of ABHD16A, ABHD12, and Lysophosphatidylserines Provides New Insights into the Pathophysiology of the Human Neurological Disorder PHARC. Biochemistry 2020, 59, 2299–2311. [Google Scholar] [CrossRef]
- Shi, X.; Li, X.; Xu, Z.; Shen, L.; Ding, Y.; Chen, S.; Mao, L.; Liu, W.; Xu, J. ABHD16A Negatively Regulates the Palmitoylation and Antiviral Function of IFITM Proteins. mBio 2022, 13, e0228922. [Google Scholar] [CrossRef]
- Perry, R.J.; Cardone, R.L.; Petersen, M.C.; Zhang, D.; Fouqueray, P.; Hallakou-Bozec, S.; Bolze, S.; Shulman, G.I.; Petersen, K.F.; Kibbey, R.G. Imeglimin Lowers Glucose Primarily by Amplifying Glucose-Stimulated Insulin Secretion in High-Fat-Fed Rodents. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E461–E470. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Chauvin, M.-A.; Bendridi, N.; Durand, A.; Meugnier, E.; Madec, A.-M.; Bernoud-Hubac, N.; Pais de Barros, J.-P.; Fontaine, É.; Acquaviva, C.; et al. Imeglimin Normalizes Glucose Tolerance and Insulin Sensitivity and Improves Mitochondrial Function in Liver of a High-Fat, High-Sucrose Diet Mice Model. Diabetes 2015, 64, 2254–2264. [Google Scholar] [CrossRef]
- Dubourg, J.; Fouqueray, P.; Thang, C.; Grouin, J.-M.; Ueki, K. Efficacy and Safety of Imeglimin Monotherapy Versus Placebo in Japanese Patients With Type 2 Diabetes (TIMES 1): A Double-Blind, Randomized, Placebo-Controlled, Parallel-Group, Multicenter Phase 3 Trial. Diabetes Care 2021, 44, 952–959. [Google Scholar] [CrossRef]
- Sanada, J.; Obata, A.; Fushimi, Y.; Kimura, T.; Shimoda, M.; Ikeda, T.; Nogami, Y.; Obata, Y.; Yamasaki, Y.; Nakanishi, S.; et al. Imeglimin Exerts Favorable Effects on Pancreatic β-Cells by Improving Morphology in Mitochondria and Increasing the Number of Insulin Granules. Sci. Rep. 2022, 12, 13220. [Google Scholar] [CrossRef]
- Hozumi, K.; Sugawara, K.; Ishihara, T.; Ishihara, N.; Ogawa, W. Effects of Imeglimin on Mitochondrial Function, AMPK Activity, and Gene Expression in Hepatocytes. Sci. Rep. 2023, 13, 746. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting Mitochondrial Fission Protects the Heart against Ischemia/Reperfusion Injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor Mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor That Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef]
- Maneechote, C.; Palee, S.; Apaijai, N.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Mitochondrial Dynamic Modulation Exerts Cardiometabolic Protection in Obese Insulin-Resistant Rats. Clin. Sci. 2019, 133, 2431–2447. [Google Scholar] [CrossRef]
- Finocchietto, P.; Perez, H.; Blanco, G.; Miksztowicz, V.; Marotte, C.; Morales, C.; Peralta, J.; Berg, G.; Poderoso, C.; Poderoso, J.J.; et al. Inhibition of Mitochondrial Fission by Drp-1 Blockade by Short-Term Leptin and Mdivi-1 Treatment Improves White Adipose Tissue Abnormalities in Obesity and Diabetes. Pharmacol. Res. 2022, 178, 106028. [Google Scholar] [CrossRef]
- Ruiz, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 (Mdivi-1) Protects Neurons against Excitotoxicity through the Modulation of Mitochondrial Function and Intracellular Ca2+ Signaling. Front. Mol. Neurosci. 2018, 11, 3. [Google Scholar] [CrossRef]
