
The Crystal Structure of Tyrosinase from Verrucomicrobium spinosum Reveals It to Be an Atypical Bacterial Tyrosinase
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning, Expression and Purification of the Pro-Tyrosinase and Core Domain of vsTyr
2.2. vsTyr Enzyme Activity Assay
2.3. Crystallization of vsTyr Core Domain
2.4. Data Collection, Structure Determination and Refinement
2.5. Prediction of the Full-Length vsTyr Structure Using AlphaFold

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unsoaked Crystals | CuSO4-Soaked Crystals | |
|---|---|---|
| Data Collection | ||
| Space group | C2 | C2 |
| a, b, c (Å), β (°) | 84.8, 63.4, 116.0, 96.8 | 86.3, 63.4, 117.3, 97.2 |
| Molecules in a. u. | 2 | 2 |
| Wavelength (Å) | 0.97626 | 0.97625 |
| Resolution (Å) b | 115.20–1.43 (1.56–1.43) | 116.37–1.64 (1.67–1.64) |
| Total reflections | 426,619 (19,225) | 508,362 (15,210) |
| Unique reflections | 62,664 (3134) | 75,699 (3073) |
| Multiplicity | 6.8 (6.1) | 6.7 (4.9) |
| Completeness (%) | 93.4 (66.8) | 98.2 (79.0) |
| ˂I/σ(I)˃ | 10.5 (1.6) | 9.8 (0.6) |
| Wilson B factor (Å2) | 23.5 | 25.8 |
| Rmerge | 0.104 (1.084) | 0.114 (2.182) |
| Rmeas | 0.112 (1.182) | 0.124 (2.441) |
| Rpim | 0.043 (0.463) | 0.047 (1.063) |
| CC1/2 | 0.99 (0.60) | 0.998 (0.362) |
| Refinement | ||
| No. of reflection in work set | 62,663 | 77,117 |
| No. of reflection in free set | 7016 | 3975 |
| Rwork | 0.1696 | 0.1907 |
| Rfree c | 0.2017 | 0.2245 |
| No. of non-hydrogen atoms | ||
| protein | 5226 | 5209 |
| solvent | 517 | 705 |
| Cu2+ | 4 | 13 |
| RMS deviations | ||
| bonds (Å) | 0.008 | 0.007 |
| angles (°) | 1.488 | 1.503 |
| Ramachandran plot regions (% residues) | ||
| favored | 98.6 | 98.7 |
| allowed | 0.8 | 0.6 |
| outliers | 0.6 | 0.6 |
| Average B factor (Å2) | ||
| protein | 16.3 | 27.1 |
| solvent | 27.3 | 36.0 |
| Cu2+ | 24.3 | 49.6 |
| PDB-ID | 8BBQ | 8BBR |
3. Results and Discussion
3.1. Production and Activity of vsTyr Variants
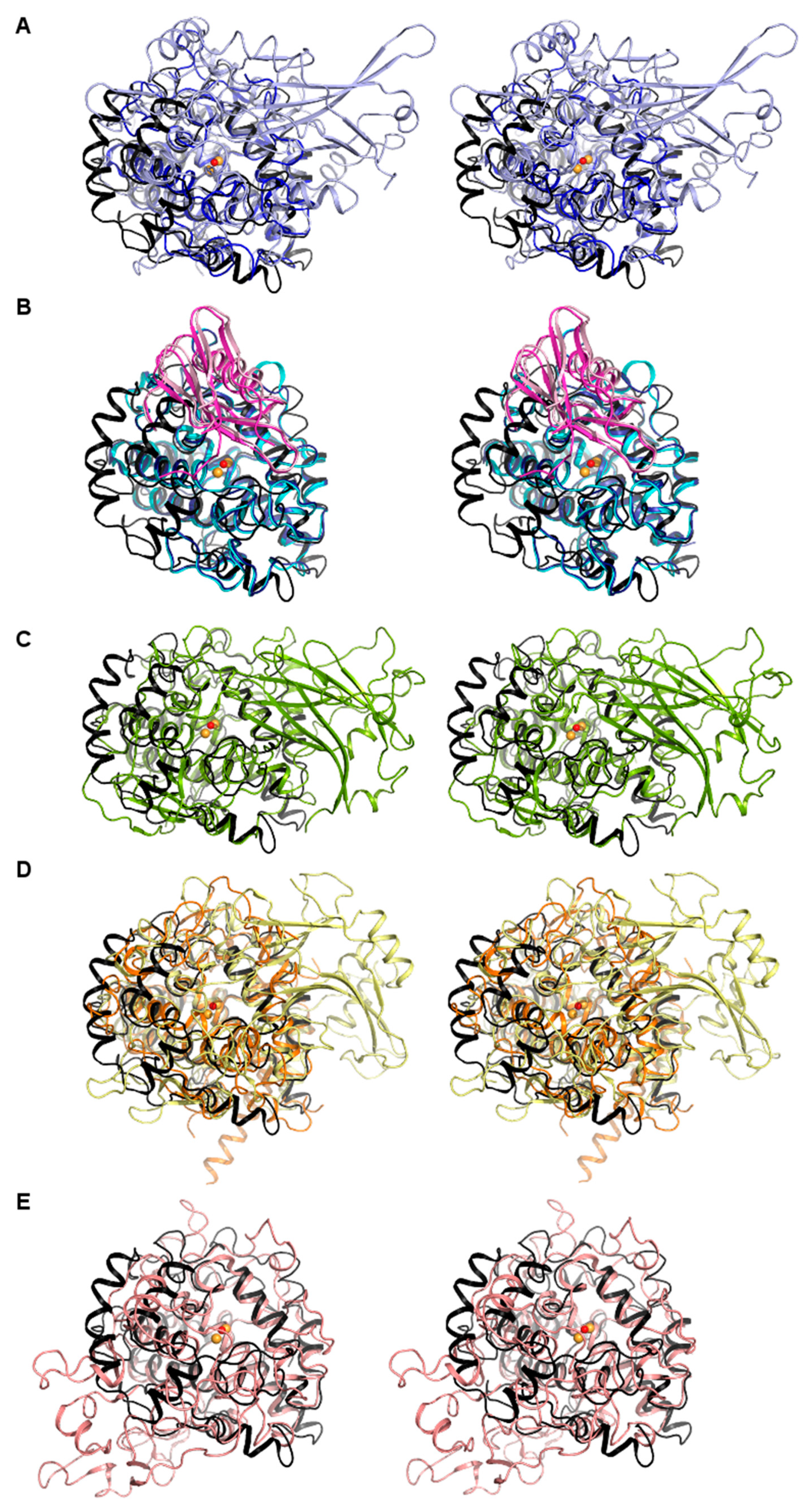
3.2. Structure of the vsTyr core Domain
| Enzyme Source Type | PDB-Id [Ref] | Enzyme Name (Organism), Abbreviation | Sequence Similarity a (% Seq-Id/ % Seq Cov/ E) | Structural Similarity b (RMSD, Å/ #Aligned Cα Atoms) | Activity Regulated via C-Term Ext or Caddie Protein | Cys–His Thioether Linkage Yes/No, (Topology Relative vsTyr) | Activity Controller Residue 1 (HisB1 + 1) | Activity Controller Residue 2 (HisB2 + 1) | Gatekeeper Residue | Additional Gatekeeper Residue c |
|---|---|---|---|---|---|---|---|---|---|---|
| Bacterial | 8BBQ | Tyrosinase (Verrucomicrobium spinosum), vsTyr | Reference sequence | Reference structure | C-term ext | Y | Asn259 | Asn263 | Leu272 | Phe453 |
| 5ZRD [26] | Tyrosinase (Burkholderia thailandensis), btTyr | 22.4/41/4 × 10−8 | 1.96/218 | C-term ext | Y (same) | Asn277 | Leu281 | Asn306 | Leu466 | |
| 3NM8 [8] | Tyrosinase (Bacillus megaterium), bmTyr | 28.6/42/5 × 10−9 | 2.11/213 | No caddie | N | Asn259 | His209 | Val218 | - | |
| 6J2U [Not publ.] | Tyrosinase (Streptomyces avermitilis), saTyr | 47.2/10/5 × 10−6 | 2.27/208 | Caddie | N | Asn191 | Val195 | Gly204 | Tyr92 | |
