
Comprehensive Genome and Transcriptome Analysis Identifies SLCO3A1 Associated with Aggressive Behavior in Pigs


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Aggressive Behavior Observation
2.2. CNVR Definition and Statistics
2.3. Comparison of the Detected CNVRs with Previous Studies
2.4. RNA Extraction and RNA-Seq
2.5. Transcriptome Data Analysis
2.6. Gene Annotation and Enrichment Analysis
2.7. CNVR Comparisons and CNV-Type Assay
2.8. Joint Analyses of Genomics and Transcriptomics Data
2.9. Statistical Analysis
3. Results
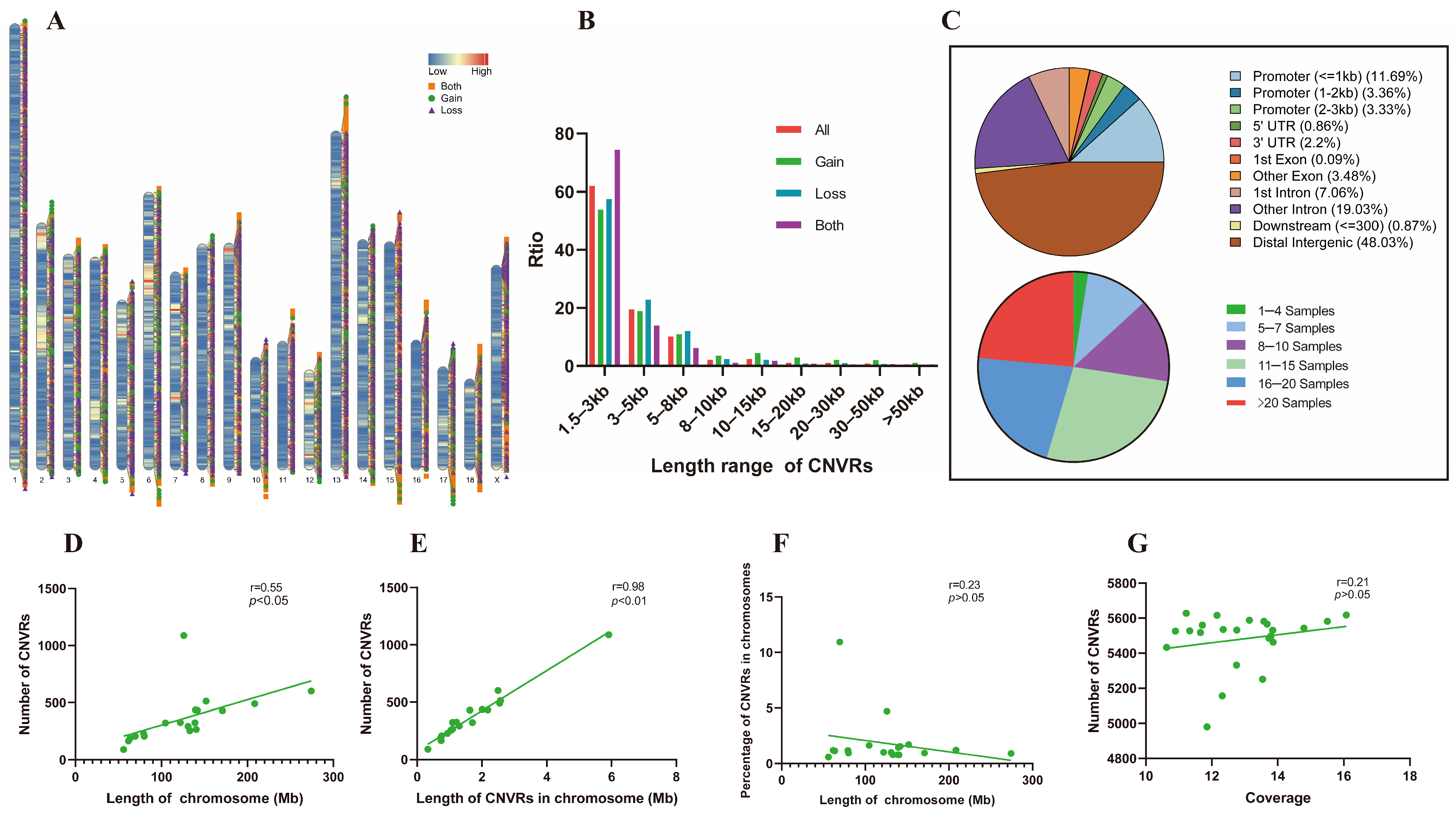
3.1. CNVR Detection and Statistics
3.2. CNVR Quantity and Chromosome Characteristics
3.3. Functional Enrichment Analysis and Comparison between the MAP and LAP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Platform | Samples | Number of CNVRs (Number of CNVRs before Remapping) | Number of Overlapped CNVRs | Percent of Overlapped CNVRs from This Study (%) | |
|---|---|---|---|---|---|---|
| [41] | Next-generation sequencing | 61 | 12,668 | 1194 | 17.38 | |
| [42] | Next-generation sequencing | 7 | 528 (540) | 256 | 3.73 | |
| [43] | Next-generation sequencing | 16 | 2614 (3118) | 618 | 9 | |
| [44] | Next-generation sequencing | 14 | 917 (1408) | 522 | 7.6 | |
| [45] | Next-generation sequencing | 49 | 2390 (3131) | 142 | 2.07 | |
| [46] | Next-generation sequencing | 240 | 3538 | 1271 | 18.5 | |
| [47] | 1 M aCGH | 12 | 709 (758) | 829 | 12.07 | |
| [12] | Porcine SNP60 | 1693 | 651 (565) | 648 | 9.43 | |
| This Study | 6869 | |||||
| Merge | 3067 | 44.6 | ||||
3.4. QTL Analysis
3.5. Analysis of Transcriptome Data from the Temporal Lobes of the MAP and LAP
3.6. Cluster Analysis of DEGs in the TL-MAP and TL-LAP
3.7. Multi-Omics Analysis Reveals a Potential Relationship between the SLCO3A1 Gene and Aggressive Behavior in Pigs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, A.J. Farm animal welfare: The five freedoms and the free market. Vet. J. 2001, 161, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Smulders, D.; Verbeke, G.; Mormede, P.; Geers, R. Validation of a behavioral observation tool to assess pig welfare. Physiol. Behav. 2006, 89, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Wieckert, D.A. Social behavior in farm animals. J. Anim. Sci. 1971, 32, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Rhim, S.J.; Son, S.H.; Hwang, H.S.; Lee, J.K.; Hong, J.K. Effects of Mixing on the Aggressive Behavior of Commercially Housed Pigs. Asian-Australas. J. Anim. Sci. 2015, 28, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.P.; Roehe, R.; D’Eath, R.B.; Ison, S.H.; Farish, M.; Jack, M.C.; Lundeheim, N.; Rydhmer, L.; Lawrence, A.B. Genetic validation of postmixing skin injuries in pigs as an indicator of aggressiveness and the relationship with injuries under more stable social conditions. J. Anim. Sci. 2009, 87, 3076–3082. [Google Scholar] [CrossRef]
- Bushby, E.V.; Dye, L.; Collins, L.M. Is Magnesium Supplementation an Effective Nutritional Method to Reduce Stress in Domestic Pigs? A Systematic Review. Front. Vet. Sci. 2020, 7, 596205. [Google Scholar] [CrossRef]
