

Assessment of Oxidative Stress-Induced Oral Epithelial Toxicity

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Optimizing Seeding Density of Cells
2.3. Induction of Oxidative Stress by H2O2 Treatment
2.3.1. Cell Viability and Growth Assay
2.3.2. Evaluation of Epithelial Cell Viability Rate with Fluorescence-Based Live–Dead Assay
2.3.3. Evaluation of Epithelial Cell Viability Rate with Fluorometric Cytotoxicity Assay (FCA)
2.4. Determination of Intracellular ROS Production and Kinetics
2.5. Real-Time Quantification of Oxidative Stress in Oral Keratinocytes Using Fluorescent Dye-Based Redox Probes
2.6. Immunohistochemistry Analysis
2.7. Statistical Analyses
3. Results
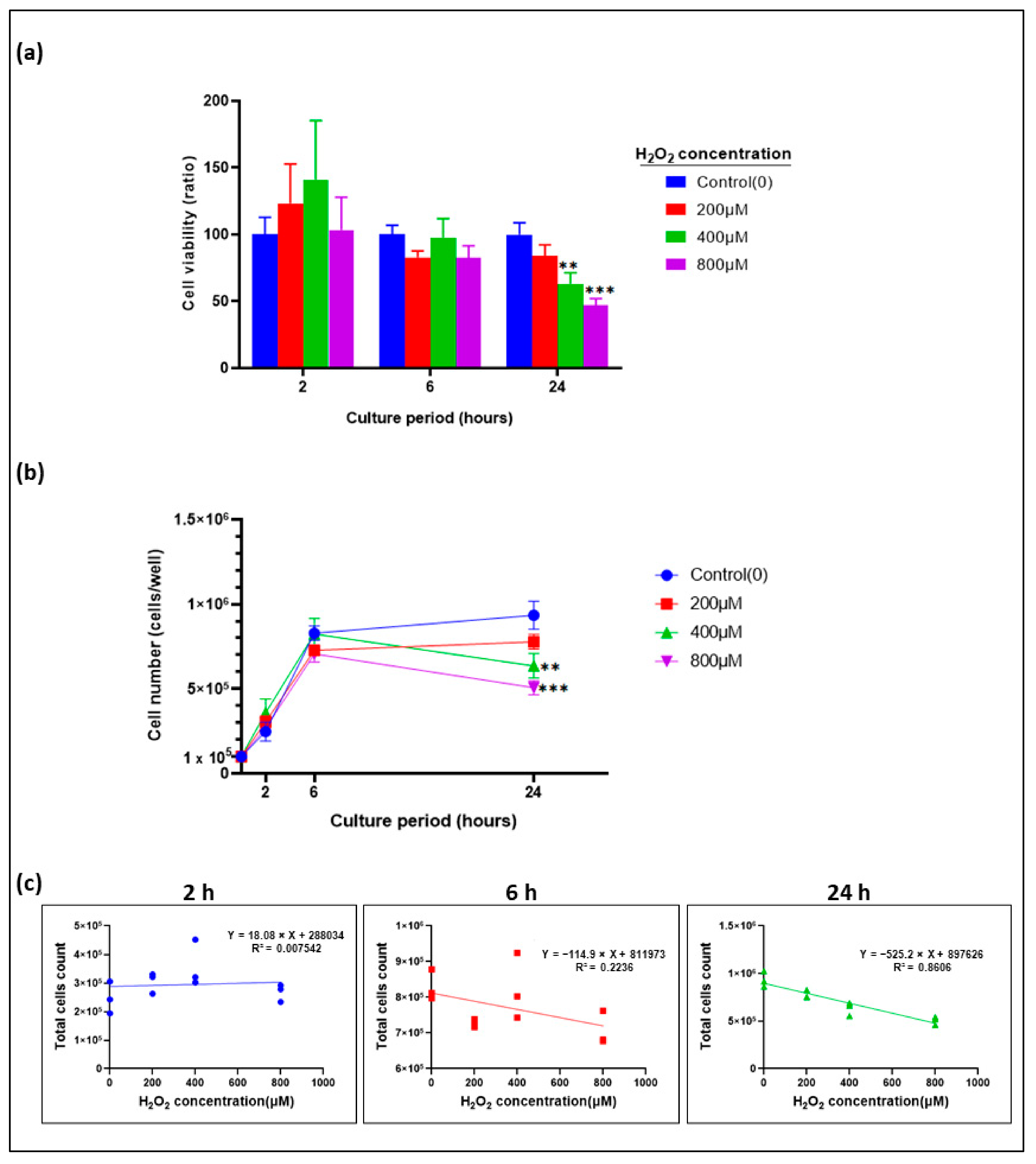
3.1. Growth Kinetics and Optimization of Cell Number for Cell Viability Assay
3.2. Epithelial Cell Proliferation inhibited by H2O2 in a Time- and Dose-Dependent Manner
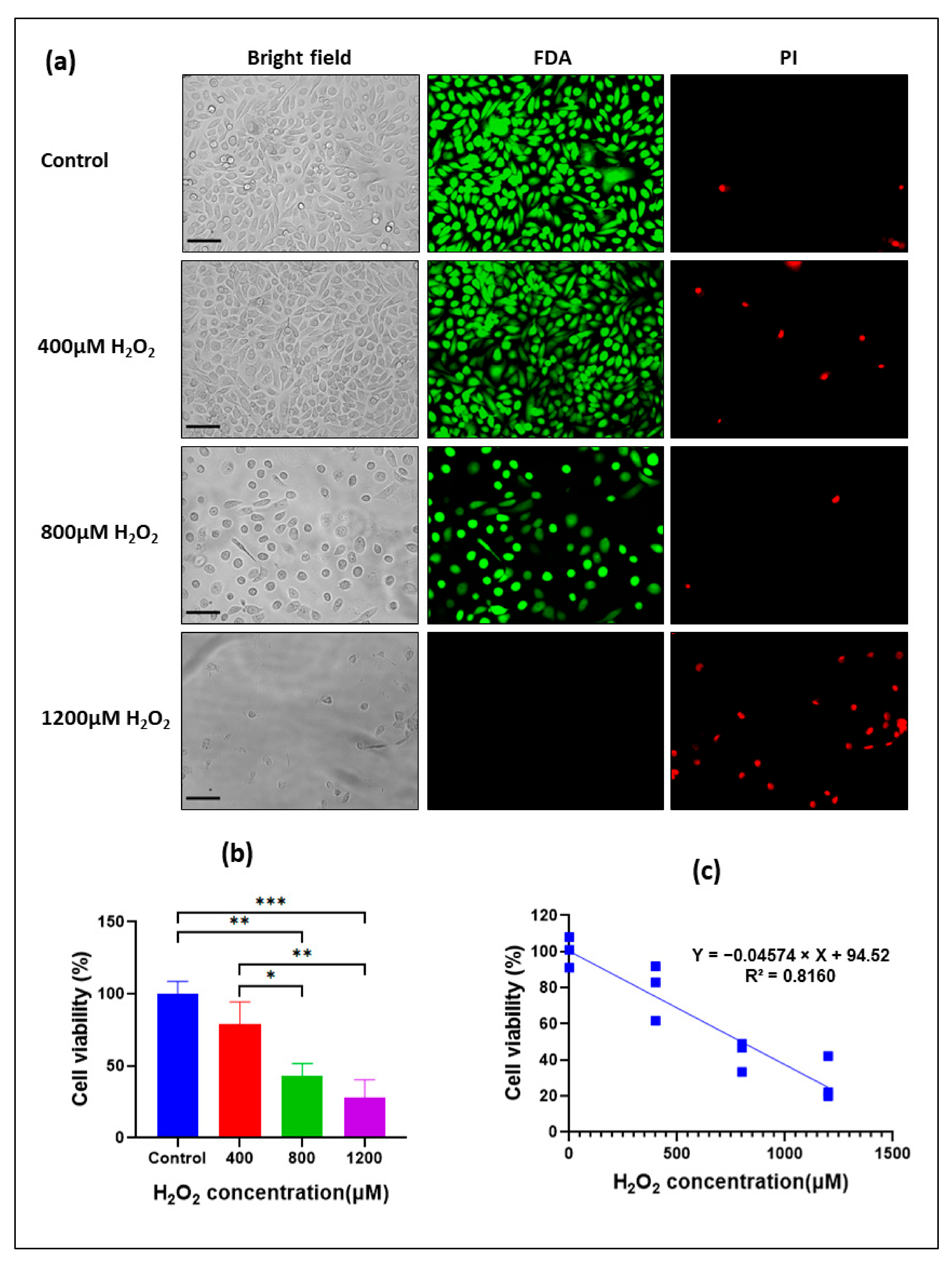
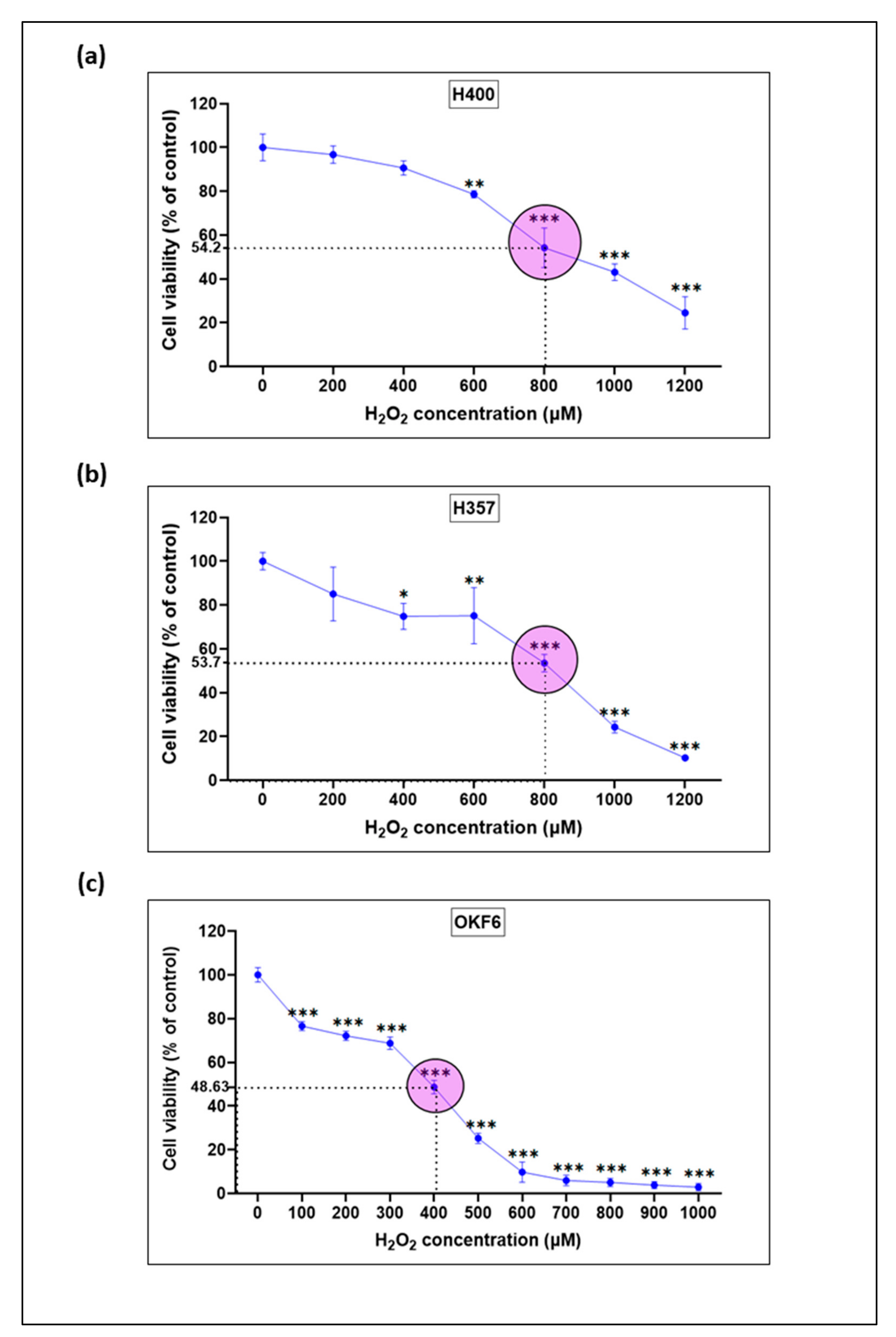
3.3. Cytotoxic Effect of H2O2 on Oral Keratinocytes
3.4. Establishment of an Oxidative Stress-Induced Mucosal Injury Model with H2O2-Treated Oral Keratinocytes
3.5. Effect of H2O2-Induced Oxidative Stress on Morphological Changes in Oral Keratinocytes
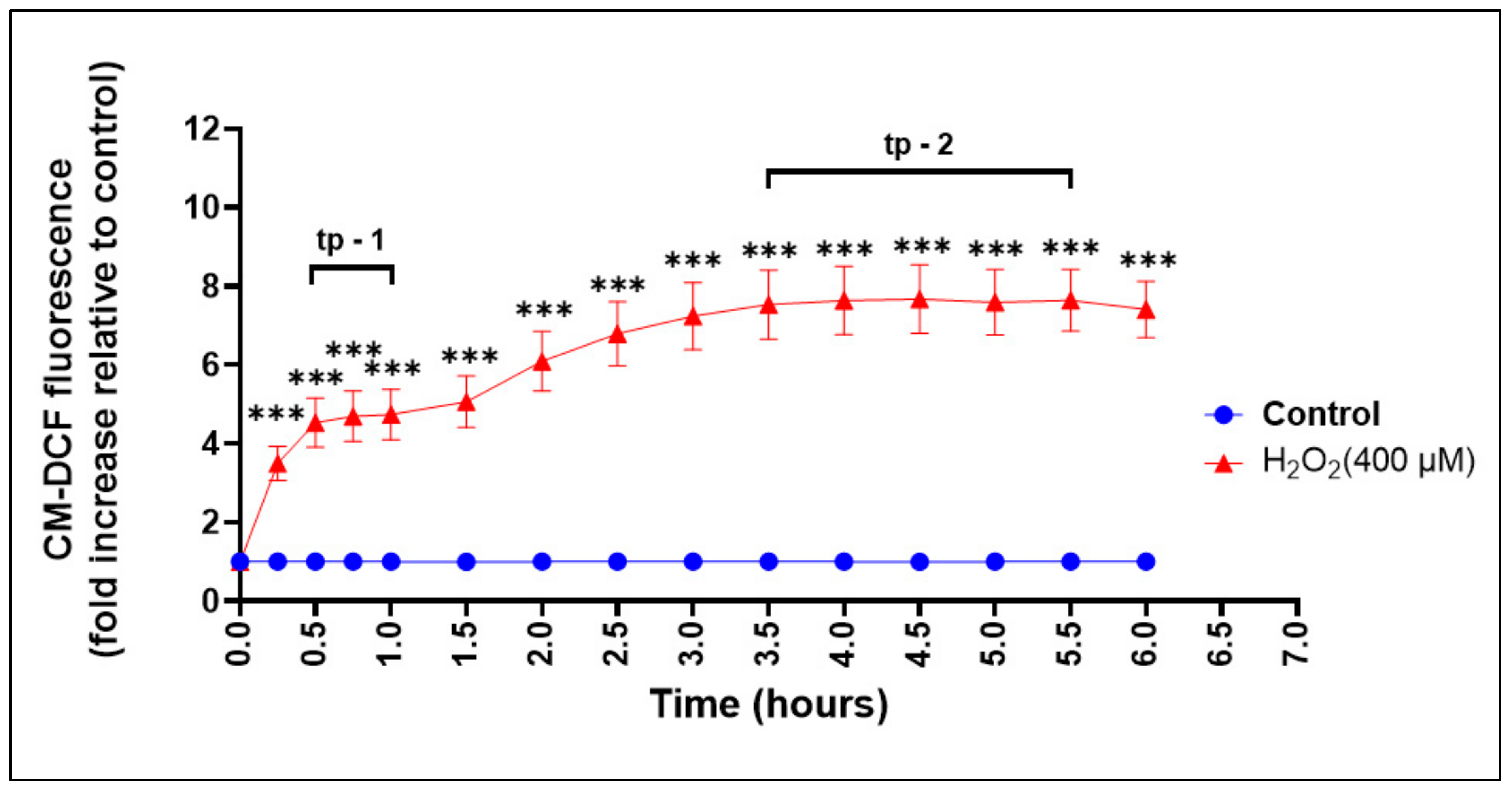
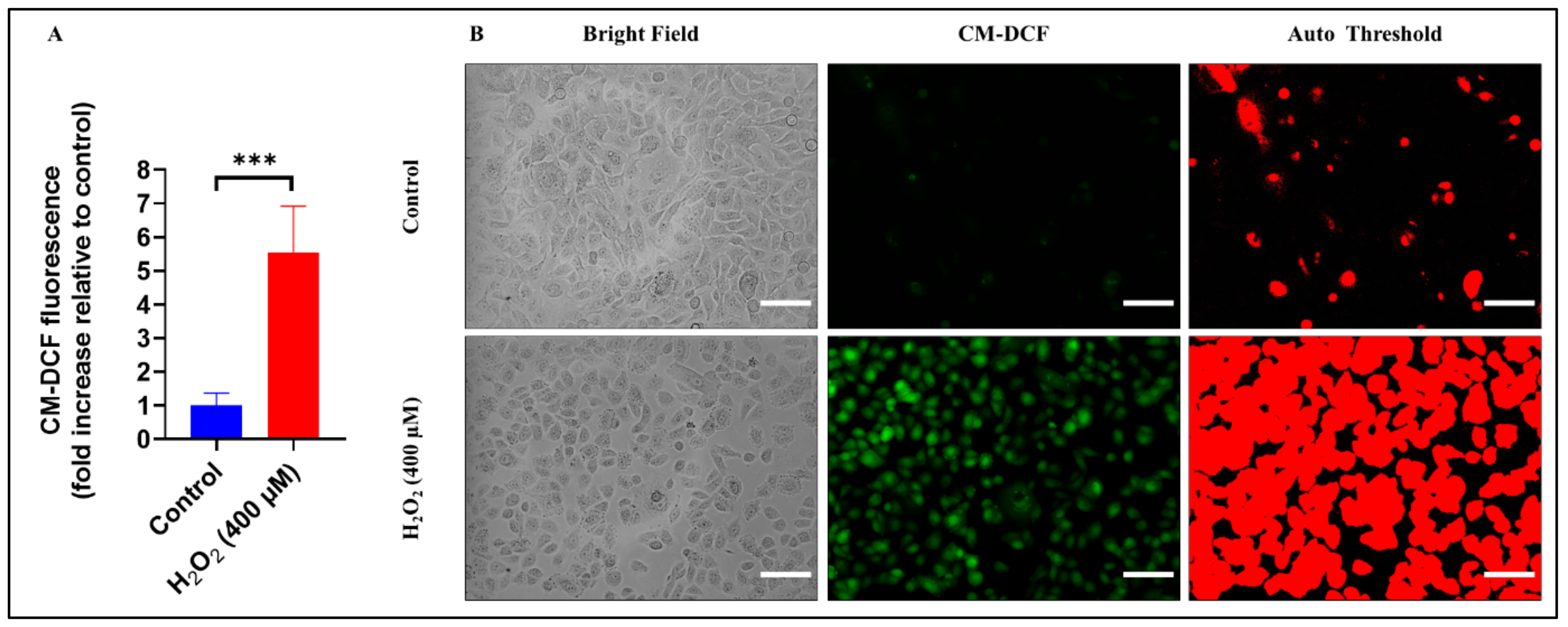
3.6. Assessment of ROS Production during Oxidative Stress-Induced Epithelial Injury In Vitro
3.7. Dynamic Real-Time Detection of the H2O2-induced Redox Changes in Oral Keratinocytes Using ROS Sensitive Fluorescent Sensors
3.8. Malondialdehyde (MDA) and Myeloperoxidase (MPO) Expression in a Mouse Model of Mucositis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sonis, S.T. Mucositis as a biological process: A new hypothesis for the development of chemotherapy-induced stomatotoxicity. Oral Oncol. 1998, 34, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.; Grant, G. Oral and intestinal mucositis—Causes and possible treatments. Aliment. Pharmacol. Ther. 2003, 18, 853–874. [Google Scholar] [CrossRef]
- Sonis, S.T. Mucositis: The impact, biology and therapeutic opportunities of oral mucositis. Oral Oncol. 2009, 45, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.I.; Campos, C.N.; Aarestrup, F.M.; Aarestrup, B.J. Oral mucositis in cancer treatment: Natural history, prevention and treatment. Mol. Clin. Oncol. 2014, 2, 337–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolini, M.; Sobue, T.; Thompson, A.; Dongari-Bagtzoglou, A. Chemotherapy induces oral mucositis in mice without additional noxious stimuli. Transl. Oncol. 2017, 10, 612–620. [Google Scholar] [CrossRef]
- Murphy, B.A.; Gilbert, J. Dysphagia in head and neck cancer patients treated with radiation: Assessment, sequelae, and rehabilitation. Semin. Radiat. Oncol. 2009, 19, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, D.I. Consequences of mucositis-induced treatment breaks and dose reductions on head and neck cancer treatment outcomes. J. Support. Oncol. 2007, 5, 23–31. [Google Scholar] [PubMed]
- Yeoh, A.; Gibson, R.; Yeoh, E.; Bowen, J.; Stringer, A.; Giam, K.; Logan, R.; Keefe, D. Radiation therapy-induced mucositis: Relationships between fractionated radiation, NF-κB, COX-1, and COX-2. Cancer Treat. Rev. 2006, 32, 645–651. [Google Scholar] [CrossRef]
- Sonis, S.T. Oral mucositis. Anticancer Drugs 2011, 22, 607–612. [Google Scholar] [CrossRef]
