
A Novel Gliotransmitter, L-β-Aminoisobutyric Acid, Contributes to Pathophysiology of Clinical Efficacies and Adverse Reactions of Clozapine


Abstract
:1. Introduction

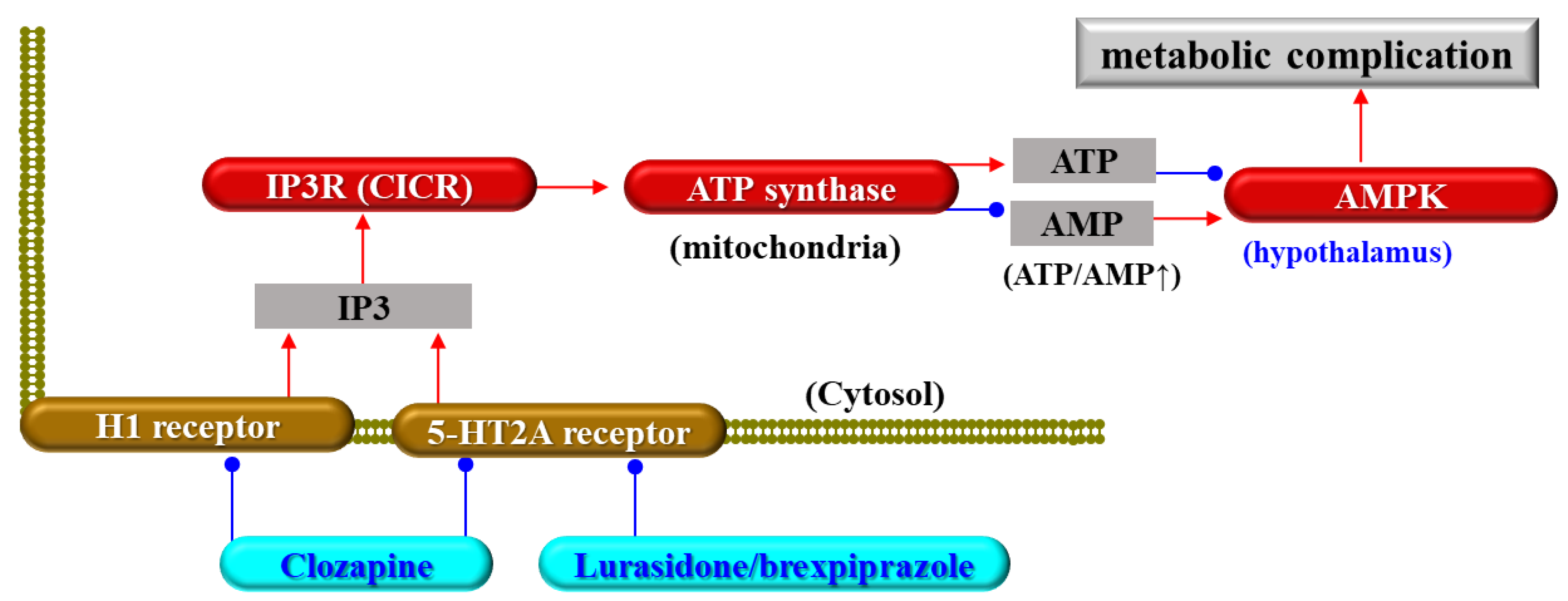
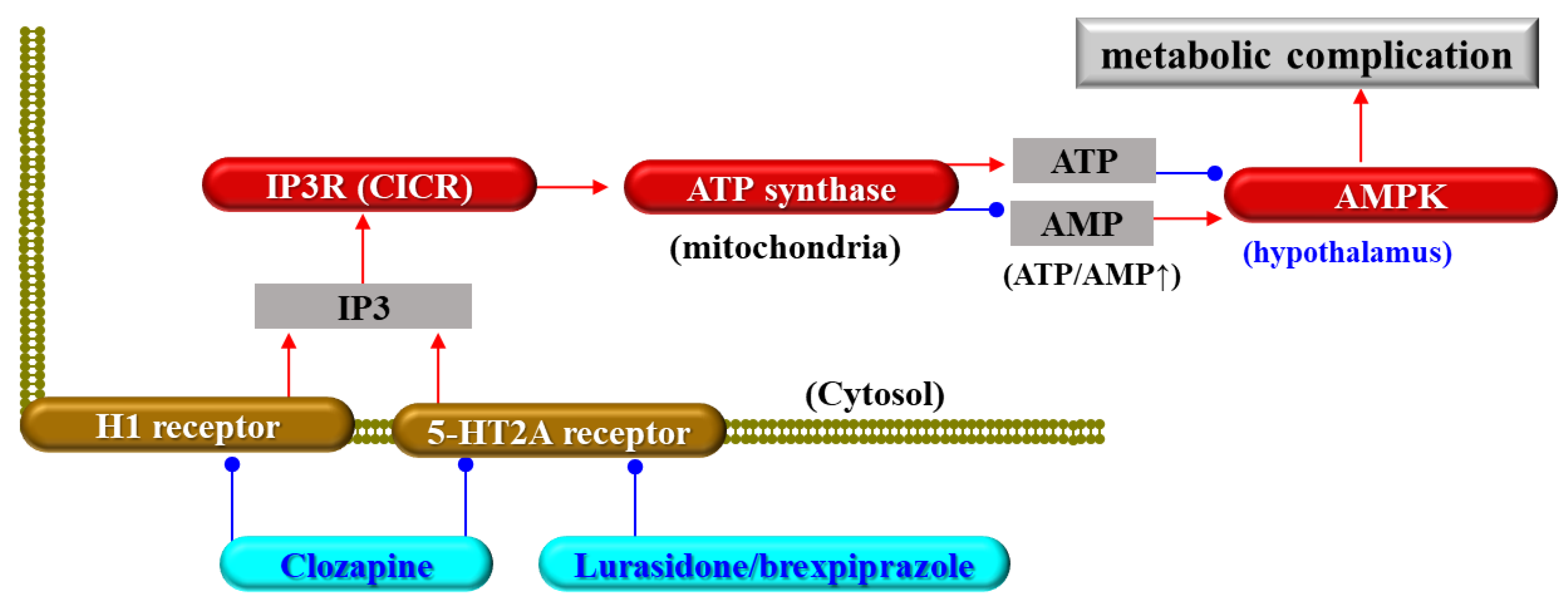{kind=link}
{kind=link}
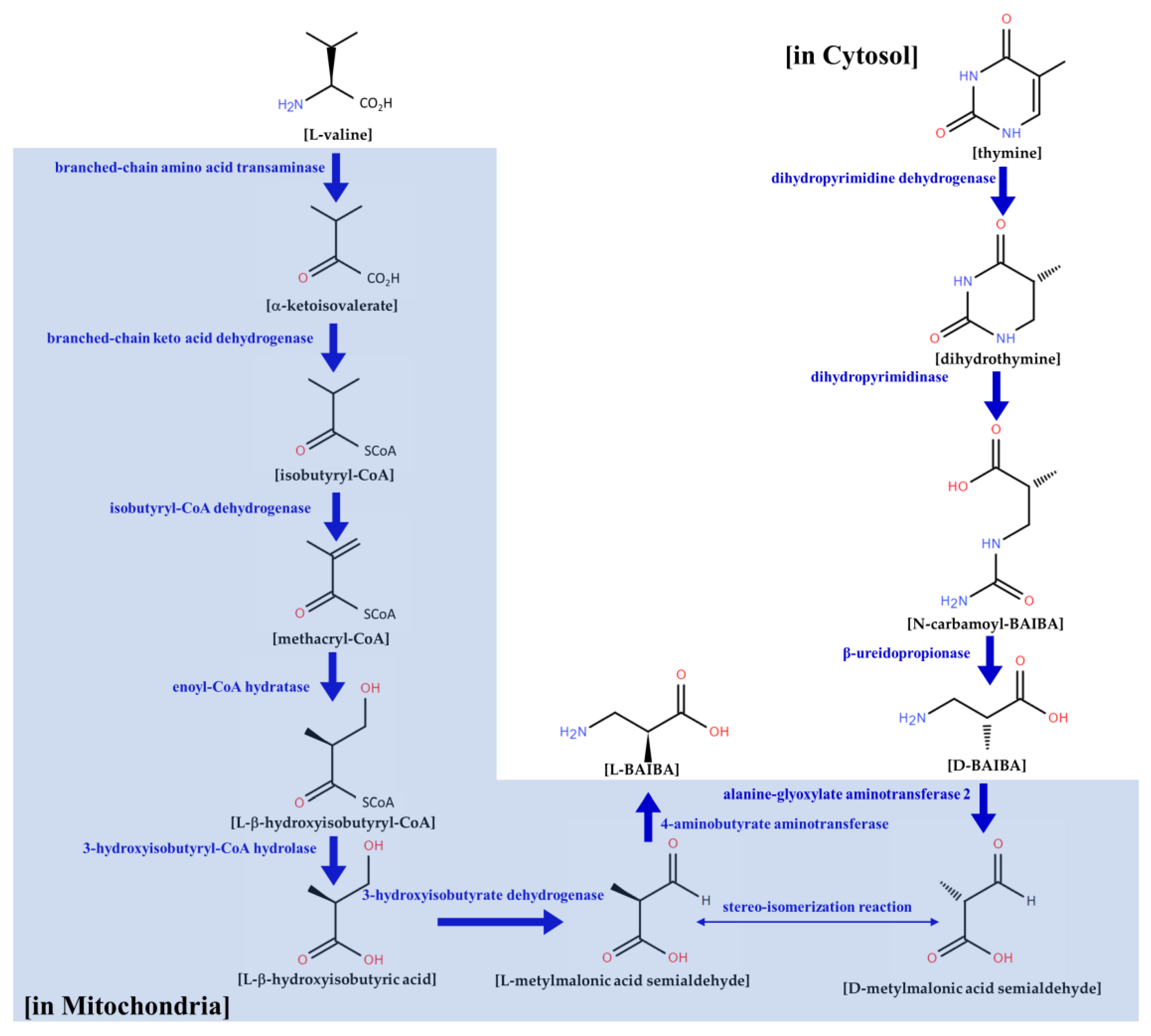{kind=link}
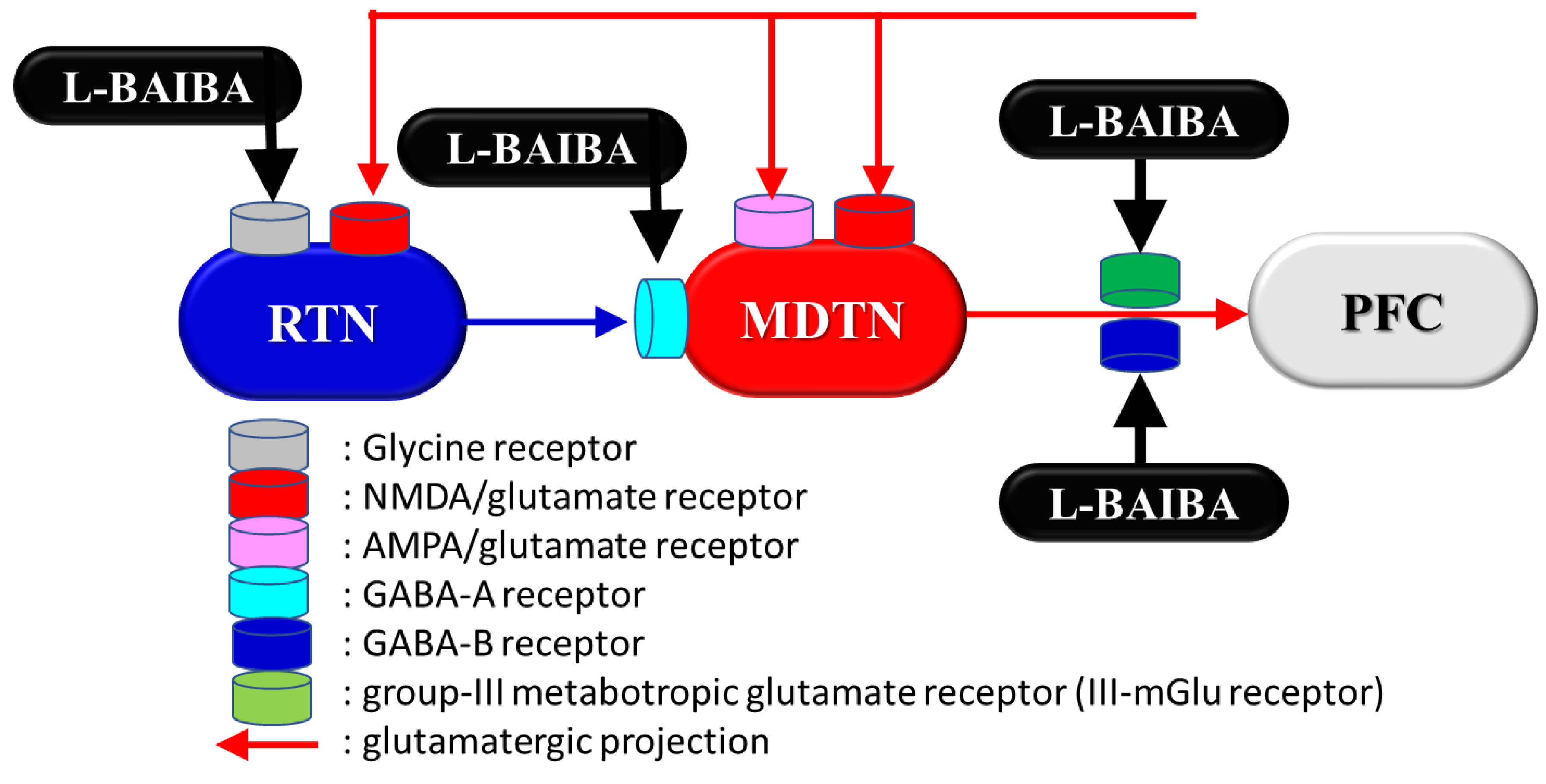{kind=link}
| Receptor | CLZ | LUR | APZ | Brex | OLZ | QTP | RIS | ZTP | HPD |
|---|---|---|---|---|---|---|---|---|---|
| 5-HT1A | 124 | 6.8 | 5.6 | 0.12 | >1000 | 432 | 423 | 471 | >1000 |
| 5-HT2A | 5.4 | 2.0 | 8.7 | 0.47 | 2.3 | 100 | 0.2 | 2.7 | 53 |
| 5-HT2C | 9.4 | 415 | 76 | 63 | 14 | >1000 | 12 | 2.6 | >1000 |
| 5-HT7 | 18.0 | 0.5 | 10.3 | 3.7 | 365 | 307 | 6.6 | 12.0 | 377 |
| H1 | 1.13 | >1000 | 27.6 | 19 | 1.2 | 11 | 20.1 | 3.21 | >1000 |
| D1 | 266 | 262 | >1000 | 160 | 100 | 712 | 244 | 71.0 | 80 |
| D2 | 157 | 1.7 | 3.3 | 0.3 | 52.3 | 245 | 3.6 | 25.0 | 0.7 |
| References | [21,22] | [23] | [24,25] | [26] | [27,28] | [29] | [25,30] | [31] | [32,33] |
2. Clozapine-Induced Metabolic Complications
3. Clozapine and TRS
3.1. Efficacy of Clozapine in TRS
3.2. Candidate Pathophysiology of TRS
3.3. Candidate Targets of Clozapine Other Than Monoamine Receptors
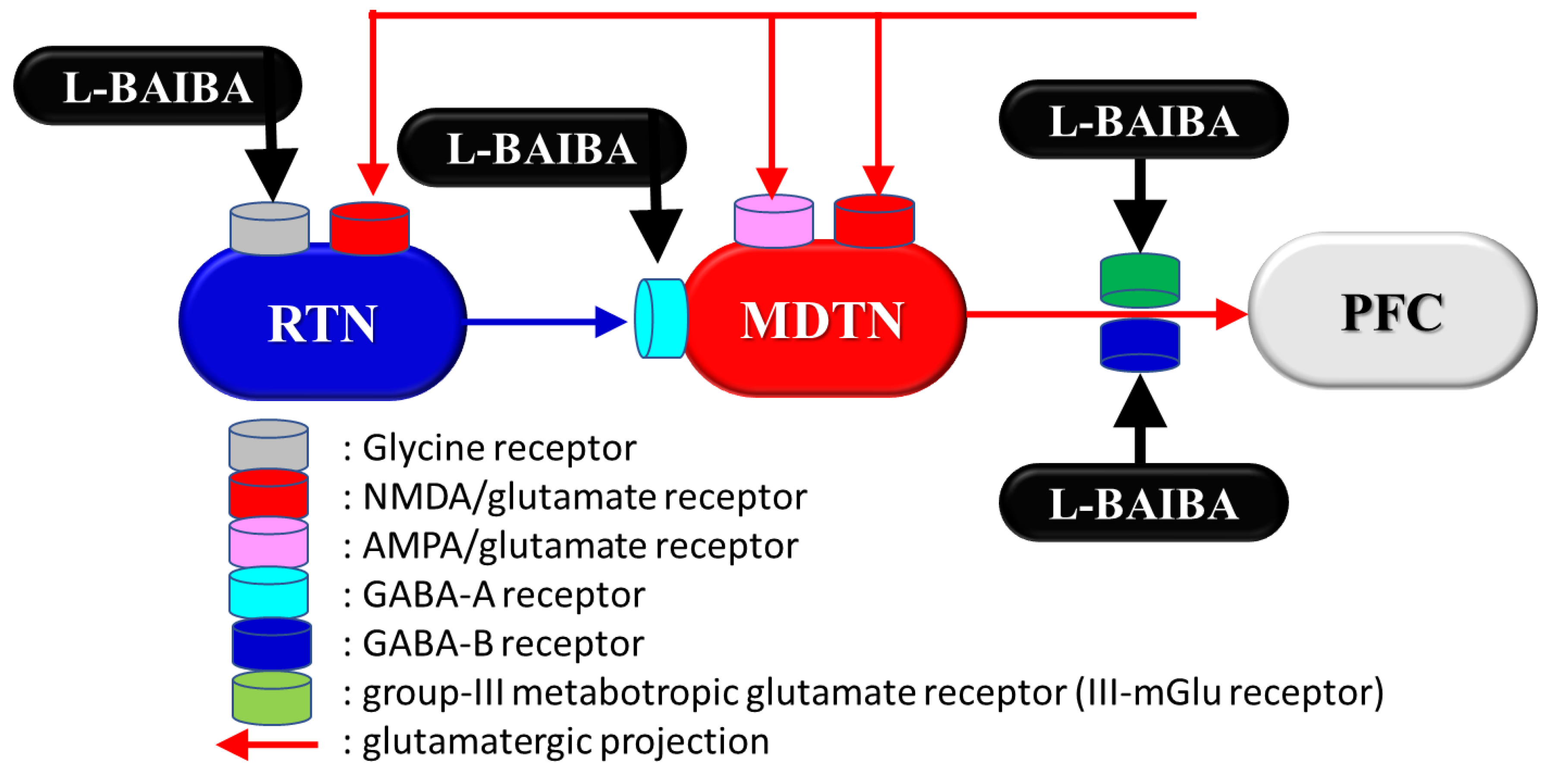
4. Impacts of L-BAIBA as the Pharmacodynamic Target of Clozapine
4.1. Impacts of L-BAIBA on Metabolic Complications Induced by Clozapine
4.2. BAIBA Enantiomer Metabolism and Distribution
4.3. BAIBA Function in the CNS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lally, J.; MacCabe, J.H. Antipsychotic medication in schizophrenia: A review. Br. Med. Bull. 2015, 114, 169–179. [Google Scholar] [CrossRef]
- Tandon, R. Schizophrenia and Other Psychotic Disorders in Diagnostic and Statistical Manual of Mental Disorders (DSM)-5: Clinical Implications of Revisions from DSM-IV. Indian J. Psychol. Med. 2014, 36, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Van Os, J. Schizofreniespectrum en andere psychotische stoornissen in de DSM-5. Tijdschr. Voor Psychiatr. 2014, 56, 167–172. [Google Scholar]
- Hu, R.J.; Malhotra, A.K.; Pickar, D. Predicting response to clozapine. CNS Drugs 1999, 11, 317–326. [Google Scholar] [CrossRef]
- Kane, J.; Honigfeld, G.; Singer, J.; Meltzer, H. Clozapine for the treatment-resistant schizophrenic: A double-blind comparison with chlorpromazine. Arch. Gen. Psychiatry 1988, 45, 789–796. [Google Scholar] [CrossRef]
- Meltzer, H.Y. Clozapine: Balancing safety with superior antipsychotic efficacy. Clin. Schizophr. Relat. Psychoses 2012, 6, 134–144. [Google Scholar] [CrossRef]
- Tiihonen, J.; Mittendorfer-Rutz, E.; Majak, M.; Mehtala, J.; Hoti, F.; Jedenius, E.; Enkusson, D.; Leval, A.; Sermon, J.; Tanskanen, A.; et al. Real-World Effectiveness of Antipsychotic Treatments in a Nationwide Cohort of 29823 Patients with Schizophrenia. JAMA Psychiatry 2017, 74, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Falkai, P.; Wobrock, T.; Lieberman, J.; Glenthoj, B.; Gattaz, W.F.; Moller, H.J.; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: Long-term treatment of schizophrenia. World J. Biol. Psychiatry 2006, 7, 5–40. [Google Scholar] [CrossRef]
- Hasan, A.; Falkai, P.; Wobrock, T.; Lieberman, J.; Glenthoj, B.; Gattaz, W.F.; Thibaut, F.; Moller, H.J.; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: Update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J. Biol. Psychiatry 2012, 13, 318–378. [Google Scholar] [CrossRef]
- Rogers, J.P.; Oldham, M.A.; Fricchione, G.; Northoff, G.; Ellen Wilson, J.; Mann, S.C.; Francis, A.; Wieck, A.; Elizabeth Wachtel, L.; Lewis, G.; et al. Evidence-based consensus guidelines for the management of catatonia: Recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 2023, 37, 327–369. [Google Scholar] [CrossRef]
- Masuda, T.; Misawa, F.; Takase, M.; Kane, J.M.; Correll, C.U. Association with Hospitalization and All-Cause Discontinuation Among Patients with Schizophrenia on Clozapine vs Other Oral Second-Generation Antipsychotics: A Systematic Review and Meta-analysis of Cohort Studies. JAMA Psychiatry 2019, 76, 1052. [Google Scholar] [CrossRef] [PubMed]
- Huhn, M.; Nikolakopoulou, A.; Schneider-Thoma, J.; Krause, M.; Samara, M.; Peter, N.; Arndt, T.; Backers, L.; Rothe, P.; Cipriani, A.; et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: A systematic review and network meta-analysis. Lancet 2019, 394, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; McCutcheon, R.A.; Brugger, S.P.; Howes, O.D. Heterogeneity and efficacy of antipsychotic treatment for schizophrenia with or without treatment resistance: A meta-analysis. Neuropsychopharmacology 2020, 45, 622–631. [Google Scholar] [CrossRef]
- Wenthur, C.J.; Lindsley, C.W. Classics in chemical neuroscience: Clozapine. ACS Chem. Neurosci. 2013, 4, 1018–1025. [Google Scholar] [CrossRef]
- Rusheen, A.E.; Gee, T.A.; Jang, D.P.; Blaha, C.D.; Bennet, K.E.; Lee, K.H.; Heien, M.L.; Oh, Y. Evaluation of electrochemical methods for tonic dopamine detection in vivo. TrAC Trends Anal. Chem. 2020, 132, 116049. [Google Scholar] [CrossRef]