| 7CIY [50] | Tyrosinase (Streptomyces castaneoglobisporus), scTyr | 46.3/7/ 3 × 10−5 | 2.29/212 | Caddie | N | Gly191 | Val195 | Gly204 | Tyr*98 (dopaquinone) | |
| Plant | 4Z11 [48] | Aurone synthase (Coreopsis grandiflora), cgAUS | 25.8/62/4 × 10−19 | 1.77/224 | C-term ext | Y | Thr256 | Arg260 | Phe273 | Ile456 |
| 6HQI [51] | Polyphenol oxidase 1 (Solanum lycopersicum), slTyr | 21.0/91/4 × 10−11 | 2.09/240 | C-term ext | ? (not resolved) | Ser242 | Ile246 | Phe270 | Leu447 | |
| 1BT1 [21] | Catechol oxidase (Ipomoea batatas), ibCO | 32.6/36/9 × 10−10 | 1.67/223 | C-term ext [52] | Y | Ile241 | Arg245 | Phe261 | - | |
| 2P3X [25] | Tyrosinase (Vitis vinifera), vvTyr | 22.6/57/8 × 10−13 | 1.66/217 | C-term ext | Y | Asn240 | Lys244 | Phe259 | - | |
| 5CE9 [20] | Tyrosinase (Juglans regia), jrTyr | 25.8/52/2 × 10−18 | 1.69/223 | C-term ext [53] | Y | Asn240 | Leu244 | Phe260 | - | |
| 6ELS [49] | Tyrosinase (Malus domestica), mdTyr | 24.3/65/1 × 10−16 | 2.01/240 | C-term ext | Y | Ala239 | Leu243 | Phe259 | Val449 | |
| Fungal | 2Y9W [54] | Tyrosinase (Agaricus bisporus), abTyr | 23.4/61/2 × 10−4 | 1.93/212 | C-term ext | Y (same) | Asn260 | Phe264 | Val283 | - |
| 3W6Q [55] | Pro-tyrosinase (Aspergillus oryzae), aoTyr | 41.8/7/0.001 | 2.63/270 | C-term ext | Y (same) | Asn329 | Asn333 | Val359 | Phe513 | |
| 6Z1S [56] | Tyrosinase-like protein (Thermothelomyces thermophila ATCC 42464), ttTyrP | 25.9/48/1 × 10−9 | 2.17/224 | No C-term ext | N | Asn292 | Tyr296 | Phe307 | - | |
| 4OUA [57] | Tyrosinase (Agaricus bisporus var. bisporus H97), abbTyr | 21.4/38/3 × 10−6 | 2.18/227 | C-term ext | Y (same) | Asp252 | Gly256 | Leu271 | Phe454 | |
| Human | 5M8L [58] | Tyrosinase-related protein 1 (Homo sapiens), hsTyrRP1 | 22.9/45/2 × 10−5 | 2.36/208 | - | N | Asn378 | Leu382 | Thr391 | - |
3.3. Di-Copper Center
3.4. Copper Site Occupancy and Additional Copper Binding Sites
3.5. The Waterkeeper Residue
3.6. Activity Controller Residues
3.7. The Gatekeeper Residue
3.8. Crystallisation Trials with vsTyr in Complex with Ligands
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Claus, H.; Decker, H. Bacterial tyrosinases. Syst. Appl. Microbiol. 2006, 29, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Fairhead, M.; Thony-Meyer, L. Bacterial tyrosinases: Old enzymes with new relevance to biotechnology. New Biotechnol. 2012, 29, 183–191. [Google Scholar] [CrossRef]
- Kanteev, M.; Goldfeder, M.; Fishman, A. Structure-function correlations in tyrosinases. Protein Sci. 2015, 24, 1360–1369. [Google Scholar] [CrossRef]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure and Function of Human Tyrosinase and Tyrosinase-Related Proteins. Chemistry 2018, 24, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lopez, J.N.; Tudela, J.; Varon, R.; Garcia-Carmona, F.; Garcia-Canovas, F. Analysis of a kinetic model for melanin biosynthesis pathway. J. Biol. Chem. 1992, 267, 3801–3810. [Google Scholar] [CrossRef]