- Ruiz, A.; Quintela-López, T.; Sánchez-Gómez, M.V.; Gaminde-Blasco, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 Disrupts Oligodendrocyte Ca2+ Homeostasis and Mitochondrial Function. Glia 2020, 68, 1743–1756. [Google Scholar] [CrossRef]
- Rosdah, A.A.; Abbott, B.M.; Langendorf, C.G.; Deng, Y.; Truong, J.Q.; Waddell, H.M.M.; Ling, N.X.Y.; Smiles, W.J.; Delbridge, L.M.D.; Liu, G.-S.; et al. A Novel Small Molecule Inhibitor of Human Drp1. Sci. Rep. 2022, 12, 21531. [Google Scholar] [CrossRef]
- Rios, L.; Pokhrel, S.; Li, S.-J.; Heo, G.; Haileselassie, B.; Mochly-Rosen, D. Targeting an Allosteric Site in Dynamin-Related Protein 1 to Inhibit Fis1-Mediated Mitochondrial Dysfunction. Nat. Commun. 2023, 14, 4356. [Google Scholar] [CrossRef]
- Haileselassie, B.; Mukherjee, R.; Joshi, A.U.; Napier, B.A.; Massis, L.M.; Ostberg, N.P.; Queliconi, B.B.; Monack, D.; Bernstein, D.; Mochly-Rosen, D. Drp1/Fis1 Interaction Mediates Mitochondrial Dysfunction in Septic Cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 130, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 Interaction Slows Progression of Amyotrophic Lateral Sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, P.; Cao, Y.; Liu, S.; Wang, W.; Li, L.; Li, J.; Jiang, Z.; Ma, Y.; Chen, S.; et al. Chemical Inhibition of Mitochondrial Fission via Targeting the DRP1-Receptor Interaction. Cell Chem. Biol. 2023, 30, 278–294.e11. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.; Tamucci, J.D.; Ng, E.L.; Liu, S.; Birk, A.V.; Szeto, H.H.; May, E.R.; Alexandrescu, A.T.; Alder, N.N. Structure-Activity Relationships of Mitochondria-Targeted Tetrapeptide Pharmacological Compounds. eLife 2022, 11, e75531. [Google Scholar] [CrossRef] [PubMed]
- Petcherski, A.; Trudeau, K.M.; Wolf, D.M.; Segawa, M.; Lee, J.; Taddeo, E.P.; Deeney, J.T.; Liesa, M. Elamipretide Promotes Mitophagosome Formation and Prevents Its Reduction Induced by Nutrient Excess in INS1 β-Cells. J. Mol. Biol. 2018, 430, 4823–4833. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.-H.; Campbell, M.D.; et al. Late-Life Restoration of Mitochondrial Function Reverses Cardiac Dysfunction in Old Mice. eLife 2020, 9, e55513. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Zhang, G.; Hall, D.; Oates, P.J.; Maity, S.; Madesh, M.; Han, X.; Sharma, K. Restoring Mitochondrial Superoxide Levels with Elamipretide (MTP-131) Protects Db/Db Mice against Progression of Diabetic Kidney Disease. J. Biol. Chem. 2020, 295, 7249–7260. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.P.; Kruse, S.E.; Percival, J.M.; Goh, J.; White, C.C.; Hopkins, H.C.; Kavanagh, T.J.; Szeto, H.H.; Rabinovitch, P.S.; Marcinek, D.J. Mitochondrial-Targeted Peptide Rapidly Improves Mitochondrial Energetics and Skeletal Muscle Performance in Aged Mice. Aging Cell 2013, 12, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Lopez, I.; Diaz-Morales, N.; Iannantuoni, F.; Lopez-Domenech, S.; de Marañon, A.M.; Abad-Jimenez, Z.; Bañuls, C.; Rovira-Llopis, S.; Herance, J.R.; Rocha, M.; et al. The Mitochondrial Antioxidant SS-31 Increases SIRT1 Levels and Ameliorates Inflammation, Oxidative Stress and Leukocyte-Endothelium Interactions in Type 2 Diabetes. Sci. Rep. 2018, 8, 15862. [Google Scholar] [CrossRef]