- Meltzer, H.Y. Update on typical and atypical antipsychotic drugs. Annu. Rev. Med. 2013, 64, 393–406. [Google Scholar] [CrossRef]
- O’Connor, W.T.; O’Shea, S.D. Clozapine and GABA transmission in schizophrenia disease models: Establishing principles to guide treatments. Pharmacol. Ther. 2015, 150, 47–80. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Lee, M.A.; Ranjan, R.; Mason, E.A.; Cola, P.A. Relapse following clozapine withdrawal: Effect of neuroleptic drugs and cyproheptadine. Psychopharmacology 1996, 124, 176–187. [Google Scholar] [CrossRef]
- Correll, C.U. From receptor pharmacology to improved outcomes: Individualising the selection, dosing, and switching of antipsychotics. Eur. Psychiatry 2010, 25 (Suppl. S2), S12–S21. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Clozapine, a fast-off-D2 antipsychotic. ACS Chem. Neurosci. 2014, 5, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Su, T.P.; Malhotra, A.K.; Hadd, K.; Breier, A.; Pickar, D. D2 dopamine receptor occupancy: A crossover comparison of risperidone with clozapine therapy in schizophrenic patients. Arch. Gen. Psychiatry 1997, 54, 972–973. [Google Scholar] [CrossRef]
- Meltzer, H.Y. The mechanism of action of novel antipsychotic drugs. Schizophr. Bull. 1991, 17, 263–287. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Horisawa, T.; Tokuda, K.; Ishiyama, T.; Ogasa, M.; Tagashira, R.; Matsumoto, K.; Nishikawa, H.; Ueda, Y.; Toma, S.; et al. Pharmacological profile of lurasidone, a novel antipsychotic agent with potent 5-hydroxytryptamine 7 (5-HT7) and 5-HT1A receptor activity. J. Pharmacol. Exp. Ther. 2010, 334, 171–181. [Google Scholar] [CrossRef]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef]
- Ghanizadeh, A.; Sahraeizadeh, A.; Berk, M. A head-to-head comparison of aripiprazole and risperidone for safety and treating autistic disorders, a randomized double blind clinical trial. Child Psychiatry Hum. Dev. 2014, 45, 185–192. [Google Scholar] [CrossRef]
- Maeda, K.; Sugino, H.; Akazawa, H.; Amada, N.; Shimada, J.; Futamura, T.; Yamashita, H.; Ito, N.; McQuade, R.D.; Mork, A.; et al. Brexpiprazole I: In vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J. Pharmacol. Exp. Ther. 2014, 350, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.; Alonso, J.M.; Andres, J.I.; Cid, J.M.; Diaz, A.; Iturrino, L.; Gil, P.; Megens, A.; Sipido, V.K.; Trabanco, A.A. Discovery of new tetracyclic tetrahydrofuran derivatives as potential broad-spectrum psychotropic agents. J. Med. Chem. 2005, 48, 1709–1712. [Google Scholar] [CrossRef]
- Bymaster, F.; Calligaro, D.; Falcone, J.; Marsh, R.; Moore, N.; Tye, N.; Seeman, P.; Wong, D.J. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 1996, 14, 87–96. [Google Scholar] [CrossRef]
- Lopez-Munoz, F.; Alamo, C. Active metabolites as antidepressant drugs: The role of norquetiapine in the mechanism of action of quetiapine in the treatment of mood disorders. Front. Psychiatry 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Rahman, T.; Toohey, N.; Mazurkiewicz, J.; Herrick-Davis, K.; Teitler, M. Risperidone irreversibly binds to and inactivates the h5-HT7 serotonin receptor. Mol. Pharmacol. 2006, 70, 1264–1270. [Google Scholar] [CrossRef]
- Schotte, A.; Janssen, P.F.; Gommeren, W.; Luyten, W.H.; Van Gompel, P.; Lesage, A.S.; De Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology 1996, 124, 57–73. [Google Scholar] [CrossRef]
- Kroeze, W.K.; Hufeisen, S.J.; Popadak, B.A.; Renock, S.M.; Steinberg, S.; Ernsberger, P.; Jayathilake, K.; Meltzer, H.Y.; Roth, B.L. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 2003, 28, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Leysen, J.E.; Janssen, P.M.; Gommeren, W.; Wynants, J.; Pauwels, P.J.; Janssen, P.A. In vitro and in vivo receptor binding and effects on monoamine turnover in rat brain regions of the novel antipsychotics risperidone and ocaperidone. Mol. Pharmacol. 1992, 41, 494–508. [Google Scholar]
- Li, X.H.; Zhong, X.M.; Lu, L.; Zheng, W.; Wang, S.B.; Rao, W.W.; Wang, S.; Ng, C.H.; Ungvari, G.S.; Wang, G.; et al. The prevalence of agranulocytosis and related death in clozapine-treated patients: A comprehensive meta-analysis of observational studies. Psychol. Med. 2020, 50, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.; Ramineni, V.; Malacova, E.; Eriksson, L.; McMahon, K.; Moudgil, V.; Scott, J.; Siskind, D. Risk factors for clozapine-induced myocarditis and cardiomyopathy: A systematic review and meta-analysis. Acta Psychiatr. Scand. 2022, 145, 442–455. [Google Scholar] [CrossRef]
- Pillinger, T.; McCutcheon, R.A.; Vano, L.; Mizuno, Y.; Arumuham, A.; Hindley, G.; Beck, K.; Natesan, S.; Efthimiou, O.; Cipriani, A.; et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: A systematic review and network meta-analysis. Lancet Psychiatry 2020, 7, 64–77. [Google Scholar] [CrossRef]