- Weller, J.E.; Camerlink, I.; Turner, S.P.; Farish, M.; Arnott, G. Socialisation and its effect on play behaviour and aggression in the domestic pig (Sus scrofa). Sci. Rep. 2019, 9, 4180. [Google Scholar] [CrossRef]
- Aviles-Rosa, E.O.; Surowiec, K.; McGlone, J. Identification of Faecal Maternal Semiochemicals in Swine (Sus scrofa) and their Effects on Weaned Piglets. Sci. Rep. 2020, 10, 5349. [Google Scholar] [CrossRef]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef]
- Iafrate, A.J.; Feuk, L.; Rivera, M.N.; Listewnik, M.L.; Donahoe, P.K.; Qi, Y.; Scherer, S.W.; Lee, C. Detection of large-scale variation in the human genome. Nat. Genet. 2004, 36, 949–951. [Google Scholar] [CrossRef]
- Fadista, J.; Nygaard, M.; Holm, L.E.; Thomsen, B.; Bendixen, C. A snapshot of CNVs in the pig genome. PLoS ONE 2008, 3, e3916. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Qiao, R.; Wei, R.; Guo, Y.; Ai, H.; Ma, J.; Ren, J.; Huang, L. A comprehensive survey of copy number variation in 18 diverse pig populations and identification of candidate copy number variable genes associated with complex traits. BMC Genom. 2012, 13, 733. [Google Scholar] [CrossRef]
- Locke, M.E.; Milojevic, M.; Eitutis, S.T.; Patel, N.; Wishart, A.E.; Daley, M.; Hill, K.A. Genomic copy number variation in Mus musculus. BMC Genom. 2015, 16, 497. [Google Scholar] [CrossRef]
- Liu, M.; Li, B.; Shi, T.; Huang, Y.; Liu, G.E.; Lan, X.; Lei, C.; Chen, H. Copy number variation of bovine SHH gene is associated with body conformation traits in Chinese beef cattle. J. Appl. Genet. 2019, 60, 199–207. [Google Scholar] [CrossRef]
- Feng, Z.; Li, X.; Cheng, J.; Jiang, R.; Huang, R.; Wang, D.; Huang, Y.; Pi, L.; Hu, L.; Chen, H. Copy Number Variation of the PIGY Gene in Sheep and Its Association Analysis with Growth Traits. Animals 2020, 10, 688. [Google Scholar] [CrossRef]
- Khatri, B.; Kang, S.; Shouse, S.; Anthony, N.; Kuenzel, W.; Kong, B.C. Copy number variation study in Japanese quail associated with stress related traits using whole genome re-sequencing data. PLoS ONE 2019, 14, e0214543. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Q.; Zhao, J.; Guo, Y.; Zhang, C.; Cheng, M.; Zhao, X.; Schinckel, A.P.; Zhou, B. Pigs’ skin lesions at weaning are primarily caused by standoff and being bullied instead of unilateral active attack at the individual level. Appl. Anim. Behav. Sci. 2022, 247, 105556. [Google Scholar] [CrossRef]
- Zhao, J.; Gao, S.; Guo, Y.; Xu, Q.; Liu, M.; Zhang, C.; Cheng, M.; Zhao, X.; Schinckel, A.P.; Zhou, B. Functionally Antagonistic Transcription Factors IRF1 and IRF2 Regulate the Transcription of the Dopamine Receptor D2 Gene Associated with Aggressive Behavior of Weaned Pigs. Biology 2022, 11, 135. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, Z.; Cai, Y.; Chen, T.; Li, C.; Fu, W.; Jiang, Y. CNVcaller: Highly efficient and widely applicable software for detecting copy number variations in large populations. Gigascience 2017, 6, gix115. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Lv, D.; Ge, Y.; Shi, J.; Weijers, D.; Yu, G.; Chen, J. RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhang, Y.; He, K.; Guo, X.; Jiang, J.; Qian, L.; Xu, J.; Che, Z.; Huang, X.; Liu, S. Transcriptomic Profiling of Fusarium pseudograminearum in Response to Carbendazim, Pyraclostrobin, Tebuconazole, and Phenamacril. J. Fungi 2023, 9, 334. [Google Scholar] [CrossRef]