- Cordero, R.J.B.; Casadevall, A. Melanin. Curr. Biol. 2020, 30, R142–R143. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ferrer, A.; Rodriguez-Lopez, J.N.; Garcia-Canovas, F.; Garcia-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys. Acta 1995, 1247, 1–11. [Google Scholar] [CrossRef]
- Sendovski, M.; Kanteev, M.; Ben-Yosef, V.S.; Adir, N.; Fishman, A. First structures of an active bacterial tyrosinase reveal copper plasticity. J. Mol. Biol. 2011, 405, 227–237. [Google Scholar] [CrossRef]
- Coates, C.J.; Costa-Paiva, E.M. Multifunctional Roles of Hemocyanins. Subcell. Biochem. 2020, 94, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Kampatsikas, I.; Rompel, A. Similar but Still Different: Which Amino Acid Residues Are Responsible for Varying Activities in Type-III Copper Enzymes? Chembiochem 2021, 22, 1161–1175. [Google Scholar] [CrossRef] [PubMed]
- Decker, H.; Schweikardt, T.; Nillius, D.; Salzbrunn, U.; Jaenicke, E.; Tuczek, F. Similar enzyme activation and catalysis in hemocyanins and tyrosinases. Gene 2007, 398, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Decker, H.; Tuczek, F. Tyrosinase/catecholoxidase activity of hemocyanins: Structural basis and molecular mechanism. Trends Biochem. Sci. 2000, 25, 392–397. [Google Scholar] [CrossRef]
- Fenoll, L.G.; Penalver, M.J.; Rodriguez-Lopez, J.N.; Varon, R.; Garcia-Canovas, F.; Tudela, J. Tyrosinase kinetics: Discrimination between two models to explain the oxidation mechanism of monophenol and diphenol substrates. Int. J. Biochem. Cell Biol. 2004, 36, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Matoba, Y.; Kumagai, T.; Yamamoto, A.; Yoshitsu, H.; Sugiyama, M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 2006, 281, 8981–8990. [Google Scholar] [CrossRef] [PubMed]
- Olivares, C.; Garcia-Borron, J.C.; Solano, F. Identification of active site residues involved in metal cofactor binding and stereospecific substrate recognition in Mammalian tyrosinase. Implications to the catalytic cycle. Biochemistry 2002, 41, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Selinheimo, E.; NiEidhin, D.; Steffensen, C.; Nielsen, J.; Lomascolo, A.; Halaouli, S.; Record, E.; O’Beirne, D.; Buchert, J.; Kruus, K. Comparison of the characteristics of fungal and plant tyrosinases. J. Biotechnol. 2007, 130, 471–480. [Google Scholar] [CrossRef] [PubMed]
- van Gastel, M.; Bubacco, L.; Groenen, E.J.; Vijgenboom, E.; Canters, G.W. EPR study of the dinuclear active copper site of tyrosinase from Streptomyces antibioticus. FEBS Lett. 2000, 474, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Panis, F.; Kampatsikas, I.; Bijelic, A.; Rompel, A. Conversion of walnut tyrosinase into a catechol oxidase by site directed mutagenesis. Sci. Rep. 2020, 10, 1659. [Google Scholar] [CrossRef] [PubMed]