- De Berardis, D.; Rapini, G.; Olivieri, L.; Di Nicola, D.; Tomasetti, C.; Valchera, A.; Fornaro, M.; Di Fabio, F.; Perna, G.; Di Nicola, M.; et al. Safety of antipsychotics for the treatment of schizophrenia: A focus on the adverse effects of clozapine. Ther. Adv. Drug Saf. 2018, 9, 237–256. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Murata, M. A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43. Int. J. Mol. Sci. 2020, 21, 7019. [Google Scholar] [CrossRef] [PubMed]
- Blackman, G.; Oloyede, E. Clozapine discontinuation withdrawal symptoms in schizophrenia. Ther. Adv. Psychopharmacol. 2021, 11, 20451253211032053. [Google Scholar] [CrossRef]
- Lander, M.; Bastiampillai, T.; Sareen, J. Review of withdrawal catatonia: What does this reveal about clozapine? Transl. Psychiatry 2018, 8, 139. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of Astroglial Connexin is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine. Cells 2020, 9, 414. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Effects of Atypical Antipsychotics, Clozapine, Quetiapine and Brexpiprazole on Astroglial Transmission Associated with Connexin43. Int. J. Mol. Sci. 2021, 22, 5623. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Motomura, E.; Shiroyama, T.; Okada, M. Impact of 5-HT7 receptor inverse agonism of lurasidone on monoaminergic tripartite synaptic transmission and pathophysiology of lower risk of weight gain. Biomed. Pharmacother. 2022, 148, 112750. [Google Scholar] [CrossRef]
- Fukuyama, K.; Motomura, E.; Okada, M. Opposing effects of clozapine and brexpiprazole on beta-aminoisobutyric acid: Pathophysiology of antipsychotics-induced weight gain. Schizophrenia 2023, 9, 8. [Google Scholar] [CrossRef]
- Fukuyama, K.; Motomura, E.; Okada, M. Enhanced L-beta-Aminoisobutyric Acid Is Involved in the Pathophysiology of Effectiveness for Treatment-Resistant Schizophrenia and Adverse Reactions of Clozapine. Biomolecules 2023, 13, 862. [Google Scholar] [CrossRef]
- Kelly, A.C.; Sheitman, B.B.; Hamer, R.M.; Rhyne, D.C.; Reed, R.M.; Graham, K.A.; Rau, S.W.; Gilmore, J.H.; Perkins, D.O.; Peebles, S.S.; et al. A naturalistic comparison of the long-term metabolic adverse effects of clozapine versus other antipsychotics for patients with psychotic illnesses. J. Clin. Psychopharmacol. 2014, 34, 441–445. [Google Scholar] [CrossRef]
- Lund, B.C.; Perry, P.J.; Brooks, J.M.; Arndt, S. Clozapine use in patients with schizophrenia and the risk of diabetes, hyperlipidemia, and hypertension: A claims-based approach. Arch. Gen. Psychiatry 2001, 58, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. Introduction: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45, S1–S2. [Google Scholar] [CrossRef]
- Taylor, S.I.; Yazdi, Z.S.; Beitelshees, A.L. Pharmacological treatment of hyperglycemia in type 2 diabetes. J. Clin. Investig. 2021, 131, e142243. [Google Scholar] [CrossRef] [PubMed]
- Popli, A.P.; Konicki, P.E.; Jurjus, G.J.; Fuller, M.A.; Jaskiw, G.E. Clozapine and associated diabetes mellitus. J. Clin. Psychiatry 1997, 58, 108–111. [Google Scholar] [CrossRef]
- Jiang, W.L.; Cai, D.B.; Yin, F.; Zhang, L.; Zhao, X.W.; He, J.; Ng, C.H.; Ungvari, G.S.; Sim, K.; Hu, M.L.; et al. Adjunctive metformin for antipsychotic-induced dyslipidemia: A meta-analysis of randomized, double-blind, placebo-controlled trials. Transl. Psychiatry 2020, 10, 117. [Google Scholar] [CrossRef]
- Zimbron, J.; Khandaker, G.M.; Toschi, C.; Jones, P.B.; Fernandez-Egea, E. A systematic review and meta-analysis of randomised controlled trials of treatments for clozapine-induced obesity and metabolic syndrome. Eur. Neuropsychopharmacol. 2016, 26, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Siskind, D.J.; Leung, J.; Russell, A.W.; Wysoczanski, D.; Kisely, S. Metformin for Clozapine Associated Obesity: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0156208. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shi, W.; Xu, J.; Huang, C.; Zhu, J. Outcomes and safety of concomitant topiramate or metformin for antipsychotics-induced obesity: A randomized-controlled trial. Ann. Gen. Psychiatry 2020, 19, 68. [Google Scholar] [CrossRef]
- Dayabandara, M.; Hanwella, R.; Ratnatunga, S.; Seneviratne, S.; Suraweera, C.; de Silva, V.A. Antipsychotic-associated weight gain: Management strategies and impact on treatment adherence. Neuropsychiatr. Dis. Treat. 2017, 13, 2231–2241. [Google Scholar] [CrossRef]
- Siskind, D.; Hahn, M.; Correll, C.U.; Fink-Jensen, A.; Russell, A.W.; Bak, N.; Broberg, B.V.; Larsen, J.; Ishoy, P.L.; Vilsboll, T.; et al. Glucagon-like peptide-1 receptor agonists for antipsychotic-associated cardio-metabolic risk factors: A systematic review and individual participant data meta-analysis. Diabetes Obes. Metab. 2019, 21, 293–302. [Google Scholar] [CrossRef]
- Wu, H.; Siafis, S.; Hamza, T.; Schneider-Thoma, J.; Davis, J.M.; Salanti, G.; Leucht, S. Antipsychotic-Induced Weight Gain: Dose-Response Meta-Analysis of Randomized Controlled Trials. Schizophr. Bull. 2022, 48, 643–654. [Google Scholar] [CrossRef]
- Fukuyama, K.; Motomura, E.; Okada, M. Therapeutic Potential and Limitation of Serotonin Type 7 Receptor Modulation. Int. J. Mol. Sci. 2023, 24, 2070. [Google Scholar] [CrossRef]
