


Are Anti-rhGAA Antibodies a Determinant of Treatment Outcome in Adults with Late-Onset Pompe Disease? A Systematic Review

 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Search Strategy
2.2. Selection Criteria
2.3. Selection Strategy
2.4. Data Extraction and Results
3. Results
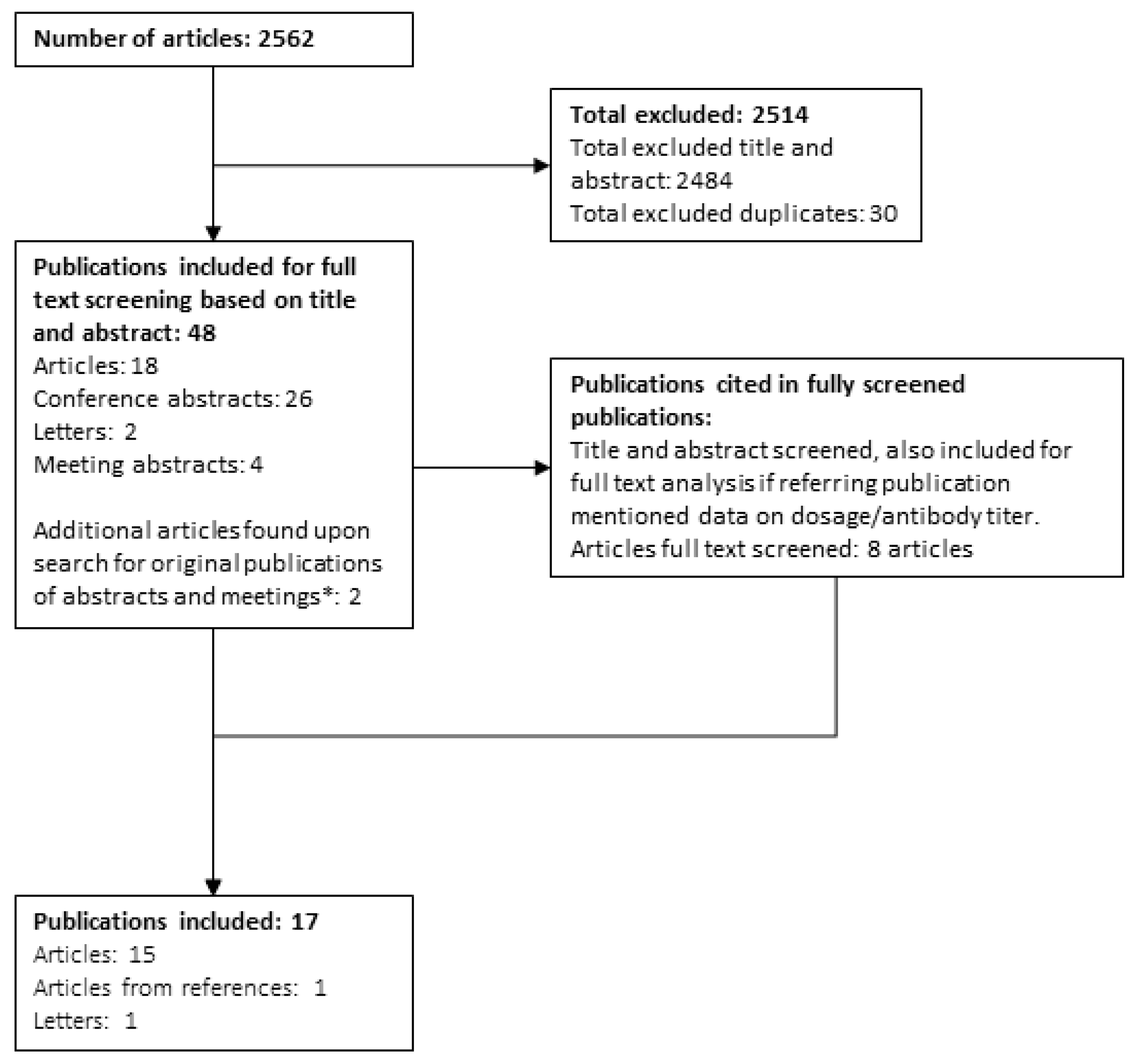
3.1. Study Selection
3.2. Study Characteristics
3.3. Definition of High Antibody Titre and High Sustained Antibody Titre (HSAT)
3.4. Data on a Group Level
3.5. Data on a Patient Level
3.6. Patients with a High Antibody Titre
3.7. Patients with a No-to-Intermediate Antibody Titre
3.8. IARs and Antibody Titres
3.9. IgE and IgM Antibodies
3.10. ERT Dosing and Antibody Formation
4. Discussion
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Kuperus, E.; Kruijshaar, M.E.; Wens, S.C.A.; de Vries, J.M.; Favejee, M.M.; van der Meijden, J.C.; Rizopoulos, D.; Brusse, E.; van Doorn, P.A.; van der Ploeg, A.T.; et al. Long-term benefit of enzyme replacement therapy in Pompe disease: A 5-year prospective study. Neurology 2017, 89, 2365–2373. [Google Scholar] [CrossRef]
- Regnery, C.; Kornblum, C.; Hanisch, F.; Vielhaber, S.; Strigl-Pill, N.; Grunert, B.; Müller-Felber, W.; Glocker, F.X.; Spranger, M.; Deschauer, M.; et al. 36 months observational clinical study of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy. J. Inherit. Metab. Dis. 2012, 35, 837–845. [Google Scholar] [CrossRef]
- Bergsma, A.J.; In ’t Groen, S.L.M.; van den Dorpel, J.J.A.; van den Hout, H.J.M.P.; van der Beek, N.A.M.E.; Schoser, B.; Toscano, A.; Musumeci, O.; Bembi, B.; Dardis, A.; et al. A genetic modifier of symptom onset in Pompe disease. EBioMedicine 2019, 43, 553–561. [Google Scholar] [CrossRef]
- Diaz-Manera, J.; Kishnani, P.S.; Kushlaf, H.; Ladha, S.; Mozaffar, T.; Straub, V.; Toscano, A.; van der Ploeg, A.T.; Berger, K.I.; Clemens, P.R.; et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): A phase 3, randomised, multicentre trial. Lancet Neurol. 2021, 20, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.; Roberts, M.; Byrne, B.J.; Sitaraman, S.; Jiang, H.; Laforêt, P.; Toscano, A.; Castelli, J.; Díaz-Manera, J.; Goldman, M.; et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): An international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol. 2021, 20, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- van der Ploeg, A.T.; Clemens, P.R.; Corzo, D.; Escolar, D.M.; Florence, J.; Groeneveld, G.J.; Herson, S.; Kishnani, P.S.; Laforet, P.; Lake, S.L.; et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 2010, 362, 1396–1406. [Google Scholar] [CrossRef]
- Bembi, B.; Pisa, F.E.; Confalonieri, M.; Ciana, G.; Fiumara, A.; Parini, R.; Rigoldi, M.; Moglia, A.; Costa, A.; Carlucci, A.; et al. Long-term observational, non-randomized study of enzyme replacement therapy in late-onset glycogenosis type II. J. Inherit. Metab. Dis. 2010, 33, 727–735. [Google Scholar] [CrossRef]
- Strothotte, S.; Strigl-Pill, N.; Grunert, B.; Kornblum, C.; Eger, K.; Wessig, C.; Deschauer, M.; Breunig, F.; Glocker, F.X.; Vielhaber, S.; et al. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J. Neurol. 2010, 257, 91–97. [Google Scholar] [CrossRef]
