


Features of Protein Unfolding Transitions and Their Relation to Domain Topology Probed by Single-Molecule FRET


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Confocal Microscopy and smFRET Analysis
3. Results and Discussion
3.1. Development of Denaturant-Induced Intra-Domain Distances
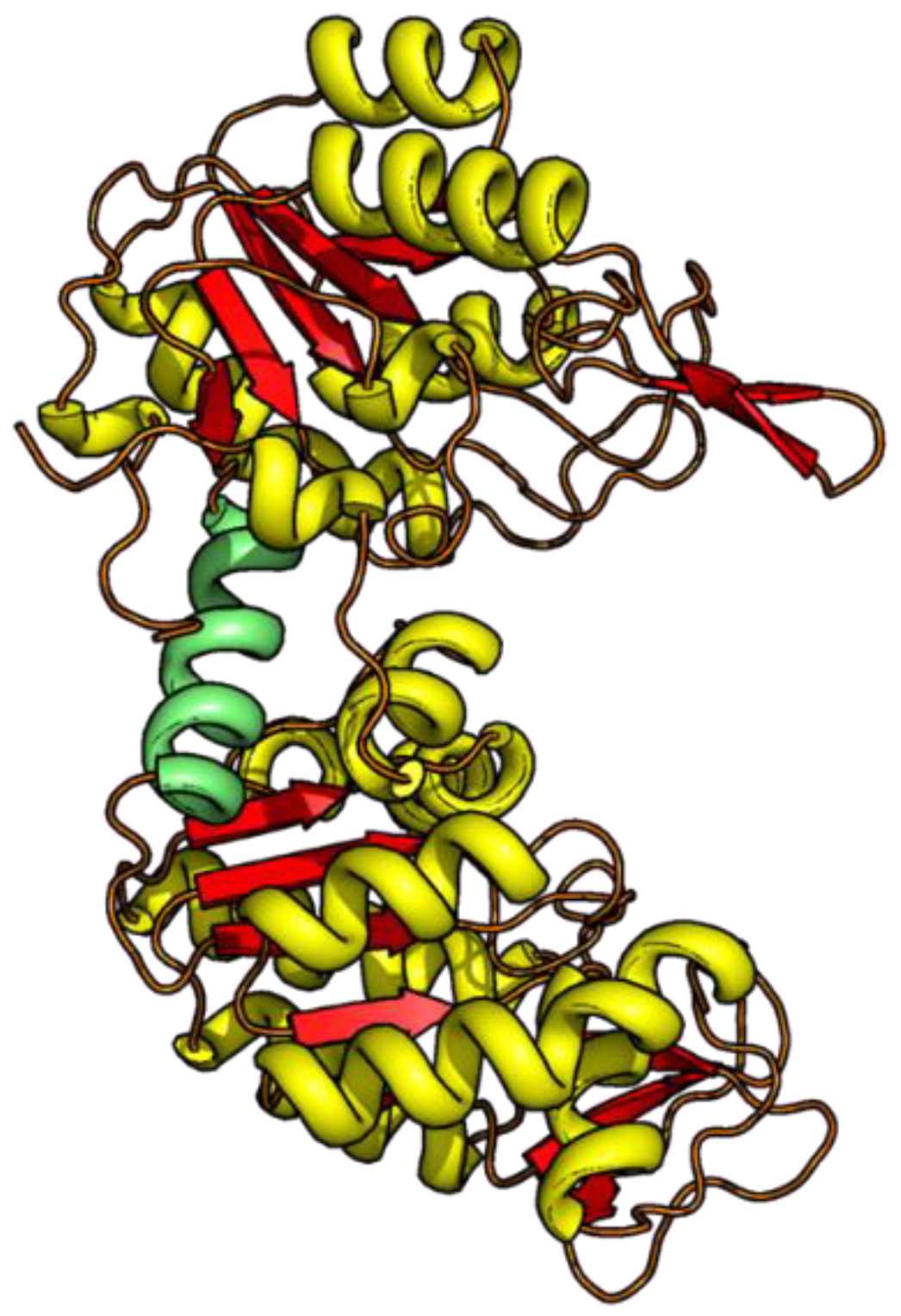
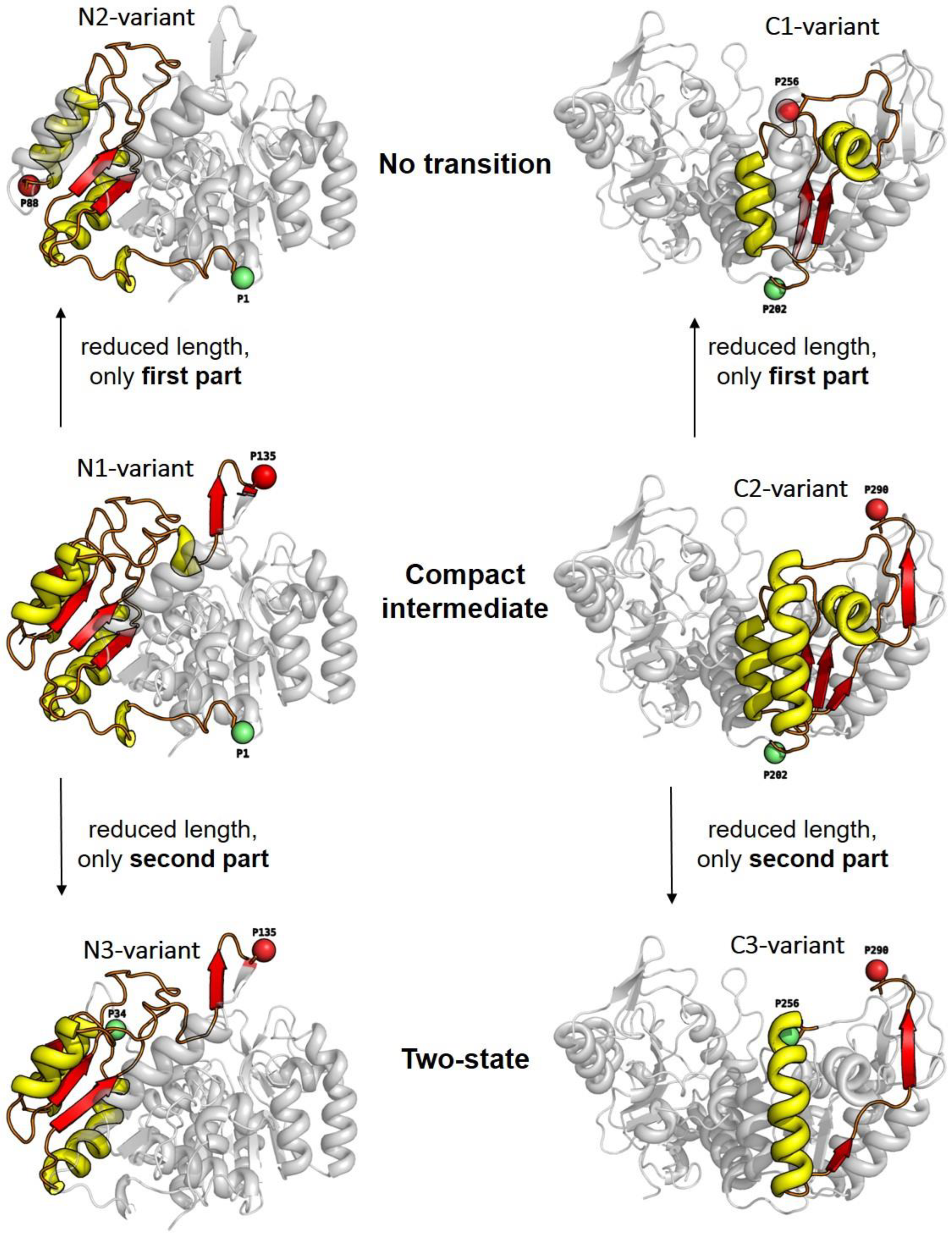
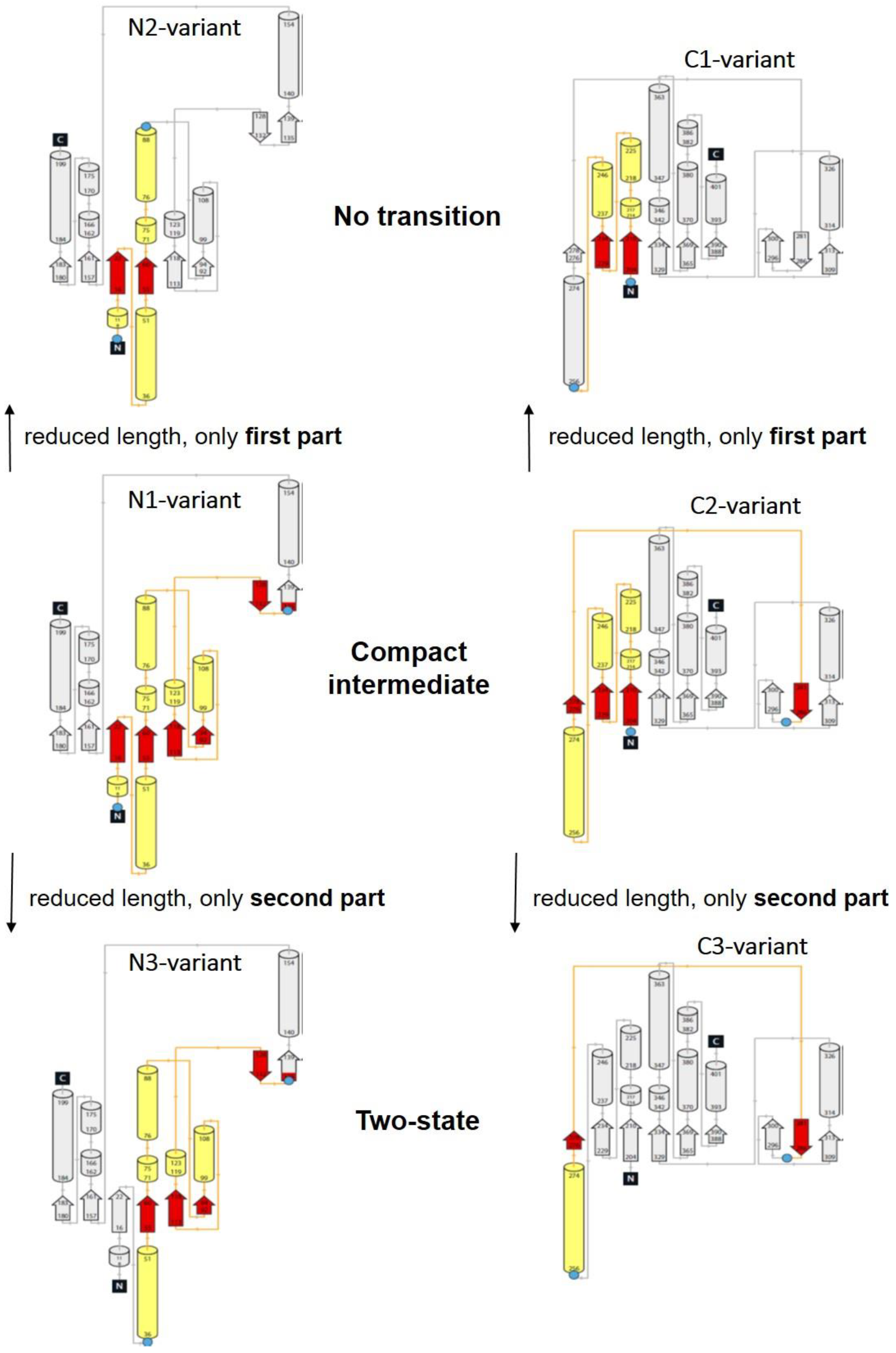
3.2. Unfolding Transitions Related to Domain Topology
- All enclosed structure elements represent, at least a part, i.e., one to four strands out of the six parallel β-strands, of the Rossmann fold, in most cases sandwiched by the accompanied α-helices.
- Significant inter-dye distance changes between the different states (native, unfolded, and, if present, compact intermediate) were only observed if one of the dye attachment positions was close to a further secondary structure element (which was not part of the Rossmann fold, but which represented one or two β-strands that were also part of the enclosed structure elements (i.e., highlighted in color)). This condition was fulfilled for the N3-and C-3 variants (TS) and for the N1-and C2-variants (CI). Importantly, it was not fulfilled for the N2- and C1-variants (NT).