- Carli, M.; Kolachalam, S.; Longoni, B.; Pintaudi, A.; Baldini, M.; Aringhieri, S.; Fasciani, I.; Annibale, P.; Maggio, R.; Scarselli, M. Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences. Pharmaceuticals (Basel) 2021, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Yoshida, S.; Zhu, G.; Hirose, S.; Kaneko, S. Biphasic actions of topiramate on monoamine exocytosis associated with both soluble N-ethylmaleimide-sensitive factor attachment protein receptors and Ca(2+)-induced Ca(2+)-releasing systems. Neuroscience 2005, 134, 233–246. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. An intimate liaison: Spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 2010, 29, 2715–2723. [Google Scholar] [CrossRef]
- Decrock, E.; De Bock, M.; Wang, N.; Gadicherla, A.K.; Bol, M.; Delvaeye, T.; Vandenabeele, P.; Vinken, M.; Bultynck, G.; Krysko, D.V.; et al. IP3, a small molecule with a powerful message. Biochim. Biophys. Acta 2013, 1833, 1772–1786. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Lopez, M. Hypothalamic AMPK as a possible target for energy balance-related diseases. Trends Pharmacol. Sci. 2022, 43, 546–556. [Google Scholar] [CrossRef]
- Fukuyama, K.; Motomura, E.; Okada, M. A Candidate Gliotransmitter, L-β-Aminoisobutyrate, Contributes to Weight Gain and Metabolic Complication Induced by Atypical Antipsychotics. Nutrients 2023, 15, 1621. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Motomura, E. Dose-Dependent Biphasic Action of Quetiapine on AMPK Signalling via 5-HT7 Receptor: Exploring Pathophysiology of Clinical and Adverse Effects of Quetiapine. Int. J. Mol. Sci. 2022, 23, 9103. [Google Scholar] [CrossRef]
- Okubo, R.; Hasegawa, T.; Fukuyama, K.; Shiroyama, T.; Okada, M. Current Limitations and Candidate Potential of 5-HT7 Receptor Antagonism in Psychiatric Pharmacotherapy. Front. Psychiatry 2021, 12, 623684. [Google Scholar] [CrossRef] [PubMed]
- Bymaster, F.P.; Nelson, D.L.; DeLapp, N.W.; Falcone, J.F.; Eckols, K.; Truex, L.L.; Foreman, M.M.; Lucaites, V.L.; Calligaro, D.O. Antagonism by olanzapine of dopamine D1, serotonin2, muscarinic, histamine H1 and alpha 1-adrenergic receptors in vitro. Schizophr. Res. 1999, 37, 107–122. [Google Scholar] [CrossRef]
- Arranz, B.; Rosel, P.; San, L.; Ramirez, N.; Duenas, R.M.; Salavert, J.; Centeno, M.; del Moral, E. Low baseline serotonin-2A receptors predict clinical response to olanzapine in first-episode schizophrenia patients. Psychiatry Res. 2007, 153, 103–109. [Google Scholar] [CrossRef]
- Deng, C.; Weston-Green, K.; Huang, X.F. The role of histaminergic H1 and H3 receptors in food intake: A mechanism for atypical antipsychotic-induced weight gain? Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Lian, J.; Su, Y.; Deng, C. Cevimeline co-treatment attenuates olanzapine-induced metabolic disorders via modulating hepatic M3 muscarinic receptor: AMPKalpha signalling pathway in female rats. J. Psychopharmacol. 2022, 36, 202–213. [Google Scholar] [CrossRef]
- Castellani, L.N.; Pereira, S.; Kowalchuk, C.; Asgariroozbehani, R.; Singh, R.; Wu, S.; Hamel, L.; Alganem, K.; Ryan, W.G.; Zhang, X.; et al. Antipsychotics impair regulation of glucose metabolism by central glucose. Mol. Psychiatry 2022, 27, 4741–4753. [Google Scholar] [CrossRef]
- Ferreira, V.; Folgueira, C.; Guillen, M.; Zubiaur, P.; Navares, M.; Sarsenbayeva, A.; Lopez-Larrubia, P.; Eriksson, J.W.; Pereira, M.J.; Abad-Santos, F.; et al. Modulation of hypothalamic AMPK phosphorylation by olanzapine controls energy balance and body weight. Metabolism 2022, 137, 155335. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; McCutcheon, R.; Agid, O.; de Bartolomeis, A.; van Beveren, N.J.; Birnbaum, M.L.; Bloomfield, M.A.; Bressan, R.A.; Buchanan, R.W.; Carpenter, W.T.; et al. Treatment-Resistant Schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group Consensus Guidelines on Diagnosis and Terminology. Am. J. Psychiatry 2017, 174, 216–229. [Google Scholar] [CrossRef]
- Howes, O.D.; Thase, M.E.; Pillinger, T. Treatment resistance in psychiatry: State of the art and new directions. Mol. Psychiatry 2022, 27, 58–72. [Google Scholar] [CrossRef]
- Siskind, D.; McCartney, L.; Goldschlager, R.; Kisely, S. Clozapine v. first- and second-generation antipsychotics in treatment-refractory schizophrenia: Systematic review and meta-analysis. Br. J. Psychiatry 2016, 209, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Siafis, S.; Fernando, P.; Falkai, P.; Honer, W.G.; Roh, A.; Siskind, D.; Leucht, S.; Hasan, A. Efficacy and safety of clozapine in psychotic disorders-a systematic quantitative meta-review. Transl. Psychiatry 2021, 11, 487. [Google Scholar] [CrossRef]
- Joo, S.W.; Kim, H.; Jo, Y.T.; Ahn, S.; Choi, Y.J.; Choi, W.; Park, S.; Lee, J. Comparative effectiveness of antipsychotic monotherapy and polypharmacy in schizophrenia patients with clozapine treatment: A nationwide, health insurance data-based study. Eur. Neuropsychopharmacol. 2022, 59, 36–44. [Google Scholar] [CrossRef]