- Angelini, C.; Semplicini, C.; Ravaglia, S.; Bembi, B.; Servidei, S.; Pegoraro, E.; Moggio, M.; Filosto, M.; Sette, E.; Crescimanno, G.; et al. Observational clinical study in juvenile-adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J. Neurol. 2012, 259, 952–958. [Google Scholar] [CrossRef]
- de Vries, J.M.; van der Beek, N.A.; Hop, W.C.; Karstens, F.P.; Wokke, J.H.; de Visser, M.; van Engelen, B.G.; Kuks, J.B.; van der Kooi, A.J.; Notermans, N.C.; et al. Effect of enzyme therapy and prognostic factors in 69 adults with Pompe disease: An open-label single-center study. Orphanet. J. Rare Dis. 2012, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- van der Ploeg, A.T.; Barohn, R.; Carlson, L.; Charrow, J.; Clemens, P.R.; Hopkin, R.J.; Kishnani, P.S.; Laforet, P.; Morgan, C.; Nations, S.; et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol. Genet. Metab. 2012, 107, 456–461. [Google Scholar] [CrossRef]
- Gungor, D.; Kruijshaar, M.E.; Plug, I.; D’Agostino, R.B.; Hagemans, M.L.; van Doorn, P.A.; Reuser, A.J.; van der Ploeg, A.T. Impact of enzyme replacement therapy on survival in adults with Pompe disease: Results from a prospective international observational study. Orphanet. J. Rare Dis. 2013, 8, 49. [Google Scholar] [CrossRef]
- Anderson, L.J.; Henley, W.; Wyatt, K.M.; Nikolaou, V.; Waldek, S.; Hughes, D.A.; Lachmann, R.H.; Logan, S. Effectiveness of enzyme replacement therapy in adults with late-onset Pompe disease: Results from the NCS-LSD cohort study. J. Inherit. Metab. Dis. 2014, 37, 945–952. [Google Scholar] [CrossRef]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Harlaar, L.; Hogrel, J.-Y.; Perniconi, B.; Kruijshaar, M.E.; Rizopoulos, D.; Taouagh, N.; Canal, A.; Brusse, E.; van Doorn, P.A.; van der Ploeg, A.T.; et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology 2019, 93, e1756–e1767. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Goldenberg, P.C.; DeArmey, S.L.; Heller, J.; Benjamin, D.; Young, S.; Bali, D.; Smith, S.A.; Li, J.S.; Mandel, H.; et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 2010, 99, 26–33. [Google Scholar] [CrossRef] [PubMed]
- van Gelder, C.M.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Plug, I.; van der Ploeg, A.T.; Reuser, A.J.J. Enzyme therapy and immune response in relation to CRIM status: The Dutch experience in classic infantile Pompe disease. J. Inherit. Metab. Dis. 2015, 38, 305–314. [Google Scholar] [CrossRef]
- Banugaria, S.G.; Prater, S.N.; Ng, Y.K.; Kobori, J.A.; Finkel, R.S.; Ladda, R.L.; Chen, Y.T.; Rosenberg, A.S.; Kishnani, P.S. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: Lessons learned from infantile Pompe disease. Genet. Med. 2011, 13, 729–736. [Google Scholar] [CrossRef]
- Desai, A.K.; Li, C.; Rosenberg, A.S.; Kishnani, P.S. Immunological challenges and approaches to immunomodulation in Pompe disease: A literature review. Ann. Transl. Med. 2019, 7, 285. [Google Scholar] [CrossRef]