- Nicolatou-Galitis, O.; Sarri, T.; Bowen, J.; Di Palma, M.; Kouloulias, V.E.; Niscola, P.; Riesenbeck, D.; Stokman, M.; Tissing, W.; Yeoh, E. Systematic review of anti-inflammatory agents for the management of oral mucositis in cancer patients. Support. Care Cancer 2013, 21, 3179–3189. [Google Scholar] [CrossRef] [Green Version]
- Biswal, B.M. Current trends in the management of oral mucositis related to cancer treatment. Malays. J. Med. Sci. MJMS 2008, 15, 4–13. [Google Scholar] [PubMed]
- Cinausero, M.; Aprile, G.; Ermacora, P.; Basile, D.; Vitale, M.G.; Fanotto, V.; Parisi, G.; Calvetti, L.; Sonis, S.T. New Frontiers in the Pathobiology and Treatment of Cancer Regimen-Related Mucosal Injury. Front. Pharmacol. 2017, 8, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonis, S.T. The pathobiology of mucositis. Nat. Rev. Cancer 2004, 4, 277–284. [Google Scholar] [CrossRef]
- Criswell, T.; Leskov, K.; Miyamoto, S.; Luo, G.; Boothman, D.A. Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene 2003, 22, 5813–5827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, F.; Yoshida, A.; Nakajima, A.; Wada-Takahashi, S.; Takahashi, S.-s.; Lee, M.C.-i. Alteration of the redox state with reactive oxygen species for 5-fluorouracil-induced oral mucositis in hamsters. PLoS ONE 2013, 8, e82834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Dasooqi, N.; Sonis, S.T.; Bowen, J.M.; Bateman, E.; Blijlevens, N.; Gibson, R.J.; Logan, R.M.; Nair, R.G.; Stringer, A.M.; Yazbeck, R.; et al. Emerging evidence on the pathobiology of mucositis. Support Care Cancer 2013, 21, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Afonso, V.; Champy, R.; Mitrovic, D.; Collin, P.; Lomri, A. Reactive oxygen species and superoxide dismutases: Role in joint diseases. Jt. Bone Spine 2007, 74, 324–329. [Google Scholar]
- Gille, J.; Joenje, H. Cell culture models for oxidative stress: Superoxide and hydrogen peroxide versus normobaric hyperoxia. Mutat. Res./DNAging 1992, 275, 405–414. [Google Scholar]
- Holbrook, N.J.; Ikeyama, S. Age-related decline in cellular response to oxidative stress: Links to growth factor signaling pathways with common defects. Biochem. Pharmacol. 2002, 64, 999–1005. [Google Scholar]
- Frippiat, C.; Dewelle, J.; Remacle, J.; Toussaint, O. Signal transduction in H2O2-induced senescence-like phenotype in human diploid fibroblasts. Free Radic. Biol. Med. 2002, 33, 1334–1346. [Google Scholar] [CrossRef]
- Kiyoshima, T.; Enoki, N.; Kobayashi, I.; Sakai, T.; Nagata, K.; Wada, H.; Fujiwara, H.; Ookuma, Y.; Sakai, H. Oxidative stress caused by a low concentration of hydrogen peroxide induces senescence-like changes in mouse gingival fibroblasts. Int. J. Mol. Med. 2012, 30, 1007–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, N.; Vicidomini, A.; McCullough, M.; Gambardella, A.; Hassona, Y.; Prime, S.S.; Colella, G. A Hyaluronic Acid-Based Compound Inhibits Fibroblast Senescence Induced by Oxidative Stress In Vitro and Prevents Oral Mucositis In Vivo. J. Cell. Physiol. 2015, 230, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Bierbaumer, L.; Schwarze, U.Y.; Gruber, R.; Neuhaus, W. Cell culture models of oral mucosal barriers: A review with a focus on applications, culture conditions and barrier properties. Tissue Barriers 2018, 6, 1479568. [Google Scholar] [CrossRef] [PubMed]