- McEvoy, J.P.; Lieberman, J.A.; Stroup, T.S.; Davis, S.M.; Meltzer, H.Y.; Rosenheck, R.A.; Swartz, M.S.; Perkins, D.O.; Keefe, R.S.; Davis, C.E.; et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am. J. Psychiatry 2006, 163, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Correll, C.U.; Howes, O.D. Treatment-Resistant Schizophrenia: Definition, Predictors, and Therapy Options. J. Clin. Psychiatry 2021, 82, MY20096AH1C. [Google Scholar] [CrossRef]
- Mouchlianitis, E.; McCutcheon, R.; Howes, O.D. Brain-imaging studies of treatment-resistant schizophrenia: A systematic review. Lancet Psychiatry 2016, 3, 451–463. [Google Scholar] [CrossRef]
- Potkin, S.G.; Kane, J.M.; Correll, C.U.; Lindenmayer, J.P.; Agid, O.; Marder, S.R.; Olfson, M.; Howes, O.D. The neurobiology of treatment-resistant schizophrenia: Paths to antipsychotic resistance and a roadmap for future research. NPJ Schizophr. 2020, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Noda, Y.; Iwata, Y.; Tsugawa, S.; Yoshida, K.; Tani, H.; Hirano, Y.; Koike, S.; Sasabayashi, D.; Katayama, H.; et al. Dopaminergic dysfunction and excitatory/inhibitory imbalance in treatment-resistant schizophrenia and novel neuromodulatory treatment. Mol. Psychiatry 2022, 27, 2950–2967. [Google Scholar] [CrossRef] [PubMed]
- Siskind, D.; Siskind, V.; Kisely, S. Clozapine Response Rates among People with Treatment-Resistant Schizophrenia: Data from a Systematic Review and Meta-Analysis. Can. J. Psychiatry 2017, 62, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, B.; Yada, Y.; So, R.; Takaki, M.; Yamada, N. The critical treatment window of clozapine in treatment-resistant schizophrenia: Secondary analysis of an observational study. Psychiatry Res. 2017, 250, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Agid, O.; Arenovich, T.; Sajeev, G.; Zipursky, R.B.; Kapur, S.; Foussias, G.; Remington, G. An algorithm-based approach to first-episode schizophrenia: Response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J. Clin. Psychiatry 2011, 72, 1439–1444. [Google Scholar] [CrossRef]
- Kahn, R.S.; Winter van Rossum, I.; Leucht, S.; McGuire, P.; Lewis, S.W.; Leboyer, M.; Arango, C.; Dazzan, P.; Drake, R.; Heres, S.; et al. Amisulpride and olanzapine followed by open-label treatment with clozapine in first-episode schizophrenia and schizophreniform disorder (OPTiMiSE): A three-phase switching study. Lancet Psychiatry 2018, 5, 797–807. [Google Scholar] [CrossRef] [PubMed]
- John, A.P.; Ko, E.K.F.; Dominic, A. Delayed Initiation of Clozapine Continues to Be a Substantial Clinical Concern. Can. J. Psychiatry 2018, 63, 526–531. [Google Scholar] [CrossRef]
- Kapur, S.; Seeman, P. Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics?: A new hypothesis. Am. J. Psychiatry 2001, 158, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Stepnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.A.; Moore, H.; Stott, L.; Holliday, N.; Javitch, J.A.; Lane, J.R.; Charlton, S.J. Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D(2) receptors. Nat. Commun. 2017, 8, 763. [Google Scholar] [CrossRef] [PubMed]
- Sahlholm, K.; Zeberg, H.; Nilsson, J.; Ogren, S.O.; Fuxe, K.; Arhem, P. The fast-off hypothesis revisited: A functional kinetic study of antipsychotic antagonism of the dopamine D2 receptor. Eur. Neuropsychopharmacol. 2016, 26, 467–476. [Google Scholar] [CrossRef]
- Kapur, S.; Seeman, P. Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. J. Psychiatry Neurosci. 2000, 25, 161–166. [Google Scholar]
- Demjaha, A.; Lappin, J.M.; Stahl, D.; Patel, M.X.; MacCabe, J.H.; Howes, O.D.; Heslin, M.; Reininghaus, U.A.; Donoghue, K.; Lomas, B.; et al. Antipsychotic treatment resistance in first-episode psychosis: Prevalence, subtypes and predictors. Psychol. Med. 2017, 47, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Lally, J.; Ajnakina, O.; Di Forti, M.; Trotta, A.; Demjaha, A.; Kolliakou, A.; Mondelli, V.; Reis Marques, T.; Pariante, C.; Dazzan, P.; et al. Two distinct patterns of treatment resistance: Clinical predictors of treatment resistance in first-episode schizophrenia spectrum psychoses. Psychol. Med. 2016, 46, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Carbon, M.; Correll, C.U. Clinical predictors of therapeutic response to antipsychotics in schizophrenia. Dialogues Clin. Neurosci. 2014, 16, 505–524. [Google Scholar] [CrossRef]
- Murray, R.M.; O’Callaghan, E.; Castle, D.J.; Lewis, S.W. A neurodevelopmental approach to the classification of schizophrenia. Schizophr. Bull. 1992, 18, 319–332. [Google Scholar] [CrossRef]