- Poelman, E.; Hoogeveen-Westerveld, M.; van den Hout, J.M.P.; Bredius, R.G.M.; Lankester, A.C.; Driessen, G.J.A.; Kamphuis, S.S.M.; Pijnappel, W.W.M.; van der Ploeg, A.T. Effects of immunomodulation in classic infantile Pompe patients with high antibody titers. Orphanet. J. Rare Dis. 2019, 14, 71. [Google Scholar] [CrossRef]
- Poelman, E.; van den Dorpel, J.; Hoogeveen-Westerveld, M.; van den Hout, J.; van der Giessen, L.J.; van der Beek, N.; Pijnappel, W.; van der Ploeg, A.T. Effects of higher and more frequent dosing of alglucosidase alfa and immunomodulation on long-term clinical outcome of classic infantile Pompe patients. J. Inherit. Metab. Dis. 2020, 43, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, N.J.; Messinger, Y.H.; Rosenberg, A.S.; Kishnani, P.S. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N. Engl. J. Med. 2009, 360, 194–195. [Google Scholar] [CrossRef] [PubMed]
- Messinger, Y.H.; Mendelsohn, N.J.; Rhead, W.; Dimmock, D.; Hershkovitz, E.; Champion, M.; Jones, S.A.; Olson, R.; White, A.; Wells, C.; et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet. Med. 2012, 14, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Banugaria, S.G.; Prater, S.N.; McGann, J.K.; Feldman, J.D.; Tannenbaum, J.A.; Bailey, C.; Gera, R.; Conway, R.L.; Viskochil, D.; Kobori, J.A.; et al. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: Lessons learned from Pompe disease. Genet. Med. 2013, 15, 123–131. [Google Scholar] [CrossRef]
- Kazi, Z.B.; Desai, A.K.; Troxler, R.B.; Kronn, D.; Packman, S.; Sabbadini, M.; Rizzo, W.B.; Scherer, K.; Abdul-Rahman, O.; Tanpaiboon, P.; et al. An immune tolerance approach using transient low-dose methotrexate in the ERT-naive setting of patients treated with a therapeutic protein: Experience in infantile-onset Pompe disease. Genet. Med. 2019, 21, 887–895. [Google Scholar] [CrossRef]
- Bronsema, K.J.; Bischoff, R.; Pijnappel, W.W.; van der Ploeg, A.T.; van de Merbel, N.C. Absolute quantification of the total and antidrug antibody-bound concentrations of recombinant human α-glucosidase in human plasma using protein G extraction and LC-MS/MS. Anal. Chem. 2015, 87, 4394–4401. [Google Scholar] [CrossRef]
- Wang, J.; Lozier, J.; Johnson, G.; Kirshner, S.; Verthelyi, D.; Pariser, A.; Shores, E.; Rosenberg, A. Neutralizing antibodies to therapeutic enzymes: Considerations for testing, prevention and treatment. Nat. Biotechnol. 2008, 26, 901–908. [Google Scholar] [CrossRef]
- de Vries, J.M.; van der Beek, N.A.; Kroos, M.A.; Ozkan, L.; van Doorn, P.A.; Richards, S.M.; Sung, C.C.; Brugma, J.D.; Zandbergen, A.A.; van der Ploeg, A.T.; et al. High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol. Genet. Metab. 2010, 101, 338–345. [Google Scholar] [CrossRef]
- Ditters, I.A.M.; Huidekoper, H.H.; Kruijshaar, M.E.; Rizopoulos, D.; Hahn, A.; Mongini, T.E.; Labarthe, F.; Tardieu, M.; Chabrol, B.; Brassier, A.; et al. Effect of alglucosidase alfa dosage on survival and walking ability in patients with classic infantile Pompe disease: A multicentre observational cohort study from the European Pompe Consortium. Lancet Child Adolesc. Health 2022, 6, 28–37. [Google Scholar] [CrossRef]