- Kaintz, C.; Mayer, R.L.; Jirsa, F.; Halbwirth, H.; Rompel, A. Site-directed mutagenesis around the CuA site of a polyphenol oxidase from Coreopsis grandiflora (cgAUS1). FEBS Lett. 2015, 589, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Bijelic, A.; Pretzler, M.; Molitor, C.; Zekiri, F.; Rompel, A. The Structure of a Plant Tyrosinase from Walnut Leaves Reveals the Importance of “Substrate-Guiding Residues” for Enzymatic Specificity. Angew. Chem. Int. Ed. Engl. 2015, 54, 14677–14680. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T.; Eicken, C.; Sacchettini, J.C.; Krebs, B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Biol. 1998, 5, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Magnus, K.A.; Hazes, B.; Ton-That, H.; Bonaventura, C.; Bonaventura, J.; Hol, W.G. Crystallographic analysis of oxygenated and deoxygenated states of arthropod hemocyanin shows unusual differences. Proteins 1994, 19, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Momoji, K.; Hirata, T.; Mikami, B. The crystal structure of a crustacean prophenoloxidase provides a clue to understanding the functionality of the type 3 copper proteins. FEBS J. 2014, 281, 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Valley, C.C.; Cembran, A.; Perlmutter, J.D.; Lewis, A.K.; Labello, N.P.; Gao, J.; Sachs, J.N. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J. Biol. Chem. 2012, 287, 34979–34991. [Google Scholar] [CrossRef] [PubMed]
- Virador, V.M.; Reyes Grajeda, J.P.; Blanco-Labra, A.; Mendiola-Olaya, E.; Smith, G.M.; Moreno, A.; Whitaker, J.R. Cloning, sequencing, purification, and crystal structure of Grenache (Vitis vinifera) polyphenol oxidase. J. Agric. Food Chem. 2010, 58, 1189–1201. [Google Scholar] [CrossRef]
- Son, H.F.; Lee, S.H.; Lee, S.H.; Kim, H.; Hong, H.; Lee, U.J.; Lee, P.G.; Kim, B.G.; Kim, K.J. Structural Basis for Highly Efficient Production of Catechol Derivatives at Acidic pH by Tyrosinase from Burkholderia thailandensis. ACS Catal. 2018, 8, 10375–10382. [Google Scholar] [CrossRef]
- Fairhead, M.; Thony-Meyer, L. Role of the C-terminal extension in a bacterial tyrosinase. FEBS J. 2010, 277, 2083–2095. [Google Scholar] [CrossRef] [PubMed]
- Axambayeva, A.S.; Zhaparova, L.R.; Shagyrova, Z.S.; Ramankulov, E.M.; Shustov, A.V. Unusual Stability of a Recombinant Verrucomicrobium spinosum Tyrosinase to Denaturing Agents and Its Use for a Production of a Protein with Adhesive Properties. Appl. Biochem. Biotechnol. 2018, 185, 736–754. [Google Scholar] [CrossRef] [PubMed]
- Espin, J.C.; van Leeuwen, J.; Wichers, H.J. Kinetic study of the activation process of a latent mushroom (Agaricus bisporus) tyrosinase by serine proteases. J. Agric. Food Chem. 1999, 47, 3509–3517. [Google Scholar] [CrossRef]
- Fling, M.; Horowitz, N.H.; Heinemann, S.F. The isolation and properties of crystalline tyrosinase from Neurospora. J. Biol. Chem. 1963, 238, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.J.; Levik, K.E.; Williams, M.A.; Ashton, A.W.; McAuley, K.E. SynchWeb: A modern interface for ISPyB. J. Appl. Crystallogr. 2015, 48, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Bunkoczi, G.; Echols, N.; McCoy, A.J.; Oeffner, R.D.; Adams, P.D.; Read, R.J. Phaser.MRage: Automated molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2276–2286. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol.Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Elfmann, C.; Stulke, J. PAE viewer: A webserver for the interactive visualization of the predicted aligned error for multimer structure predictions and crosslinks. Nucleic Acids Res. 2023, 51, W404–W410. [Google Scholar] [CrossRef]