- Sobue, T.; Bertolini, M.; Thompson, A.; Peterson, D.E.; Diaz, P.I.; Dongari-Bagtzoglou, A. Chemotherapy-induced oral mucositis and associated infections in a novel organotypic model. Mol. Oral Microbiol. 2018, 33, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Sonis, S.; Andreotta, P.; Lyng, G. On the pathogenesis of mTOR inhibitor-associated stomatitis (mIAS)—Studies using an organotypic model of the oral mucosa. Oral Dis. 2017, 23, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Tschachojan, V.; Schroer, H.; Averbeck, N.; Mueller-Klieser, W. Carbon ions and X-rays induce pro-inflammatory effects in 3D oral mucosa models with and without PBMCs. Oncol. Rep. 2014, 32, 1820–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambros, M.P.; Parsa, C.; Mulamalla, H.; Orlando, R.; Lau, B.; Huang, Y.; Pon, D.; Chow, M. Identifying cell and molecular stress after radiation in a three-dimensional (3-D) model of oral mucositis. Biochem. Biophys. Res. Commun. 2011, 405, 102–106. [Google Scholar] [CrossRef]
- Tobita, T.; Izumi, K.; Feinberg, S.E. Development of an in vitro model for radiation-induced effects on oral keratinocytes. Int. J. Oral Maxillofac. Surg. 2010, 39, 364–370. [Google Scholar]
- Abbott, A. Biology’s New Dimension; Nature Publishing Group: New York, NY, USA, 2003. [Google Scholar]
- Yiannis, C.; Huang, K.; Tran, A.N.; Zeng, C.; Dao, E.; Baselyous, O.; Mithwani, M.A.; Paolini, R.; Cirillo, N.; Yap, T. Protective effect of kava constituents in an in vitro model of oral mucositis. J. Cancer Res. Clin. Oncol. 2020, 146, 1801–1811. [Google Scholar] [CrossRef]
- Lin, K.-Y.; Chung, C.-H.; Ciou, J.-S.; Su, P.-F.; Wang, P.-W.; Shieh, D.-B.; Wang, T.-C. Molecular damage and responses of oral keratinocyte to hydrogen peroxide. BMC Oral Health 2019, 19, 10. [Google Scholar] [CrossRef] [Green Version]
- Picciolo, G.; Mannino, F.; Irrera, N.; Minutoli, L.; Altavilla, D.; Vaccaro, M.; Oteri, G.; Squadrito, F.; Pallio, G. Reduction of oxidative stress blunts the NLRP3 inflammatory cascade in LPS stimulated human gingival fibroblasts and oral mucosal epithelial cells. Biomed. Pharmacother. 2022, 146, 112525. [Google Scholar] [CrossRef] [PubMed]
- Picciolo, G.; Mannino, F.; Irrera, N.; Altavilla, D.; Minutoli, L.; Vaccaro, M.; Arcoraci, V.; Squadrito, V.; Picciolo, G.; Squadrito, F.; et al. PDRN, a natural bioactive compound, blunts inflammation and positively reprograms healing genes in an “in vitro” model of oral mucositis. Biomed. Pharmacother. 2021, 138, 111538. [Google Scholar] [CrossRef] [PubMed]
- Picciolo, G.; Pallio, G.; Altavilla, D.; Vaccaro, M.; Oteri, G.; Irrera, N.; Squadrito, F. β-Caryophyllene Reduces the Inflammatory Phenotype of Periodontal Cells by Targeting CB2 Receptors. Biomedicines 2020, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Prime, S.S.; Nixon, S.V.; Crane, I.J.; Stone, A.; Matthews, J.B.; Maitland, N.J.; Remnant, L.; Powell, S.K.; Game, S.M.; Scully, C. The behaviour of human oral squamous cell carcinoma in cell culture. J. Pathol. 1990, 160, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human Keratinocytes That Express hTERT and Also Bypass a p16INK4a-Enforced Mechanism That Limits Life Span Become Immortal yet Retain Normal Growth and Differentiation Characteristics. Mol. Cell. Biol. 2000, 20, 1436–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongari-Bagtzoglou, A.; Kashleva, H. Candida albicans triggers interleukin-8 secretion by oral epithelial cells. Microb. Pathog. 2003, 34, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Dongari-Bagtzoglou, A.; Kashleva, H. Granulocyte-macrophage colony-stimulating factor responses of oral epithelial cells to Candida albicans. Oral Microbiol. Immunol. 2003, 18, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Feucht, E.; DeSanti, C.; Weinberg, A. Selective induction of human beta-defensin mRNAs by Actinobacillus actinomycetemcomitans in primary and immortalized oral epithelial cells. Oral Microbiol. Immunol. 2003, 18, 359–363. [Google Scholar] [CrossRef]