- Yang, F.; Wang, T.; Yan, P.; Li, W.; Kong, J.; Zong, Y.; Chao, X.; Li, W.; Zhao, X.; Wang, J. Identification of pyroptosis-related subtypes and establishment of prognostic model and immune characteristics in asthma. Front. Immunol. 2022, 13, 937832. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Hou, L.; Yin, Y.; Wang, B.; Liu, C.; Zhou, W.; Niu, P.; Li, Q.; Huang, R.; Li, P. Genome-wide association study reveals new QTL and functional candidate genes for the number of ribs and carcass length in pigs. Anim. Genet. 2023, 54, 435–445. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ran, X.Q.; Pan, H.; Huang, S.H.; Liu, C.; Niu, X.; Li, S.; Wang, J.F. Copy number variations of MTHFSD gene across pig breeds and its association with litter size traits in Chinese indigenous Xiang pig. J. Anim. Physiol. Anim. Nutr. 2018, 102, 1320–1327. [Google Scholar] [CrossRef]
- Shabalin, A.A. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Argos, M.; Tong, L.; Pierce, B.L.; Rakibuz-Zaman, M.; Ahmed, A.; Islam, T.; Rahman, M.; Paul-Brutus, R.; Rahaman, R.; Roy, S.; et al. Genome-wide association study of smoking behaviours among Bangladeshi adults. J. Med. Genet. 2014, 51, 327–333. [Google Scholar] [CrossRef]
- Zheng, X.; Zhao, P.; Yang, K.; Ning, C.; Wang, H.; Zhou, L.; Liu, J. CNV analysis of Meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J. Anim. Sci. Biotechnol. 2020, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Revilla, M.; Puig-Oliveras, A.; Castello, A.; Crespo-Piazuelo, D.; Paludo, E.; Fernandez, A.I.; Ballester, M.; Folch, J.M. A global analysis of CNVs in swine using whole genome sequence data and association analysis with fatty acid composition and growth traits. PLoS ONE 2017, 12, e0177014. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.; Madsen, O.; Megens, H.J.; Frantz, L.A.; Bosse, M.; Bastiaansen, J.W.; Crooijmans, R.P.; Groenen, M.A. Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication. BMC Genom. 2013, 14, 449. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.; Madsen, O.; Megens, H.J.; Frantz, L.A.; Bosse, M.; Crooijmans, R.P.; Groenen, M.A. Copy number variation in the speciation of pigs: A possible prominent role for olfactory receptors. BMC Genom. 2015, 16, 330. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, C.; Yang, K.; Liu, J.; Zhang, Y.; Wang, Y.; Xu, X.; Michal, J.J.; Jiang, Z.; Liu, B. Genome Wide Distributions and Functional Characterization of Copy Number Variations between Chinese and Western Pigs. PLoS ONE 2015, 10, e0131522. [Google Scholar] [CrossRef] [PubMed]