- Bourque, J.; Lakis, N.; Champagne, J.; Stip, E.; Lalonde, P.; Lipp, O.; Mendrek, A. Clozapine and visuospatial processing in treatment-resistant schizophrenia. Cogn. Neuropsychiatry 2013, 18, 615–630. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Balletta, R.; Giordano, S.; Buonaguro, E.F.; Latte, G.; Iasevoli, F. Differential cognitive performances between schizophrenic responders and non-responders to antipsychotics: Correlation with course of the illness, psychopathology, attitude to the treatment and antipsychotics doses. Psychiatry Res. 2013, 210, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhu, Y.; Fan, F.; Chen, S.; Hong, Y.; Cui, Y.; Luo, X.; Tan, S.; Wang, Z.; Shang, L.; et al. Hippocampus and cognitive domain deficits in treatment-resistant schizophrenia: A comparison with matched treatment-responsive patients and healthy controls(✰,✰✰, bigstar, bigstar bigstar). Psychiatry Res. Neuroimaging 2020, 297, 111043. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.S.; Chan, H.Y.; Peng, Y.C.; Chen, W.J. Severity in sustained attention impairment and clozapine-resistant schizophrenia: A retrospective study. BMC Psychiatry 2019, 19, 220. [Google Scholar] [CrossRef]
- Ito, Y.; Murata, M.; Taku, O.; Fukuyama, K.; Motomura, E.; Dohi, K.; Okada, M. Developed catatonia with rhabdomyolysis and exacerbated cardiac failure upon switching from clozapine to olanzapine owing to cardiomyopathy during clozapine medication—A case report. Asian J. Psychiatr. 2023, 80, 103376. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.O.; Bustillo, J.R. Glutamatergic dysfunction in Schizophrenia. Transl. Psychiatry 2022, 12, 500. [Google Scholar] [CrossRef]
- Wang, H.Y.; MacDonald, M.L.; Borgmann-Winter, K.E.; Banerjee, A.; Sleiman, P.; Tom, A.; Khan, A.; Lee, K.C.; Roussos, P.; Siegel, S.J.; et al. mGluR5 hypofunction is integral to glutamatergic dysregulation in schizophrenia. Mol. Psychiatry 2020, 25, 750–760. [Google Scholar] [CrossRef]
- Gonzalez-Maeso, J.; Ang, R.L.; Yuen, T.; Chan, P.; Weisstaub, N.V.; Lopez-Gimenez, J.F.; Zhou, M.; Okawa, Y.; Callado, L.F.; Milligan, G.; et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 2008, 452, 93–97. [Google Scholar] [CrossRef]
- Fell, M.J.; Svensson, K.A.; Johnson, B.G.; Schoepp, D.D. Evidence for the role of metabotropic glutamate (mGlu)2 not mGlu3 receptors in the preclinical antipsychotic pharmacology of the mGlu2/3 receptor agonist (-)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY404039). J. Pharmacol. Exp. Ther. 2008, 326, 209–217. [Google Scholar] [CrossRef]
- Mizukami, K.; Sasaki, M.; Ishikawa, M.; Iwakiri, M.; Hidaka, S.; Shiraishi, H.; Iritani, S. Immunohistochemical localization of gamma-aminobutyric acid(B) receptor in the hippocampus of subjects with schizophrenia. Neurosci. Lett. 2000, 283, 101–104. [Google Scholar] [CrossRef]
- Ishikawa, M.; Mizukami, K.; Iwakiri, M.; Asada, T. Immunohistochemical and immunoblot analysis of gamma-aminobutyric acid B receptor in the prefrontal cortex of subjects with schizophrenia and bipolar disorder. Neurosci. Lett. 2005, 383, 272–277. [Google Scholar] [CrossRef]
- Bortolato, M.; Frau, R.; Orru, M.; Piras, A.P.; Fa, M.; Tuveri, A.; Puligheddu, M.; Gessa, G.L.; Castelli, M.P.; Mereu, G.; et al. Activation of GABA(B) receptors reverses spontaneous gating deficits in juvenile DBA/2J mice. Psychopharmacology 2007, 194, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Micoulaud-Franchi, J.A.; Aramaki, M.; Geoffroy, P.A.; Richieri, R.; Cermolacce, M.; Faget, C.; Ystad, S.; Kronland-Martinet, R.; Lancon, C.; Vion-Dury, J. Effects of clozapine on perceptual abnormalities and sensory gating: A preliminary cross-sectional study in schizophrenia. J. Clin. Psychopharmacol. 2015, 35, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Beas, B.S.; Setlow, B.; Bizon, J.L. Effects of acute administration of the GABA(B) receptor agonist baclofen on behavioral flexibility in rats. Psychopharmacology 2016, 233, 2787–2797. [Google Scholar] [CrossRef]
- Okada, M.; Kawano, Y.; Fukuyama, K.; Motomura, E.; Shiroyama, T. Candidate Strategies for Development of a Rapid-Acting Antidepressant Class That Does Not Result in Neuropsychiatric Adverse Effects: Prevention of Ketamine-Induced Neuropsychiatric Adverse Reactions. Int. J. Mol. Sci. 2020, 21, 7951. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Huang, J.; Zhou, Q.X.; Yang, C.X.; Yang, C.P.; Mei, W.Y.; Zhang, L.; Zhang, Q.; Hu, L.; Hu, Y.Q.; et al. ZFP804A mutant mice display sex-dependent schizophrenia-like behaviors. Mol. Psychiatry 2021, 26, 2514–2532. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Hong, C.J.; Huang, Y.H.; Tsai, S.J. The effects of glycine transporter I inhibitor, N-methylglycine (sarcosine), on ketamine-induced alterations in sensorimotor gating and regional brain c-Fos expression in rats. Neurosci. Lett. 2010, 469, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.C.; McKinnon, R.A.; Miners, J.O.; Bastiampillai, T. Binding of clozapine to the GABAB receptor: Clinical and structural insights. Mol. Psychiatry 2020, 25, 1910–1919. [Google Scholar] [CrossRef]
- Roth BL, D.J. Psychoactive Drug Screening Program (PDSP). 2017. Available online: https://pdsp.unc.edu/databases/pdsp.php?knowID=0&kiKey=&receptorDD=&receptor=&speciesDD=&species=&sourcesDD=&source=&hotLigandDD=&hotLigand=&testLigandDD=&testFreeRadio=testFreeRadio&testLigand=clozapine&referenceDD=&reference=&KiGreater=&KiLess=&kiAllRadio=all&doQuery=Submit+Query (accessed on 27 July 2023).