- Dongari-Bagtzoglou, A.; Kashleva, H.; Villar, C.C. Bioactive interleukin-1α is cytolytically released from Candida albicans-infected oral epithelial cells. Med. Mycol. 2004, 42, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Hassona, Y.; Cirillo, N.; Lim, K.P.; Herman, A.; Mellone, M.; Thomas, G.J.; Pitiyage, G.N.; Parkinson, E.K.; Prime, S.S. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-β. Carcinogenesis 2013, 34, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Dalley, A.J.; Pitty, L.P.; Major, A.G.; AbdulMajeed, A.A.; Farah, C.S. Expression of ABCG 2 and B mi-1 in oral potentially malignant lesions and oral squamous cell carcinoma. Cancer Med. 2014, 3, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Celentano, A.; McCullough, M.; Cirillo, N. Glucocorticoids reduce chemotherapeutic effectiveness on OSCC cells via glucose-dependent mechanisms. J. Cell. Physiol. 2019, 234, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Cross, C.E.; Tolba, M.F.; Rondelli, C.M.; Xu, M.; Abdel-Rahman, S.Z. Oxidative stress alters miRNA and gene expression profiles in villous first trimester trophoblasts. BioMed Res. Int. 2015, 2015, 257090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hseu, Y.C.; Yang, T.Y.; Li, M.L.; Rajendran, P.; Mathew, D.C.; Tsai, C.H.; Lin, R.W.; Lee, C.C.; Yang, H.L. Chalcone flavokawain A attenuates TGF-β1-induced fibrotic pathology via inhibition of ROS/Smad3 signaling pathways and induction of Nrf2/ARE-mediated antioxidant genes in vascular smooth muscle cells. J. Cell. Mol. Med. 2019, 23, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Man, W.; Ming, D.; Fang, D.; Chao, L.; Jing, C. Dimethyl sulfoxide attenuates hydrogen peroxide-induced injury in cardiomyocytes via heme oxygenase-1. J. Cell. Biochem. 2014, 115, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Royack, G.; Nguyen, M.; Tong, D.; Poot, M.; Oda, D. Response of human oral epithelial cells to oxidative damage and the effect of vitamin E. Oral Oncol. 2000, 36, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Keston, A.S.; Brandt, R. The fluorometric analysis of ultramicro quantities of hydrogen peroxide. Anal. Biochem. 1965, 11, 1–5. [Google Scholar] [CrossRef]
- Ro, S.-H.; Nam, M.; Jang, I.; Park, H.-W.; Park, H.; Semple, I.A.; Kim, M.; Kim, J.S.; Park, H.; Einat, P. Sestrin2 inhibits uncoupling protein 1 expression through suppressing reactive oxygen species. Proc. Natl. Acad. Sci. USA 2014, 111, 7849–7854. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar]
- Mohammed, A.I.; Celentano, A.; Paolini, R.; Low, J.T.; McCullough, M.J.; O’ Reilly, L.A.; Cirillo, N. Characterization of a novel dual murine model of chemotherapy-induced oral and intestinal mucositis. Sci. Rep. 2023, 13, 1396. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Cardaci, S.; Jerby, L.; MacKenzie, E.D.; Sciacovelli, M.; Johnson, T.I.; Gaude, E.; King, A.; Leach, J.D.G.; Edrada-Ebel, R.; et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun. 2015, 6, 6001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2007, 26, 1101–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altiok, N.; Ersoz, M.; Karpuz, V.; Koyuturk, M. Ginkgo biloba extract regulates differentially the cell death induced by hydrogen peroxide and simvastatin. Neurotoxicology 2006, 27, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-S.; Marbán, E. Physiological Levels of Reactive Oxygen Species Are Required to Maintain Genomic Stability in Stem Cells. Stem Cells 2010, 28, 1178–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, R.M.; Suzuki, Y.J. Cell Proliferation, Reactive Oxygen and Cellular Glutathione. Dose-Response 2005, 3, 425–442. [Google Scholar] [CrossRef]