- Miret-Casals, L.; Sebastián, D.; Brea, J.; Rico-Leo, E.M.; Palacín, M.; Fernández-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278.e4. [Google Scholar] [CrossRef]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial Fusion Exploits a Therapeutic Vulnerability of Pancreatic Cancer. JCI Insight 2019, 5, e126915. [Google Scholar] [CrossRef]
- Dang, X.; Zhang, L.; Franco, A.; Li, J.; Rocha, A.G.; Devanathan, S.; Dolle, R.E.; Bernstein, P.R.; Dorn, G.W. Discovery of 6-Phenylhexanamide Derivatives as Potent Stereoselective Mitofusin Activators for the Treatment of Mitochondrial Diseases. J. Med. Chem. 2020, 63, 7033–7051. [Google Scholar] [CrossRef]
- Franco, A.; Dang, X.; Walton, E.K.; Ho, J.N.; Zablocka, B.; Ly, C.; Miller, T.M.; Baloh, R.H.; Shy, M.E.; Yoo, A.S.; et al. Burst Mitofusin Activation Reverses Neuromuscular Dysfunction in Murine CMT2A. eLife 2020, 9, e61119. [Google Scholar] [CrossRef]
- Franco, A.; Dang, X.; Zhang, L.; Molinoff, P.B.; Dorn, G.W. Mitochondrial Dysfunction and Pharmacodynamics of Mitofusin Activation in Murine Charcot-Marie-Tooth Disease Type 2A. J. Pharmacol. Exp. Ther. 2022, 383, 137–148. [Google Scholar] [CrossRef]
- Zacharioudakis, E.; Agianian, B.; Kumar Mv, V.; Biris, N.; Garner, T.P.; Rabinovich-Nikitin, I.; Ouchida, A.T.; Margulets, V.; Nordstrøm, L.U.; Riley, J.S.; et al. Modulating Mitofusins to Control Mitochondrial Function and Signaling. Nat. Commun. 2022, 13, 3775. [Google Scholar] [CrossRef]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological Advances in Mitochondrial Therapy. EBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Grzeszczak, W. Imeglimin: A New Antidiabetic Drug with Potential Future in the Treatment of Patients with Type 2 Diabetes. Endokrynol. Pol. 2022, 73, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Fauzi, M.; Murakami, T.; Fujimoto, H.; Botagarova, A.; Sakaki, K.; Kiyobayashi, S.; Ogura, M.; Inagaki, N. Preservation Effect of Imeglimin on Pancreatic β-Cell Mass: Noninvasive Evaluation Using 111In-Exendin-4 SPECT/CT Imaging and the Perspective of Mitochondrial Involvements. Front. Endocrinol. 2022, 13, 1010825. [Google Scholar] [CrossRef]
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial Protein Interaction Landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; De Rasmo, D.; Signorile, A.; Corcelli, A.; Lobasso, S. Beneficial Effects of SS-31 Peptide on Cardiac Mitochondrial Dysfunction in Tafazzin Knockdown Mice. Sci. Rep. 2022, 12, 19847. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Bianchini, E.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Natural Compounds Modulating Mitochondrial Functions. Evid.-Based Complement. Altern. Med. 2015, 2015, 527209. [Google Scholar] [CrossRef]
- Waseem, M.; Kaushik, P.; Dutta, S.; Chakraborty, R.; Hassan, M.I.; Parvez, S. Modulatory Role of Quercetin in Mitochondrial Dysfunction in Titanium Dioxide Nanoparticle-Induced Hepatotoxicity. ACS Omega 2022, 7, 3192–3202. [Google Scholar] [CrossRef]
- Qiu, L.; Luo, Y.; Chen, X. Quercetin Attenuates Mitochondrial Dysfunction and Biogenesis via Upregulated AMPK/SIRT1 Signaling Pathway in OA Rats. Biomed. Pharmacother. 2018, 103, 1585–1591. [Google Scholar] [CrossRef]