- Miyazawa, A.; Kanahara, N.; Shiko, Y.; Ozawa, Y.; Kawasaki, Y.; Komatsu, H.; Masumo, Y.; Nakata, Y.; Iyo, M. The cortical silent period in schizophrenia: A systematic review and meta-analysis focusing on disease stage and antipsychotic medication. J. Psychopharmacol. 2022, 36, 479–488. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Vellucci, L.; Barone, A.; Manchia, M.; De Luca, V.; Iasevoli, F.; Correll, C.U. Clozapine’s multiple cellular mechanisms: What do we know after more than fifty years? A systematic review and critical assessment of translational mechanisms relevant for innovative strategies in treatment-resistant schizophrenia. Pharmacol. Ther. 2022, 236, 108236. [Google Scholar] [CrossRef]
- Tanahashi, S.; Ueda, Y.; Nakajima, A.; Yamamura, S.; Nagase, H.; Okada, M. Novel delta1-receptor agonist KNT-127 increases the release of dopamine and L-glutamate in the striatum, nucleus accumbens and median pre-frontal cortex. Neuropharmacology 2012, 62, 2057–2067. [Google Scholar] [CrossRef]
- Crumpler, H.R.; Dent, C.E.; Harris, H.; Westall, R.G. beta-Aminoisobutyric acid (alpha-methyl-beta-alanine); a new amino-acid obtained from human urine. Nature 1951, 167, 307–308. [Google Scholar] [CrossRef]
- Roberts, L.D.; Bostrom, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. beta-Aminoisobutyric acid induces browning of white fat and hepatic beta-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef]
- Jung, T.W.; Hwang, H.J.; Hong, H.C.; Yoo, H.J.; Baik, S.H.; Choi, K.M. BAIBA attenuates insulin resistance and inflammation induced by palmitate or a high fat diet via an AMPK-PPARdelta-dependent pathway in mice. Diabetologia 2015, 58, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.X.; Zhao, M.X.; Shu, X.D.; Xiong, X.Q.; Wang, J.J.; Gao, X.Y.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. beta-aminoisobutyric acid attenuates hepatic endoplasmic reticulum stress and glucose/lipid metabolic disturbance in mice with type 2 diabetes. Sci. Rep. 2016, 6, 21924. [Google Scholar] [CrossRef]
- Zheng, W.; Li, X.B.; Tang, Y.L.; Xiang, Y.Q.; Wang, C.Y.; de Leon, J. Metformin for Weight Gain and Metabolic Abnormalities Associated with Antipsychotic Treatment: Meta-Analysis of Randomized Placebo-Controlled Trials. J. Clin. Psychopharmacol. 2015, 35, 499–509. [Google Scholar] [CrossRef] [PubMed]
- de Silva, V.A.; Suraweera, C.; Ratnatunga, S.S.; Dayabandara, M.; Wanniarachchi, N.; Hanwella, R. Metformin in prevention and treatment of antipsychotic induced weight gain: A systematic review and meta-analysis. BMC Psychiatry 2016, 16, 341. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, Q.E.; Cai, D.B.; Yang, X.H.; Ungvari, G.S.; Ng, C.H.; Wu, R.R.; Xiang, Y.T. Combination of Metformin and Lifestyle Intervention for Antipsychotic-Related Weight Gain: A Meta-Analysis of Randomized Controlled Trials. Pharmacopsychiatry 2019, 52, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Motomura, E.; Okada, M. Brexpiprazole reduces 5-HT7 receptor function on astroglial transmission systems. Int. J. Mol. Sci. 2022, 23, 6571. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Effects of an Atypical Antipsychotic, Zotepine, on Astroglial L-Glutamate Release through Hemichannels: Exploring the Mechanism of Mood-Stabilising Antipsychotic Actions and Antipsychotic-Induced Convulsion. Pharmaceuticals 2021, 14, 1116. [Google Scholar] [CrossRef]
- Vemula, H.; Kitase, Y.; Ayon, N.J.; Bonewald, L.; Gutheil, W.G. Gaussian and linear deconvolution of LC-MS/MS chromatograms of the eight aminobutyric acid isomers. Anal. Biochem. 2017, 516, 75–85. [Google Scholar] [CrossRef]
- Solem, E.; Jellum, E.; Eldjarn, L. The absolute configuration of β-aminoisobutyric acid in human serum and urine. Clinica Chimica Acta 1974, 50, 393–403. [Google Scholar] [CrossRef]
- Lee, I.S.; Nishikimi, M.; Inoue, M.; Muragaki, Y.; Ooshima, A. Specific expression of alanine-glyoxylate aminotransferase 2 in the epithelial cells of Henle’s loop. Nephron 1999, 83, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Cline, R.E.; Henderson, R.B.; Fink, R.M. Metabolism of thymine (methyl-C14 or -2-C14) by rat liver in vitro. J. Biol. Chem. 1956, 221, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.R.; Struys, E.; Kok, R.M.; Roe, D.S.; Harris, R.A.; Jakobs, C. Methylmalonic semialdehyde dehydrogenase deficiency: Psychomotor delay and methylmalonic aciduria without metabolic decompensation. Mol. Genet. Metab. 1998, 65, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Pollitt, R.J.; Green, A.; Smith, R. Excessive excretion of beta-alanine and of 3-hydroxypropionic, R- and S-3-aminoisobutyric, R- and S-3-hydroxyisobutyric and S-2-(hydroxymethyl)butyric acids probably due to a defect in the metabolism of the corresponding malonic semialdehydes. J. Inherit. Metab. Dis. 1985, 8, 75–79. [Google Scholar] [CrossRef]
- Kakimoto, Y.; Kanazawa, A.; Taniguchi, K.; Sano, I. Beta-aminoisobutyrate-alpha-ketoglutarate transaminase in relation to beta-aminoisobutyric aciduria. Biochim. Biophys. Acta 1968, 156, 374–380. [Google Scholar] [CrossRef]
- Kamei, Y.; Hatazawa, Y.; Uchitomi, R.; Yoshimura, R.; Miura, S. Regulation of Skeletal Muscle Function by Amino Acids. Nutrients 2020, 12, 261. [Google Scholar] [CrossRef]
- Tamaki, N.; Kaneko, M.; Kikugawa, M.; Fujimoto, S. Evaluation of interconversion between (R)- and (S)-enantiomers of beta-aminoisobutyrate. Biochim. Biophys. Acta 1990, 1035, 117–119. [Google Scholar] [CrossRef]
- Koenig, M.K.; Hodgeman, R.; Riviello, J.J.; Chung, W.; Bain, J.; Chiriboga, C.A.; Ichikawa, K.; Osaka, H.; Tsuji, M.; Gibson, K.M.J.N. Phenotype of GABA-transaminase deficiency. Neurology 2017, 88, 1919–1924. [Google Scholar] [CrossRef]
- van Gennip, A.H.; Kamerling, J.P.; de Bree, P.K.; Wadman, S.K. Linear relationship between the R- and S-enantiomers of a beta-aminoisobutyric acid in human urine. Clin. Chim. Acta 1981, 116, 261–267. [Google Scholar] [CrossRef]
- Armstrong, M.D.; Yates, K.; Kakimoto, Y.; Taniguchi, K.; Kappe, T. Excretion of β-aminoisobutyric acid by man. J. Biol. Chem. 1963, 238, 1447–1455. [Google Scholar] [CrossRef]