- Desai, A.K.; Kazi, Z.B.; Bali, D.S.; Kishnani, P.S. Characterization of immune response in Cross-Reactive Immunological Material (CRIM)-positive infantile Pompe disease patients treated with enzyme replacement therapy. Mol. Genet. Metab. Rep. 2019, 20, 100475. [Google Scholar] [CrossRef]
- van Kooten, H.A.; Ditters, I.A.M.; Hoogeveen-Westerveld, M.; Jacobs, E.H.; van den Hout, J.M.P.; van Doorn, P.A.; Pijnappel, W.; van der Ploeg, A.T.; van der Beek, N. Antibodies against recombinant human alpha-glucosidase do not seem to affect clinical outcome in childhood onset Pompe disease. Orphanet. J. Rare Dis. 2022, 17, 31. [Google Scholar] [CrossRef]
- Lipinski, S.E.; Lipinski, M.J.; Burnette, A.; Platts-Mills, T.A.; Wilson, W.G. Desensitization of an adult patient with Pompe disease and a history of anaphylaxis to alglucosidase alfa. Mol. Genet. Metab. 2009, 98, 319–321. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Papadimas, G.K.; Spengos, K.; Manta, P. Pretreatment antibodies against acid α-glycosidase in a patient with late-onset Pompe disease. Muscle Nerve 2012, 45, 452. [Google Scholar] [CrossRef]
- Patel, T.T.; Banugaria, S.G.; Case, L.E.; Wenninger, S.; Schoser, B.; Kishnani, P.S. The impact of antibodies in late-onset Pompe disease: A case series and literature review. Mol. Genet. Metab. 2012, 106, 301–309. [Google Scholar] [CrossRef]
- Lin, D.S.; Chiang, M.F.; Ho, C.S.; Hsiao, C.D.; Lin, C.Y.; Wang, N.L.; Chuang, C.K.; Huang, Y.W.; Chang, P.C.; Liu, H.L. Low-frequency enzyme replacement therapy in late-onset Pompe disease. Muscle Nerve 2013, 47, 612–613. [Google Scholar] [CrossRef]
- Schneider, I.; Deschauer, M.; Hanisch, F. Enzyme replacement therapy and antibodies in late-onset Pompe disease. Mol. Genet. Metab. Rep. 2014, 1, 232–234. [Google Scholar] [CrossRef]
- Case, L.E.; Bjartmar, C.; Morgan, C.; Casey, R.; Charrow, J.; Clancy, J.P.; Dasouki, M.; DeArmey, S.; Nedd, K.; Nevins, M.; et al. Safety and efficacy of alternative alglucosidase alfa regimens in Pompe disease. Neuromuscul. Disord. NMD 2015, 25, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Gallay, L.; Petiot, P.; Durieu, I.; Streichenberger, N.; Berard, F. SWORD: A simplified desensitization protocol for enzyme replacement therapy in adult Pompe disease. Neuromuscul. Disord. 2016, 26, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Masat, E.; Laforêt, P.; De Antonio, M.; Corre, G.; Perniconi, B.; Taouagh, N.; Mariampillai, K.; Amelin, D.; Mauhin, W.; Hogrel, J.Y.; et al. Long-term exposure to Myozyme results in a decrease of anti-drug antibodies in late-onset Pompe disease patients. Sci. Rep. 2016, 6, 36182. [Google Scholar] [CrossRef] [PubMed]
- de Vries, J.M.; Kuperus, E.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Wens, S.C.A.; Stok, M.; van der Beek, N.A.M.E.; Kruijshaar, M.E.; Rizopoulos, D.; van Doorn, P.A.; et al. Pompe disease in adulthood: Effects of antibody formation on enzyme replacement therapy. Genet. Med. 2017, 19, 90. [Google Scholar] [CrossRef]