- Reaves, T.A.; Chin, A.C.; Parkos, C.A. Neutrophil transepithelial migration: Role of toll-like receptors in mucosal inflammation. Mem. Inst. Oswaldo Cruz 2005, 100, 191–198. [Google Scholar]
- Deng, J.-S.; Chi, C.-S.; Huang, S.-S.; Shie, P.-H.; Lin, T.-H.; Huang, G.-J. Antioxidant, analgesic, and anti-inflammatory activities of the ethanolic extracts of Taxillus liquidambaricola. J. Ethnopharmacol. 2011, 137, 1161–1171. [Google Scholar]
- Azevedo, O.G.R.; Oliveira, R.A.C.; Oliveira, B.C.; Zaja-Milatovic, S.; Araújo, C.V.; Wong, D.V.T.; Costa, T.B.; Lucena, H.B.M.; Lima-Júnior, R.C.P.; Ribeiro, R.A. Apolipoprotein E COG 133 mimetic peptide improves 5-fluorouracil-induced intestinal mucositis. BMC Gastroenterol. 2012, 12, 35. [Google Scholar]
- De Ávila, P.H.M.; de Ávila, R.I.; dos Santos Filho, E.X.; Cunha Bastos, C.C.; Batista, A.C.; Mendonça, E.F.; Serpa, R.C.; Marreto, R.N.; da Cruz, A.F.; Lima, E.M.; et al. Mucoadhesive formulation of Bidens pilosa L. (Asteraceae) reduces intestinal injury from 5-fluorouracil-induced mucositis in mice. Toxicol. Rep. 2015, 2, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Sonis, S.T. New thoughts on the initiation of mucositis. Oral Dis. 2010, 16, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Sonis, S.T. Pathobiology of oral mucositis: Novel insights and opportunities. J. Support. Oncol. 2007, 5, 3–11. [Google Scholar] [PubMed]
- Lalla, R.V.; Sonis, S.T.; Peterson, D.E. Management of oral mucositis in patients who have cancer. Dent. Clin. N. Am. 2008, 52, 61–77. [Google Scholar]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Lingen, M.; Sonis, S.T.; Walsh-Reitz, M.M.; Toback, F.G. Role of AMP-18 in oral mucositis. Oral Oncol. 2011, 47, 831–839. [Google Scholar] [PubMed]
- Wu, X.; Chen, P.; Sonis, S.T.; Lingen, M.W.; Berger, A.; Toback, F.G. A novel peptide to treat oral mucositis blocks endothelial and epithelial cell apoptosis. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, e409–e415. [Google Scholar] [PubMed] [Green Version]
- Bell, S.; Howard, A.; Wilson, J.; Abbot, E.; Smith, W.; Townes, C.; Hirst, B.; Hall, J. Streptococcus pyogenes infection of tonsil explants is associated with a human β-defensin 1 response from control but not recurrent acute tonsillitis patients. Mol. Oral Microbiol. 2012, 27, 160–171. [Google Scholar] [PubMed]
- Maggioni, D.; Garavello, W.; Rigolio, R.; Pignataro, L.; Gaini, R.; Nicolini, G. Apigenin impairs oral squamous cell carcinoma growth in vitro inducing cell cycle arrest and apoptosis. Int. J. Oncol. 2013, 43, 1675–1682. [Google Scholar] [CrossRef] [Green Version]
- Seto, K.; Shoda, J.; Horibe, T.; Warabi, E.; Ishige, K.; Yamagata, K.; Kohno, M.; Yanagawa, T.; Bukawa, H.; Kawakami, K. Interleukin-4 receptor α-based hybrid peptide effectively induces antitumor activity in head and neck squamous cell carcinoma. Oncol. Rep. 2013, 29, 2147–2153. [Google Scholar]