- Keel, B.N.; Nonneman, D.J.; Lindholm-Perry, A.K.; Oliver, W.T.; Rohrer, G.A. A Survey of Copy Number Variation in the Porcine Genome Detected From Whole-Genome Sequence. Front. Genet. 2019, 10, 737. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, J.; Wang, H.; Kang, H.; Zhang, Q.; Liu, J.F. Improved Detection and Characterization of Copy Number Variations Among Diverse Pig Breeds by Array CGH. G3 Genes Genomes Genet. 2015, 5, 1253–1261. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, F.; Luo, L.; Wu, W.; Dai, J.; Zhong, J.; Lin, X.; Chai, C.; Ding, P.; Liang, L.; et al. Single-cell atlas of domestic pig cerebral cortex and hypothalamus. Sci. Bull. 2021, 66, 1448–1461. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, Z.; Sun, Y.; Wang, H.; Wang, C.; Yu, S.; Liu, J.; Zhang, Y.; Fan, B.; Li, K.; et al. Analysis of genome-wide copy number variations in Chinese indigenous and western pig breeds by 60 K SNP genotyping arrays. PLoS ONE 2014, 9, e106780. [Google Scholar] [CrossRef]
- Xie, J.; Li, R.; Li, S.; Ran, X.; Wang, J.; Jiang, J.; Zhao, P. Identification of Copy Number Variations in Xiang and Kele Pigs. PLoS ONE 2016, 11, e0148565. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, Y.; Liu, S.; Meng, Q. Genome-Wide Assessment Characteristics of Genes Overlapping Copy Number Variation Regions in Duroc Purebred Population. Front. Genet. 2021, 12, 753748. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, J.; Fu, W.; Jiang, L.; Ding, X.; Liu, J.F.; Zhang, Q. A genome-wide detection of copy number variations using SNP genotyping arrays in swine. BMC Genom. 2012, 13, 273. [Google Scholar] [CrossRef]
- Froesel, M.; Gacoin, M.; Clavagnier, S.; Hauser, M.; Goudard, Q.; Ben Hamed, S. Socially meaningful visual context either enhances or inhibits vocalisation processing in the macaque brain. Nat. Commun. 2022, 13, 4886. [Google Scholar] [CrossRef] [PubMed]
- Pinel, J.P.; Treit, D.; Rovner, L.I. Temporal Lobe Aggression in Rats. Science 1977, 197, 1088–1089. [Google Scholar] [CrossRef] [PubMed]
- Nakaji, P.; Meltzer, H.S.; Singel, S.A.; Alksne, J.F. Improvement of aggressive and antisocial behavior after resection of temporal lobe tumors. Pediatrics 2003, 112, e430. [Google Scholar] [CrossRef] [PubMed]
- Bellino, S.; Bozzatello, P.; Badino, C.; Mantelli, E.; Rocca, P. Efficacy of Polyunsaturated Fatty Acids (PUFAs) on Impulsive Behaviours and Aggressiveness in Psychiatric Disorders. Int. J. Mol. Sci. 2021, 22, 620. [Google Scholar] [CrossRef]
- Singer, L.T.; Min, M.O.; Momotaz, H.; Powers, G.; Minnes, S.; Bearer, C.F. Association of fatty acid ethyl esters in meconium with behavior during childhood. Drug Alcohol Depend. 2021, 218, 108437. [Google Scholar] [CrossRef]
- LA, H.-C. Effects of tryptophan on depression and aggression in STZ-D mice. Diabetes 1991, 40, 1598–1602. [Google Scholar] [CrossRef]
- Wolkers, C.P.; Serra, M.; Szawka, R.E.; Urbinati, E.C. The time course of aggressive behaviour in juvenile matrinxa Brycon amazonicus fed with dietary L-tryptophan supplementation. J. Fish Biol. 2014, 84, 45–57. [Google Scholar] [CrossRef]
- Lepage, O.; Larson, E.T.; Mayer, I.; Winberg, S. Serotonin, but not melatonin, plays a role in shaping dominant-subordinate relationships and aggression in rainbow trout. Horm. Behav. 2005, 48, 233–242. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, J.; Xu, Q.; Gao, S.; Liu, M.; Zhang, C.; Schinckel, A.P.; Zhou, B. Identification of functional single nucleotide polymorphisms in the porcine SLC6A4 gene associated with aggressive behavior in weaned pigs after mixing. J. Anim. Sci. 2022, 100, skac131. [Google Scholar] [CrossRef]