- Filosto, M.; Cotti Piccinelli, S.; Ravaglia, S.; Servidei, S.; Moggio, M.; Musumeci, O.; Donati, M.A.; Pegoraro, E.; Di Muzio, A.; Maggi, L.; et al. Assessing the Role of Anti rh-GAA in Modulating Response to ERT in a Late-Onset Pompe Disease Cohort from the Italian GSDII Study Group. Adv. Ther. 2019, 36, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Simon, E.; Carrasco-Rozas, A.; Gallardo, E.; Gonzalez-Quereda, L.; Alonso-Perez, J.; Belmonte, I.; Pedrosa-Hernandez, I.; Montiel, E.; Segovia, S.; Suarez-Calvet, X.; et al. Study of the effect of anti-rhGAA antibodies at low and intermediate titers in late onset Pompe patients treated with ERT. Mol. Genet. Metab. 2019, 128, 129–136. [Google Scholar] [CrossRef]
- Alandy-Dy, J.; Wencel, M.; Hall, K.; Simon, J.; Chen, Y.; Valenti, E.; Yang, J.; Bali, D.; Lakatos, A.; Goyal, N.; et al. Variable clinical features and genotype-phenotype correlations in 18 patients with late-onset Pompe disease. Ann. Transl. Med. 2019, 7, 276. [Google Scholar] [CrossRef]
- Winkler, M.; von Landenberg, C.; Kuchenbecker, K.; Reimann, J.; Kornblum, C. Long-term effects of enzyme replacement therapy in an elderly cohort of late-onset Pompe disease. Neuromuscul. Disord. 2022, 32, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, P. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Chien, Y.H.; Benjamin, E.; Schoser, B.; Kishnani, P.; Mozaffar, T.; Díaz-Manera, J.; Johnson, F.; Das, S.S.; Nair, A.; Williams, H.; et al. Immunogenicity of cipaglucosidase alfa/miglustat versus alglucosidase alfa/placebo in late-onset Pompe disease (LOPD): A phase III, randomized study (PROPEL). Mol. Genet. Metab. 2022, 135, S30. [Google Scholar] [CrossRef]
- Brooks, D.A.; Kakavanos, R.; Hopwood, J.J. Significance of immune response to enzyme-replacement therapy for patients with a lysosomal storage disorder. Trends Mol. Med. 2003, 9, 450–453. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; McVie-Wylie, A.; Jiang, C.; Thurberg, B.L.; Raben, N.; Mattaliano, R.J.; Cheng, S.H. Carbohydrate-remodelled acid alpha-glucosidase with higher affinity for the cation-independent mannose 6-phosphate receptor demonstrates improved delivery to muscles of Pompe mice. Biochem. J. 2005, 389 Pt 3, 619–628. [Google Scholar] [CrossRef]
- Winkel, L.P.F.; Kamphoven, J.H.J.; van den Hout, H.J.M.P.; Severijnen, L.A.; van Doorn, P.A.; Reuser, A.J.J.; van der Ploeg, A.T. Morphological changes in muscle tissue of patients with infantile Pompe’s disease receiving enzyme replacement therapy. Muscle Nerve 2003, 27, 743–751. [Google Scholar] [CrossRef]
- Raben, N.; Danon, M.; Gilbert, A.L.; Dwivedi, S.; Collins, B.; Thurberg, B.L.; Mattaliano, R.J.; Nagaraju, K.; Plotz, P.H. Enzyme replacement therapy in the mouse model of Pompe disease. Mol. Genet. Metab. 2003, 80, 159–169. [Google Scholar] [CrossRef]