- Gejyo, F.; Kinoshita, Y.; Ikenaka, T. Identification of beta-aminoisobutyric acid in uremic serum. Clin. Chim. Acta 1976, 70, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Stautemas, J.; Van Kuilenburg, A.B.P.; Stroomer, L.; Vaz, F.; Blancquaert, L.; Lefevere, F.B.D.; Everaert, I.; Derave, W. Acute Aerobic Exercise Leads to Increased Plasma Levels of R- and S-beta-Aminoisobutyric Acid in Humans. Front. Physiol. 2019, 10, 1240. [Google Scholar] [CrossRef] [PubMed]
- Tanianskii, D.A.; Jarzebska, N.; Birkenfeld, A.L.; O’Sullivan, J.F.; Rodionov, R.N. Beta-Aminoisobutyric Acid as a Novel Regulator of Carbohydrate and Lipid Metabolism. Nutrients 2019, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, T.; Asanuma, A.; Yanagisawa, K.; Anzai, K.; Goto, S. Taurine and beta-alanine act on both GABA and glycine receptors in Xenopus oocyte injected with mouse brain messenger RNA. Brain Res. 1988, 464, 97–105. [Google Scholar] [CrossRef]
- Schmieden, V.; Betz, H. Pharmacology of the inhibitory glycine receptor: Agonist and antagonist actions of amino acids and piperidine carboxylic acid compounds. Mol. Pharmacol. 1995, 48, 919–927. [Google Scholar]
- de la Fuente Revenga, M.; Ibi, D.; Cuddy, T.; Toneatti, R.; Kurita, M.; Ijaz, M.K.; Miles, M.F.; Wolstenholme, J.T.; Gonzalez-Maeso, J. Chronic clozapine treatment restrains via HDAC2 the performance of mGlu2 receptor agonism in a rodent model of antipsychotic activity. Neuropsychopharmacology 2019, 44, 443–454. [Google Scholar] [CrossRef]
- Wu, Y.; Blichowski, M.; Daskalakis, Z.J.; Wu, Z.; Liu, C.C.; Cortez, M.A.; Snead, O.C., 3rd. Evidence that clozapine directly interacts on the GABAB receptor. Neuroreport 2011, 22, 637–641. [Google Scholar] [CrossRef]
- Goh, K.K.; Wu, T.H.; Chen, C.H.; Lu, M.L. Efficacy of N-methyl-D-aspartate receptor modulator augmentation in schizophrenia: A meta-analysis of randomised, placebo-controlled trials. J. Psychopharmacol. 2021, 35, 236–252. [Google Scholar] [CrossRef]
- Marchi, M.; Galli, G.; Magarini, F.M.; Mattei, G.; Galeazzi, G.M. Sarcosine as an add-on treatment to antipsychotic medication for people with schizophrenia: A systematic review and meta-analysis of randomized controlled trials. Expert. Opin. Drug Metab. Toxicol. 2021, 17, 483–493. [Google Scholar] [CrossRef]
- Singh, S.P.; Singh, V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs 2011, 25, 859–885. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.A.; Uhlrich, D.; Bickford, M.E. Cortical function: A view from the thalamus. Neuron 2005, 45, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Groh, A.; Bokor, H.; Mease, R.A.; Plattner, V.M.; Hangya, B.; Stroh, A.; Deschenes, M.; Acsady, L. Convergence of cortical and sensory driver inputs on single thalamocortical cells. Cereb. Cortex 2014, 24, 3167–3179. [Google Scholar] [CrossRef] [PubMed]
- Cruz, K.G.; Leow, Y.N.; Le, N.M.; Adam, E.; Huda, R.; Sur, M. Cortical-subcortical interactions in goal-directed behavior. Physiol. Rev. 2023, 103, 347–389. [Google Scholar] [CrossRef]
- Ahissar, E.; Oram, T. Thalamic relay or cortico-thalamic processing? Old question, new answers. Cereb. Cortex 2015, 25, 845–848. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone Sub-Chronically Activates Serotonergic Transmission via Desensitization of 5-HT1A and 5-HT7 Receptors in Dorsal Raphe Nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Ueda, Y. Lurasidone inhibits NMDA receptor antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br. J. Pharmacol. 2019, 176, 4002–4018. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br. J. Pharmacol. 2020, 177, 2143–2162. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Age-Dependent and Sleep/Seizure-Induced Pathomechanisms of Autosomal Dominant Sleep-Related Hypermotor Epilepsy. Int. J. Mol. Sci. 2020, 21, 8142. [Google Scholar] [CrossRef]
- Okada, M. Can rodent models elucidate the pathomechanisms of genetic epilepsy? Br. J. Pharmacol. 2022, 179, 1620–1639. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Nakano, T.; Shiroyama, T.; Okada, M. Chronic Administrations of Guanfacine on Mesocortical Catecholaminergic and Thalamocortical Glutamatergic Transmissions. Int. J. Mol. Sci. 2021, 22, 4122. [Google Scholar] [CrossRef]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Okubo, R.; Fukuyama, K. Vortioxetine Subchronically Activates Serotonergic Transmission via Desensitization of Serotonin 5-HT1A Receptor with 5-HT3 Receptor Inhibition in Rats. Int. J. Mol. Sci. 2019, 20, 6235. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K. Interaction between Mesocortical and Mesothalamic Catecholaminergic Transmissions Associated with NMDA Receptor in the Locus Coeruleus. Biomolecules 2020, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Oka, T.; Nakamoto, M.; Fukuyama, K.; Shiroyama, T. Astroglial Connexin43 as a Potential Target for a Mood Stabiliser. Int. J. Mol. Sci. 2020, 22, 339. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Matsumoto, R.; Yamamoto, Y.; Fukuyama, K. Effects of Subchronic Administrations of Vortioxetine, Lurasidone, and Escitalopram on Thalamocortical Glutamatergic Transmission Associated with Serotonin 5-HT7 Receptor. Int. J. Mol. Sci. 2021, 22, 1351. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Motomura, E.; Okada, M. A Novel Gliotransmitter, L-β-Aminoisobutyric Acid, Contributes to Pathophysiology of Clinical Efficacies and Adverse Reactions of Clozapine. Biomolecules 2023, 13, 1288. https://doi.org/10.3390/biom13091288
Fukuyama K, Motomura E, Okada M. A Novel Gliotransmitter, L-β-Aminoisobutyric Acid, Contributes to Pathophysiology of Clinical Efficacies and Adverse Reactions of Clozapine. Biomolecules. 2023; 13(9):1288. https://doi.org/10.3390/biom13091288
Chicago/Turabian StyleFukuyama, Kouji, Eishi Motomura, and Motohiro Okada. 2023. "A Novel Gliotransmitter, L-β-Aminoisobutyric Acid, Contributes to Pathophysiology of Clinical Efficacies and Adverse Reactions of Clozapine" Biomolecules 13, no. 9: 1288. https://doi.org/10.3390/biom13091288
APA StyleFukuyama, K., Motomura, E., & Okada, M. (2023). A Novel Gliotransmitter, L-β-Aminoisobutyric Acid, Contributes to Pathophysiology of Clinical Efficacies and Adverse Reactions of Clozapine. Biomolecules, 13(9), 1288. https://doi.org/10.3390/biom13091288