- Konig, J.; Seithel, A.; Gradhand, U.; Fromm, M.F. Pharmacogenomics of human OATP transporters. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2006, 372, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K. Organic anion transporting polypeptide (OATP)1B1 and OATP1B3 as important regulators of the pharmacokinetics of substrate drugs. Biol. Pharm. Bull. 2015, 38, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Takeda, F.; Oda, M.; Terasaki, M.; Kubota, A.; Asada, K.; Ichimura, Y.; Kojima, H.; Saitoh, H. Downregulated expression of organic anion transporting polypeptide (Oatp) 2b1 in the small intestine of rats with acute kidney injury. Drug Metab. Pharmacokinet. 2021, 40, 100411. [Google Scholar] [CrossRef]
- Lauterbach, E.C. Psychotropic drug effects on gene transcriptomics relevant to Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 38, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.D.; Gao, B.; Sidler Pfandler, M.A.; Zhang-Fu, W.; Leuthold, S.; Hagenbuch, B.; Folkers, G.; Meier, P.J.; Stieger, B. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am. J. Physiol. Cell Physiol. 2007, 292, C795–C806. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Ohashi, I.; Saito, T.; Nagai, J.; Ida, K.; Naruto, T.; Wada, T.; Kurosawa, K. Deletion of UBE3A in brothers with Angelman syndrome at the breakpoint with an inversion at 15q11.2. Am. J. Med. Genet. A 2014, 164A, 2873–2878. [Google Scholar] [CrossRef]
- Wang, K.S.; Liu, X.; Zhang, Q.; Zeng, M. ANAPC1 and SLCO3A1 are associated with nicotine dependence: Meta-analysis of genome-wide association studies. Drug Alcohol Depend. 2012, 124, 325–332. [Google Scholar] [CrossRef]
- Chen, C.; Liu, C.; Xiong, X.; Fang, S.; Yang, H.; Zhang, Z.; Ren, J.; Guo, Y.; Huang, L. Copy number variation in the MSRB3 gene enlarges porcine ear size through a mechanism involving miR-584-5p. Genet. Sel. Evol. 2018, 50, 72. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Yang, H.; Xu, Q.; Liu, M.; Chao, X.; Chen, J.; Zhou, B.; Liu, Y. Comprehensive Genome and Transcriptome Analysis Identifies SLCO3A1 Associated with Aggressive Behavior in Pigs. Biomolecules 2023, 13, 1381. https://doi.org/10.3390/biom13091381
Zhang C, Yang H, Xu Q, Liu M, Chao X, Chen J, Zhou B, Liu Y. Comprehensive Genome and Transcriptome Analysis Identifies SLCO3A1 Associated with Aggressive Behavior in Pigs. Biomolecules. 2023; 13(9):1381. https://doi.org/10.3390/biom13091381
Chicago/Turabian StyleZhang, Chunlei, Huan Yang, Qinglei Xu, Mingzheng Liu, Xiaohuan Chao, Jiahao Chen, Bo Zhou, and Yang Liu. 2023. "Comprehensive Genome and Transcriptome Analysis Identifies SLCO3A1 Associated with Aggressive Behavior in Pigs" Biomolecules 13, no. 9: 1381. https://doi.org/10.3390/biom13091381
APA StyleZhang, C., Yang, H., Xu, Q., Liu, M., Chao, X., Chen, J., Zhou, B., & Liu, Y. (2023). Comprehensive Genome and Transcriptome Analysis Identifies SLCO3A1 Associated with Aggressive Behavior in Pigs. Biomolecules, 13(9), 1381. https://doi.org/10.3390/biom13091381