- Agirre, J.; Atanasova, M.; Bagdonas, H.; Ballard, C.B.; Basle, A.; Beilsten-Edmands, J.; Borges, R.J.; Brown, D.G.; Burgos-Marmol, J.J.; Berrisford, J.M.; et al. The CCP4 suite: Integrative software for macromolecular crystallography. Acta Crystallogr. D Struct. Biol. 2023, 79, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., 3rd; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Kaintz, C.; Mauracher, S.G.; Rompel, A. Type-3 copper proteins: Recent advances on polyphenol oxidases. Adv. Protein Chem. Struct. Biol. 2014, 97, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Molitor, C.; Mauracher, S.G.; Rompel, A. Aurone synthase is a catechol oxidase with hydroxylase activity and provides insights into the mechanism of plant polyphenol oxidases. Proc. Natl. Acad. Sci. USA 2016, 113, E1806–E1815. [Google Scholar] [CrossRef] [PubMed]
- Kampatsikas, I.; Bijelic, A.; Pretzler, M.; Rompel, A. A Peptide-Induced Self-Cleavage Reaction Initiates the Activation of Tyrosinase. Angew. Chem. Int. Ed. Engl. 2019, 58, 7475–7479. [Google Scholar] [CrossRef] [PubMed]
- Matoba, Y.; Oda, K.; Muraki, Y.; Masuda, T. The basicity of an active-site water molecule discriminates between tyrosinase and catechol oxidase activity. Int. J. Biol. Macromol. 2021, 183, 1861–1870. [Google Scholar] [CrossRef]
- Kampatsikas, I.; Bijelic, A.; Rompel, A. Biochemical and structural characterization of tomato polyphenol oxidases provide novel insights into their substrate specificity. Sci. Rep. 2019, 9, 4022. [Google Scholar] [CrossRef]
- Gerdemann, C.; Eicken, C.; Galla, H.J.; Krebs, B. Comparative modeling of the latent form of a plant catechol oxidase using a molluskan hemocyanin structure. J. Inorg. Biochem. 2002, 89, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Zekiri, F.; Molitor, C.; Mauracher, S.G.; Michael, C.; Mayer, R.L.; Gerner, C.; Rompel, A. Purification and characterization of tyrosinase from walnut leaves (Juglans regia). Phytochemistry 2014, 101, 5–15. [Google Scholar] [CrossRef]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef]
- Fujieda, N.; Yabuta, S.; Ikeda, T.; Oyama, T.; Muraki, N.; Kurisu, G.; Itoh, S. Crystal structures of copper-depleted and copper-bound fungal pro-tyrosinase: Insights into endogenous cysteine-dependent copper incorporation. J. Biol. Chem. 2013, 288, 22128–22140. [Google Scholar] [CrossRef] [PubMed]
- Nikolaivits, E.; Valmas, A.; Dedes, G.; Topakas, E.; Dimarogona, M. Considerations Regarding Activity Determinants of Fungal Polyphenol Oxidases Based on Mutational and Structural Studies. Appl. Environ. Microbiol. 2021, 87, e00396-21. [Google Scholar] [CrossRef] [PubMed]