- Chen, W.-J.; Cheng, Y.; Li, W.; Dong, X.-K.; Wei, J.; Yang, C.-H.; Jiang, Y.-H. Quercetin Attenuates Cardiac Hypertrophy by Inhibiting Mitochondrial Dysfunction Through SIRT3/PARP-1 Pathway. Front. Pharmacol. 2021, 12, 739615. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Gao, Y.; Li, L.; Tang, C.; Wen, G.; Zhou, Y.; Zhou, M.; Mao, L.; Fan, Y. Protective Effects of Quercetin on Mitochondrial Biogenesis in Experimental Traumatic Brain Injury via the Nrf2 Signaling Pathway. PLoS ONE 2016, 11, e0164237. [Google Scholar] [CrossRef]
- Nhu, N.T.; Li, Q.; Liu, Y.; Xu, J.; Xiao, S.-Y.; Lee, S.-D. Effects of Mdivi-1 on Neural Mitochondrial Dysfunction and Mitochondria-Mediated Apoptosis in Ischemia-Reperfusion Injury After Stroke: A Systematic Review of Preclinical Studies. Front. Mol. Neurosci. 2021, 14, 778569. [Google Scholar] [CrossRef]
- So, E.C.; Hsing, C.-H.; Liang, C.-H.; Wu, S.-N. The Actions of Mdivi-1, an Inhibitor of Mitochondrial Fission, on Rapidly Activating Delayed-Rectifier K+ Current and Membrane Potential in HL-1 Murine Atrial Cardiomyocytes. Eur. J. Pharmacol. 2012, 683, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related Protein 1 (Drp1)-mediated Diastolic Dysfunction in Myocardial Ischemia-reperfusion Injury: Therapeutic Benefits of Drp1 Inhibition to Reduce Mitochondrial Fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhang, Z.; Li, S.; Wang, W.; Du, T.; Fang, Q.; Wang, Y.; Wang, D.W. Mdivi-1 Alleviates Cardiac Fibrosis Post Myocardial Infarction at Infarcted Border Zone, Possibly via Inhibition of Drp1-Activated Mitochondrial Fission and Oxidative Stress. Arch. Biochem. Biophys. 2022, 718, 109147. [Google Scholar] [CrossRef] [PubMed]
- Grohm, J.; Kim, S.-W.; Mamrak, U.; Tobaben, S.; Cassidy-Stone, A.; Nunnari, J.; Plesnila, N.; Culmsee, C. Inhibition of Drp1 Provides Neuroprotection in Vitro and in Vivo. Cell Death Differ. 2012, 19, 1446–1458. [Google Scholar] [CrossRef]
- Tokuyama, T.; Hirai, A.; Shiiba, I.; Ito, N.; Matsuno, K.; Takeda, K.; Saito, K.; Mii, K.; Matsushita, N.; Fukuda, T.; et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules 2020, 10, 450. [Google Scholar] [CrossRef]
- Filichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 Mitochondrial Translocation Provides Neural Protection in Dopaminergic System in a Parkinson’s Disease Model Induced by MPTP. Sci. Rep. 2016, 6, 32656. [Google Scholar] [CrossRef]
- Gueven, N.; Ravishankar, P.; Eri, R.; Rybalka, E. Idebenone: When an Antioxidant Is Not an Antioxidant. Redox Biol. 2021, 38, 101812. [Google Scholar] [CrossRef]
- Tinker, R.J.; Lim, A.Z.; Stefanetti, R.J.; McFarland, R. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol. Diagn. Ther. 2021, 25, 181–206. [Google Scholar] [CrossRef]
- Jaber, S.M.; Ge, S.X.; Milstein, J.L.; VanRyzin, J.W.; Waddell, J.; Polster, B.M. Idebenone Has Distinct Effects on Mitochondrial Respiration in Cortical Astrocytes Compared to Cortical Neurons Due to Differential NQO1 Activity. J. Neurosci. 2020, 40, 4609–4619. [Google Scholar] [CrossRef]
- Heitz, F.D.; Erb, M.; Anklin, C.; Robay, D.; Pernet, V.; Gueven, N. Idebenone Protects against Retinal Damage and Loss of Vision in a Mouse Model of Leber’s Hereditary Optic Neuropathy. PLoS ONE 2012, 7, e45182. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).