- Uchida, K.; Veeramachaneni, R.; Huey, B.; Bhattacharya, A.; Schmidt, B.L.; Albertson, D.G. Investigation of HOXA9 promoter methylation as a biomarker to distinguish oral cancer patients at low risk of neck metastasis. BMC Cancer 2014, 14, 353. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Mancini, M.; Sonis, S.T.; Fernandez-Martinez, J.; Liu, J.; Cohen, E.E.; Toback, F.G. A Novel Peptide for Simultaneously Enhanced Treatment of Head and Neck Cancer and Mitigation of Oral Mucositis. PLoS ONE 2016, 11, e0152995. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, R.; Deshmukh, S. A new therapeutic approach to treat oral mucositis using specific MMP blockers in an osmotically active solution. J. Cancer Res. Treat. 2013, 1, 4–11. [Google Scholar]
- Jones, K.H.; Senft, J.A. An improved method to determine cell viability by simultaneous staining with fluorescein diacetate-propidium iodide. J. Histochem. Cytochem. 1985, 33, 77–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, R.; Nygren, P.; Ekberg, M.; Slater, L. Chemotherapeutic drug sensitivity testing of human leukemia cells in vitro using a semiautomated fluorometric assay. Leukemia 1990, 4, 567–571. [Google Scholar] [PubMed]
- Larsson, R.; Kristensen, J.; Sandberg, C.; Nygren, P. Laboratory determination of chemotherapeutic drug resistance in tumor cells from patients with leukemia, using a fluorometric microculture cytotoxicity assay (FMCA). Int. J. Cancer 1992, 50, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, R.D. An overview of chemosensitivity testing. In Chemosensitivity; Springer: Berlin/Heidelberg, Germany, 2005; pp. 3–18. [Google Scholar]
- Rubinstein, L.V.; Shoemaker, R.H.; Paull, K.D.; Simon, R.M.; Tosini, S.; Skehan, P.; Scudiero, D.A.; Monks, A.; Boyd, M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990, 82, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Nygren, P.; Csoka, K.; Botling, J.; Nilsson, K.; Larsson, R. Anti-cancer drug characterisation using a human cell line panel representing defined types of drug resistance. Br. J. Cancer 1996, 74, 888–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindhagen, E.; Rickardson, L.; Elliott, G.; Leoni, L.; Nygren, P.; Larsson, R.; Åleskog, A. Pharmacological profiling of novel non-COX-inhibiting indole-pyran analogues of etodolac reveals high solid tumour activity of SDX-308 in vitro. Investig. New Drugs 2007, 25, 297–303. [Google Scholar] [CrossRef]
- Wickström, M.; Haglund, C.; Lindman, H.; Nygren, P.; Larsson, R.; Gullbo, J. The novel alkylating prodrug J1: Diagnosis directed activity profile ex vivo and combination analyses in vitro. Investig. New Drugs 2008, 26, 195–204. [Google Scholar]
- Datta, K.; Babbar, P.; Srivastava, T.; Sinha, S.; Chattopadhyay, P. p53 dependent apoptosis in glioma cell lines in response to hydrogen peroxide induced oxidative stress. Int. J. Biochem. Cell Biol. 2002, 34, 148–157. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, H. Setting up the oxidative cell mode with hippocampal cell of primary culture induced by H2O2. Wei Sheng Yan Jiu = J. Hyg. Res. 2014, 43, 719. [Google Scholar]
- Zhou, X.; Zhang, G.-y.; Wang, J.; Lu, S.-l.; Cao, J.; Sun, L.-z. A novel bridge between oxidative stress and immunity: The interaction between hydrogen peroxide and human leukocyte antigen G in placental trophoblasts during preeclampsia. Am. J. Obstet. Gynecol. 2012, 206, 447.e7–447.e16. [Google Scholar]