- Looking at the domain topologies, differences between the two-state and compact intermediate transitions seem to be related to which position within the Rossmann fold one of the two dye attachment sites was located in. A more upstream position of the dye attachment site appeared to reveal the compact intermediate, whereas a more downstream attachment site did not (position 1 versus position 34 for the N-variants and position 202 versus position 256 for the C-variants).
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schuler, B.; Eaton, W.A. Protein Folding Studied by Single-Molecule Fret. Curr. Opin. Struct. Biol. 2008, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Eaton, W.A. Protein Folding Transition Path Times from Single Molecule Fret. Curr. Opin. Struct. Biol. 2018, 48, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Deniz, A.A.; Mukhopadhyay, S.; Lemke, E.A. Single-Molecule Biophysics: At the Interface of Biology, Physics and Chemistry. J. R. Soc. Interface 2008, 5, 15–45. [Google Scholar] [CrossRef] [PubMed]
- Cerminara, M.; Schöne, A.; Ritter, I.; Gabba, M.; Fitter, J. Mapping Multiple Distances in a Multidomain Protein for the Identification of Folding Intermediates. Biophys. J. 2020, 118, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Ratner, V.; Kahana, E.; Eichler, M.; Haas, E. A General Strategy for Site-Specific Double Labeling of Globular Proteins for Kinetic Fret Studies. Bioconjug. Chem. 2002, 13, 1163–1170. [Google Scholar] [CrossRef]
- Lerner, E.; Hilzenrat, G.; Amir, D.; Tauber, E.; Garini, Y.; Haas, E. Preparation of Homogeneous Samples of Double-Labelled Protein Suitable for Single-Molecule Fret Measurements. Anal. Bioanal. Chem. 2013, 405, 5983–5991. [Google Scholar] [CrossRef]
- Zosel, F.; Holla, A.; Schuler, B. Labeling of Proteins for Single-Molecule Fluorescence Spectroscopy. Methods Mol. Biol. 2022, 2376, 207–233. [Google Scholar] [CrossRef]
- Voelz, V.A.; Jager, M.; Yao, S.; Chen, Y.; Zhu, L.; Waldauer, S.A.; Bowman, G.R.; Friedrichs, M.; Bakajin, O.; Lapidus, L.J.; et al. Slow Unfolded-State Structuring in Acyl-Coa Binding Protein Folding Revealed by Simulation and Experiment. J. Am. Chem. Soc. 2012, 134, 12565–12577. [Google Scholar] [CrossRef]
- Lindhoud, S.; Pirchi, M.; Westphal, A.H.; Haran, G.; van Mierlo, C.P. Gradual Folding of an Off-Pathway Molten Globule Detected at the Single-Molecule Level. J. Mol. Biol. 2015, 427, 3148–3157. [Google Scholar] [CrossRef]
- Dingfelder, F.; Benke, S.; Nettels, D.; Schuler, B. Mapping an Equilibrium Folding Intermediate of the Cytolytic Pore Toxin Clya with Single-Molecule Fret. J. Phys. Chem. B 2018, 122, 11251–11261. [Google Scholar] [CrossRef]
- Watson, H.; Walker, N.; Shaw, P.; Bryant, T.; Wendell, P.; Fothergill, L.; Perkins, R.; Conroy, S.; Dobson, M.; Tuite, M. Sequence and Structure of Yeast Phosphoglycerate Kinase. EMBO J. 1982, 1, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. Scop—A Structural Classification of Proteins Database for the Investigation of Sequences and Structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Missiakas, D.; Betton, J.M.; Minard, P.; Yon, J.M. Unfolding-Refolding of the Domains in Yeast Phosphoglycerate Kinase: Comparison with the Isolated Engineered Domains. Biochemistry 1990, 29, 8683–8689. [Google Scholar] [CrossRef]
- Osváth, S.; Köhler, G.; Závodszky, P.; Fidy, J. Asymmetric Effect of Domain Interactions on the Kinetics of Folding in Yeast Phosphoglycerate Kinase. Protein Sci. 2005, 14, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, T.; Schlesinger, R.; Gabba, M.; Fitter, J. Native and Unfolded States of Phosphoglycerate Kinase Studied by Single-Molecule Fret. ChemPhysChem Eur. J. Chem. Phys. Phys. Chem. 2011, 12, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.K.; Zaychikov, E.; Bräuchle, C.; Lamb, D.C. Pulsed Interleaved Excitation. Biophys. J. 2005, 89, 3508–3522. [Google Scholar] [CrossRef] [PubMed]
- Schrimpf, W.; Barth, A.; Hendrix, J.; Lamb, C.D. Pam: A Framework for Integrated Analysis of Imaging, Single-Molecule, and Ensemble Fluorescence Data. Biophys. J. 2018, 114, 1518–1528. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger Inc.: New York, NY, USA, 2020.