- Chien, Y.H.; Lee, N.C.; Thurberg, B.L.; Chiang, S.C.; Zhang, X.K.; Keutzer, J.; Huang, A.C.; Wu, M.H.; Huang, P.H.; Tsai, F.J.; et al. Pompe disease in infants: Improving the prognosis by newborn screening and early treatment. Pediatrics 2009, 124, e1116–e1125. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.F.; Hofbauer, C.J.; Horling, F.M.; Allacher, P.; Wolfsegger, M.J.; Oldenburg, J.; Male, C.; Windyga, J.; Tiede, A.; Schwarz, H.P.; et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood 2013, 121, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Hunley, T.E.; Corzo, D.; Dudek, M.; Kishnani, P.; Amalfitano, A.; Chen, Y.T.; Richards, S.M.; Phillips, J.A., 3rd; Fogo, A.B.; Tiller, G.E. Nephrotic syndrome complicating alpha-glucosidase replacement therapy for Pompe disease. Pediatrics 2004, 114, e532–e535. [Google Scholar] [CrossRef]
- Capanoglu, M.; Dibek Misirlioglu, E.; Azkur, D.; Vezir, E.; Guvenir, H.; Gunduz, M.; Toyran, M.; Civelek, E.; Kocabas, C.N. IgE-Mediated Hypersensitivity and Desensitisation with Recombinant Enzymes in Pompe Disease and Type I and Type VI Mucopolysaccharidosis. Int. Arch. Allergy Immunol. 2016, 169, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Kroos, M.A.; Pomponio, R.J.; Hagemans, M.L.; Keulemans, J.L.; Phipps, M.; DeRiso, M.; Palmer, R.E.; Ausems, M.G.; Van der Beek, N.A.; Van Diggelen, O.P.; et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007, 68, 110–115. [Google Scholar] [CrossRef]
- van der Wal, E.; Bergsma, A.J.; Pijnenburg, J.M.; van der Ploeg, A.T.; Pijnappel, W. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32-13T>G GAA Splicing Variant in Pompe Disease. Mol. Ther. Nucleic Acids 2017, 7, 90–100. [Google Scholar] [CrossRef]
- Berrier, K.L.; Kazi, Z.B.; Prater, S.N.; Bali, D.S.; Goldstein, J.; Stefanescu, M.C.; Rehder, C.W.; Botha, E.G.; Ellaway, C.; Bhattacharya, K.; et al. CRIM-negative infantile Pompe disease: Characterization of immune responses in patients treated with ERT monotherapy. Genet. Med. 2015, 17, 912–918. [Google Scholar] [CrossRef]
- Nayak, S.; Doerfler, P.A.; Porvasnik, S.L.; Cloutier, D.D.; Khanna, R.; Valenzano, K.J.; Herzog, R.W.; Byrne, B.J. Immune responses and hypercoagulation in ERT for Pompe disease are mutation and rhGAA dose dependent. PLoS ONE 2014, 9, e98336. [Google Scholar] [CrossRef]

{kind=link}
| Author (Year) | Title | Study Design | Sample Size * | Outcome(s) |
|---|---|---|---|---|
| Lipinski, S.E. (2009) [33] | Desensitization of an adult patient with Pompe disease and a history of anaphylaxis to alglucosidase alfa | Case report | One patient | IgE antibodies, IgG antibodies, intradermal skin testing results, reactions during infusion, management for reactions, complement and tryptase testing |
| De Vries, J.M. (2010) [29] | High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa | Case report | One patient (three reference patients) | Muscle strength (MRC, HHD), 6MWT, FVC, acid alfa-glucosidase activity in leucocytes and fibroblasts, inhibition of alglucosidase alfa uptake, IARs |
| Van Der Ploeg, A.T. (2010) [7] | A randomized study of alglucosidase alfa in late-onset Pompe’s disease | Randomised controlled trial | 60 patients | Distance walked (6-MWT), percentage of predicted FVC, quantitative muscle testing arm (% of predicted), maximum inspiratory pressure (% of predicted), maximum expiratory pressure (% of predicted), SF36 score, antibody titre |