- Mauracher, S.G.; Molitor, C.; Al-Oweini, R.; Kortz, U.; Rompel, A. Latent and active abPPO4 mushroom tyrosinase cocrystallized with hexatungstotellurate(VI) in a single crystal. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 2301–2315. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure of Human Tyrosinase Related Protein 1 Reveals a Binuclear Zinc Active Site Important for Melanogenesis. Angew. Chem. Int. Ed. Engl. 2017, 56, 9812–9815. [Google Scholar] [CrossRef]
- Pretzler, M.; Rompel, A. What causes the different functionality in type-III-copper enzymes? A state of the art perspective. Inorganica Chim. Acta 2018, 481, 25–31. [Google Scholar] [CrossRef]
- Matoba, Y.; Bando, N.; Oda, K.; Noda, M.; Higashikawa, F.; Kumagai, T.; Sugiyama, M. A molecular mechanism for copper transportation to tyrosinase that is assisted by a metallochaperone, caddie protein. J. Biol. Chem. 2011, 286, 30219–30231. [Google Scholar] [CrossRef]
- Cuff, M.E.; Miller, K.I.; van Holde, K.E.; Hendrickson, W.A. Crystal structure of a functional unit from Octopus hemocyanin. J. Mol. Biol. 1998, 278, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Mauracher, S.G.; Molitor, C.; Al-Oweini, R.; Kortz, U.; Rompel, A. Crystallization and preliminary X-ray crystallographic analysis of latent isoform PPO4 mushroom (Agaricus bisporus) tyrosinase. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Goldfeder, M.; Kanteev, M.; Isaschar-Ovdat, S.; Adir, N.; Fishman, A. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 2014, 5, 4505. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Jiang, H.; Deng, J. Crystal structure of Manduca sexta prophenoloxidase provides insights into the mechanism of type 3 copper enzymes. Proc. Natl. Acad. Sci. USA 2009, 106, 17002–17006. [Google Scholar] [CrossRef]
- Kampatsikas, I.; Pretzler, M.; Rompel, A. Identification of Amino Acid Residues Responsible for C-H Activation in Type-III Copper Enzymes by Generating Tyrosinase Activity in a Catechol Oxidase. Angew. Chem. Int. Ed. Engl. 2020, 59, 20940–20945. [Google Scholar] [CrossRef]
- Perbandt, M.; Guthohrlein, E.W.; Rypniewski, W.; Idakieva, K.; Stoeva, S.; Voelter, W.; Genov, N.; Betzel, C. The structure of a functional unit from the wall of a gastropod hemocyanin offers a possible mechanism for cooperativity. Biochemistry 2003, 42, 6341–6346. [Google Scholar] [CrossRef]
- Decker, H.; Schweikardt, T.; Tuczek, F. The first crystal structure of tyrosinase: All questions answered? Angew. Chem. Int. Ed. Engl. 2006, 45, 4546–4550. [Google Scholar] [CrossRef] [PubMed]
- Prexler, S.M.; Frassek, M.; Moerschbacher, B.M.; Dirks-Hofmeister, M.E. Catechol Oxidase versus Tyrosinase Classification Revisited by Site-Directed Mutagenesis Studies. Angew. Chem. Int. Ed. Engl. 2019, 58, 8757–8761. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, Y.; Deng, J.; Jiang, H. The structure of a prophenoloxidase (PPO) from Anopheles gambiae provides new insights into the mechanism of PPO activation. BMC Biol. 2016, 14, 2. [Google Scholar] [CrossRef]
- Pilhofer, M.; Ladinsky, M.S.; McDowall, A.W.; Petroni, G.; Jensen, G.J. Microtubules in bacteria: Ancient tubulins build a five-protofilament homolog of the eukaryotic cytoskeleton. PLoS Biol. 2011, 9, e1001213. [Google Scholar] [CrossRef]