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. Pdbsum: Structural Summaries of Pdb Entries. Protein Sci. Publ. Protein Soc. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Höfig, H.; Gabba, M.; Poblete, S.; Kempe, D.; Fitter, J. Inter-Dye Distance Distributions Studied by a Combination of Single-Molecule Fret-Filtered Lifetime Measurements and a Weighted Accessible Volume (Wav) Algorithm. Molecules 2014, 19, 19269–19291. [Google Scholar] [CrossRef]
- Lapidus, L.J.; Steinbach, P.L.; Eaton, W.A.; Szabo, A.; Hofrichter, J. Effects of Chain Stiffness on the Dynamics of Loop Formation in Polypeptides. Appendix: Testing a 1-Dimensional Diffusion Model for Peptide Dynamics. J. Phys. Chem. B 2002, 106, 11628–11640. [Google Scholar] [CrossRef]
- Ptitsyn, O.B.; Uversky, V.N. The Molten Globule Is a 3rd Thermodynamical State of Protein Molecules. FEBS Lett. 1994, 341, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Osvath, S.; Herenyi, L.; Zavodszky, P.; Fidy, J.; Köhler, G. Hierarchic Finite Level Energy Landscape Model—To Describe the Refolding Kinetics of Phosphoglycerate Kinase. J. Biol. Chem. 2006, 281, 24375–24380. [Google Scholar] [CrossRef] [PubMed]
- Ptitsyn, O.; Pain, R.; Semisotnov, G.; Zerovnik, E.; Razgulyaev, O. Evidence for a Molten Globule State as a General Intermediate in Protein Folding. FEBS Lett. 1990, 262, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. Compact Intermediate States in Protein Folding. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 495–522. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N-Domain | C-Domain | |||||
|---|---|---|---|---|---|---|
| Label Positions | 1–135 N1-Variant | 1–88 N2-Variant | 34–135 N3-Variant | 202–256 C1-Variant | 202–290 C2-Variant | 256–290 C3-Variant |
| # Residues | 135 | 88 | 101 | 54 | 88 | 34 |
| Native RDA (0 M) | 48 Å | 51 Å | 38 Å | 51 Å | 57 Å | 31 Å |
| Intermediate RDA (0.6–0.7 M) | 43 Å | - | - | - | 45 Å | - |
| Unfolded RDA (≥1 M) | 75 Å | 52 Å | 65 Å | 51 Å | 63 Å | 54 Å |
| Polymer | 76 Å | 61 Å | 65 Å | 48 Å | 61 Å | 38 Å |
| Type of transition | CI | NT | TS | NT | CI | TS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustorff, N.; Fitter, J. Features of Protein Unfolding Transitions and Their Relation to Domain Topology Probed by Single-Molecule FRET. Biomolecules 2023, 13, 1280. https://doi.org/10.3390/biom13091280
Bustorff N, Fitter J. Features of Protein Unfolding Transitions and Their Relation to Domain Topology Probed by Single-Molecule FRET. Biomolecules. 2023; 13(9):1280. https://doi.org/10.3390/biom13091280
Chicago/Turabian StyleBustorff, Nuno, and Jörg Fitter. 2023. "Features of Protein Unfolding Transitions and Their Relation to Domain Topology Probed by Single-Molecule FRET" Biomolecules 13, no. 9: 1280. https://doi.org/10.3390/biom13091280
APA StyleBustorff, N., & Fitter, J. (2023). Features of Protein Unfolding Transitions and Their Relation to Domain Topology Probed by Single-Molecule FRET. Biomolecules, 13(9), 1280. https://doi.org/10.3390/biom13091280