- Moll, S.J.; Jones, C.J.; Crocker, I.P.; Baker, P.N.; Heazell, A.E. Epidermal growth factor rescues trophoblast apoptosis induced by reactive oxygen species. Apoptosis 2007, 12, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Fukushima, K.; Takao, T.; Seki, H.; Takeda, S.; Wake, N. Oxidative stress produced by xanthine oxidase induces apoptosis in human extravillous trophoblast cells. J. Reprod. Dev. 2013, 59, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [PubMed] [Green Version]
- Acebedo, A.R.; Amor, E.C.; Jacinto, S.D. Apoptosis Inducing Activity of HPLC Fraction from Voacanga globosa (Blanco) Merr. on the Human Colon Carcinoma Cell Line, HCT116. Asian Pac. J. Cancer Prev. 2014, 15, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Doonan, F.; Cotter, T.G. Morphological assessment of apoptosis. Methods 2008, 44, 200–204. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Patel, V.; Cotrim, A.; Leelahavanichkul, K.; Molinolo, A.A.; Mitchell, J.B.; Gutkind, J.S. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell 2012, 11, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhang, Q.; Yu, W.; Chang, B.; Le, A.D. Oral Mucositis: An Update on Innate Immunity and New Interventional Targets. J. Dent. Res. 2020, 99, 1122–1130. [Google Scholar] [CrossRef]
- Rtibi, K.; Selmi, S.; Grami, D.; Amri, M.; Sebai, H.; Marzouki, L. Contribution of oxidative stress in acute intestinal mucositis induced by 5 fluorouracil (5-FU) and its pro-drug capecitabine in rats. Toxicol. Mech. Methods 2018, 28, 262–267. [Google Scholar] [CrossRef]
- Russi, E.G.; Raber-Durlacher, J.E.; Sonis, S.T. Local and Systemic Pathogenesis and Consequences of Regimen-Induced Inflammatory Responses in Patients with Head and Neck Cancer Receiving Chemoradiation. Mediat. Inflamm. 2014, 2014, 518261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.; Sangha, S.; Pan, M.; Shin, D.H.; Park, H.; Mohammed, A.I.; Cirillo, N. Oxidative Stress and Chemoradiation-Induced Oral Mucositis: A Scoping Review of In Vitro, In Vivo and Clinical Studies. Int. J. Mol. Sci. 2022, 23, 4863. [Google Scholar] [PubMed]
- Rhee, S.G.; Kang, S.W.; Jeong, W.; Chang, T.-S.; Yang, K.-S.; Woo, H.A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 2005, 17, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Vilhardt, F.; van Deurs, B. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J. 2004, 23, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice-Evans, C.; Halliwell, B.; Lunt, G.; Davies, K.J. Oxidative stress: The paradox of aerobic life. In Biochemical Society Symposia; Portland Press Limited: London, UK, 1995; pp. 1–31. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.; Cross, C.E. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med. 1992, 119, 598–620. [Google Scholar]
- Waddington, R.J.; Moseley, R.; Embery, G. Periodontal Disease Mechanisms: Reactive oxygen species: A potential role in the pathogenesis of periodontal diseases. Oral Dis. 2000, 6, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Tanaka, H.; Okada, T.; Konishi, H.; Takahashi, M.; Ito, M.; Asai, J. Effect of reactive oxygen species on the elastin mRNA expression in cultured human dermal fibroblasts. Free Radic. Biol. Med. 1997, 23, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lee, E.-J.; Lee, J.H.; Park, I.-C.; Lee, S.-J.; Hahn, H.J.; Ahn, K.J.; An, S.; An, I.