- Rivas-Marin, E.; Devos, D.P. The Paradigms They Are a-Changin’: Past, present and future of PVC bacteria research. Antonie van Leeuwenhoek 2018, 111, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laiho, A.; Toronen, P.; Salgado, M. DALI shines a light on remote homologs: One hundred discoveries. Protein Sci. 2023, 32, e4519. [Google Scholar] [CrossRef] [PubMed]
- Molitor, C.; Mauracher, S.G.; Pargan, S.; Mayer, R.L.; Halbwirth, H.; Rompel, A. Latent and active aurone synthase from petals of C. grandiflora: A polyphenol oxidase with unique characteristics. Planta 2015, 242, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Pluvinage, B.; Grondin, J.M.; Amundsen, C.; Klassen, L.; Moote, P.E.; Xiao, Y.; Thomas, D.; Pudlo, N.A.; Anele, A.; Martens, E.C.; et al. Molecular basis of an agarose metabolic pathway acquired by a human intestinal symbiont. Nat. Commun. 2018, 9, 1043. [Google Scholar] [CrossRef] [PubMed]
- Hehemann, J.H.; Kelly, A.G.; Pudlo, N.A.; Martens, E.C.; Boraston, A.B. Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc. Natl. Acad. Sci. USA 2012, 109, 19786–19791. [Google Scholar] [CrossRef]
- Abidi, W.; Zouhir, S.; Caleechurn, M.; Roche, S.; Krasteva, P.V. Architecture and regulation of an enterobacterial cellulose secretion system. Sci. Adv. 2021, 7, eabd8049. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Tian, X.; Li, X.; She, Q. Identification and structural analysis of a carbohydrate-binding module specific to alginate, a representative of a new family, CBM96. J. Biol. Chem. 2023, 299, 102854. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.L.; McNamara, J.T.; Fischer, M.; Rich, J.; Chen, H.M.; Withers, S.G.; Zimmer, J. Observing cellulose biosynthesis and membrane translocation in crystallo. Nature 2016, 531, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Guan, C.; Cui, T.; Tarentino, A.L.; Plummer, T.H., Jr.; Van Roey, P. Active site and oligosaccharide recognition residues of peptide-N4-(N-acetyl-beta-D-glucosaminyl)asparagine amidase F. J. Biol. Chem. 1995, 270, 29493–29497. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fekry, M.; Dave, K.K.; Badgujar, D.; Hamnevik, E.; Aurelius, O.; Dobritzsch, D.; Danielson, U.H. The Crystal Structure of Tyrosinase from Verrucomicrobium spinosum Reveals It to Be an Atypical Bacterial Tyrosinase. Biomolecules 2023, 13, 1360. https://doi.org/10.3390/biom13091360
Fekry M, Dave KK, Badgujar D, Hamnevik E, Aurelius O, Dobritzsch D, Danielson UH. The Crystal Structure of Tyrosinase from Verrucomicrobium spinosum Reveals It to Be an Atypical Bacterial Tyrosinase. Biomolecules. 2023; 13(9):1360. https://doi.org/10.3390/biom13091360
Chicago/Turabian StyleFekry, Mostafa, Khyati K. Dave, Dilip Badgujar, Emil Hamnevik, Oskar Aurelius, Doreen Dobritzsch, and U. Helena Danielson. 2023. "The Crystal Structure of Tyrosinase from Verrucomicrobium spinosum Reveals It to Be an Atypical Bacterial Tyrosinase" Biomolecules 13, no. 9: 1360. https://doi.org/10.3390/biom13091360