-S.; Cha, H.J. Oridonin protects HaCaT keratinocytes against hydrogen peroxide-induced oxidative stress by altering microRNA expression. Int. J. Mol. Med. 2014, 33, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Park, W.H. The effects of exogenous H2O2 on cell death, reactive oxygen species and glutathione levels in calf pulmonary artery and human umbilical vein endothelial cells. Int. J. Mol. Med. 2013, 31, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Piao, M.J.; Keum, Y.S.; Kim, H.S.; Hyun, J.W. Fucoxanthin Protects Cultured Human Keratinocytes against Oxidative Stress by Blocking Free Radicals and Inhibiting Apoptosis. Biomol. Ther. 2013, 21, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Yeom, M.; Shin, S.; Jeon, K.; Park, D.; Jung, E. Protective Effects of Aqueous Extract of Mentha suaveolens against Oxidative Stress-Induced Damages in Human Keratinocyte HaCaT Cells. Evid. -Based Complement. Altern. Med. 2019, 2019, 5045491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransy, C.; Vaz, C.; Lombès, A.; Bouillaud, F. Use of H2O2 to Cause Oxidative Stress, the Catalase Issue. Int. J. Mol. Sci. 2020, 21, 9149. [Google Scholar] [CrossRef] [PubMed]
- Lima, V.; Brito, G.; Cunha, F.d.Q.; Rebouças, C.; Falcão, B.; Augusto, R.; Souza, M.; Leitão, B.; Ribeiro, R. Effects of the tumour necrosis factor-α inhibitors pentoxifylline and thalidomide in short-term experimental oral mucositis in hamsters. Eur. J. Oral Sci. 2005, 113, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Kolli, V.K.; Abraham, P.; Isaac, B.; Kasthuri, N. Preclinical efficacy of melatonin to reduce methotrexate-induced oxidative stress and small intestinal damage in rats. Dig. Dis. Sci. 2013, 58, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Brennan, M.L.; Shen, Z.; MacPherson, J.C.; Schmitt, D.; Molenda, C.E.; Hazen, S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem. 2002, 277, 46116–46122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Ai, G.; Zhong, L.; Mai, L.; Chen, J.-N.; Liu, Y.; Li, Y.; Huang, X.; Su, Z.; Zhan, J.Y.-X. Protective Effects of Oxyberberine in 5-Fluorouracil-Induced Intestinal Mucositis in the Mice Model. Evid.-Based Complement. Altern. Med. 2022, 2022, 1238358. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A.A.d.; Araújo, L.d.S.; Medeiros, C.A.C.X.d.; Leitão, R.F.d.C.; Brito, G.A.d.C.; Costa, D.V.d.S.; Guerra, G.C.B.; Garcia, V.B.; Lima, M.L.d.S.; Araújo Junior, R.F.d. Protective effect of angiotensin II receptor blocker against oxidative stress and inflammation in an oral mucositis experimental model. J. Oral Pathol. Med. 2018, 47, 972–984. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, A.I.; Sangha, S.; Nguyen, H.; Shin, D.H.; Pan, M.; Park, H.; McCullough, M.J.; Celentano, A.; Cirillo, N. Assessment of Oxidative Stress-Induced Oral Epithelial Toxicity. Biomolecules 2023, 13, 1239. https://doi.org/10.3390/biom13081239
Mohammed AI, Sangha S, Nguyen H, Shin DH, Pan M, Park H, McCullough MJ, Celentano A, Cirillo N. Assessment of Oxidative Stress-Induced Oral Epithelial Toxicity. Biomolecules. 2023; 13(8):1239. https://doi.org/10.3390/biom13081239
Chicago/Turabian StyleMohammed, Ali I., Simran Sangha, Huynh Nguyen, Dong Ha Shin, Michelle Pan, Hayoung Park, Michael J. McCullough, Antonio Celentano, and Nicola Cirillo. 2023. "Assessment of Oxidative Stress-Induced Oral Epithelial Toxicity" Biomolecules 13, no. 8: 1239. https://doi.org/10.3390/biom13081239
APA StyleMohammed, A. I., Sangha, S., Nguyen, H., Shin, D. H., Pan, M., Park, H., McCullough, M. J., Celentano, A., & Cirillo, N. (2023). Assessment of Oxidative Stress-Induced Oral Epithelial Toxicity. Biomolecules, 13(8), 1239. https://doi.org/10.3390/biom13081239