| Papadopoulos (2012) [34] | Pre-treatment antibodies against acid a-glucosidase in a patient with Late-onset Pompe disease | Case report | One patient | Antibody titre |
| Patel, T.T. (2012) [35] | The impact of antibodies in late-onset Pompe disease: A case series and literature review | Retrospective, cohort study, literature review | 60 patients | Antibody titres, FVC in upright position, 6MWT, supine-to-stand, 4-stair climb, gait speed, MRC, SF36, physical component summary |
| Regnery, C (2012) [3] | 36 months observational clinical study of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy | Prospective, cohort study | 38 patients | Walton Gardner Medwin score, arm function test, FVC, 6MWT, MRC, timed tests (modified Gowers, 10 m walk, four-stair climb), SF-36, CK levels, antibody titres, side effects of ERT |
| Lin (2013) [36] | Low-frequency enzyme replacement therapy in late-onset Pompe disease | Case report | One patient | FVC, 6MWT, WGM scale |
| Schneider, I. (2014) [37] | Enzyme replacement therapy and antibodies in late-onset Pompe disease | Cohort study | 10 patients | Anti-rhGAA antibody titres, 6MWT, VC, non-invasive ventilation, wheelchair dependency |
| Case, L.E. (2015) [38] | Safety and efficacy of alternative alglucosidase alfa regimens in Pompe disease | Randomised, cohort study | Four patients | Treatment efficacy: GMFM-66, Pompe PEDI, MMT, ventilator use, PCS (MOSSF-36). Safety: AEs, vital signs, physical examinations, ECGs, haematology, chemistry, urinalysis and antibodies, anti-rhGAA antibody formation, inhibitory antibody formation, IgE, serum tryptase, complement activation, skin testing. |
| Gallay, L. (2016) [39] | SWORD: A simplified desensitization protocol for enzyme replacement therapy in adult Pompe disease | Case report | One patient | Successful ERT therapy **, IARs |
| Masat, E. (2016) [40] | Long-term exposure to Myozyme results in a decrease of anti-drug antibodies in late-onset Pompe disease patients | Prospective, cohort study | 28 patients | Anti-rhGAA antibody titres, anti-rhGAA IgG subclasses, IgM, IgE, non-neutralising IgG, PBMC reactivity to rhGAA, T cell reactivity to rhGAA, cytokine and chemokine secretion and upregulation, FVC, 6MWT |
| De Vries, J.M. (2017) [41] | Pompe disease in adulthood: effects of antibody formation on enzyme replacement therapy | Prospective, cohort study | 73 patients | Antibody titre, neutralising antibodies, IARs, effect of GAA variants on antibody formation, effect of antibodies on clinical outcome |
| Filosto, M. (2019) [42] | Assessing the Role of Anti rh-GAA in Modulating Response to ERT in a Late-Onset Pompe Disease Cohort from the Italian GSDII Study Group | Prospective, cohort study | 64 patients | Antibody titre, MRC sum score, 6MWT, GSGC score, FVC |
| Fernandez-Simon (2019) [43] | Study of the effect of anti-rhGAA antibodies at low and intermediate titers in late onset Pompe patients treated with ERT | Prospective, cohort study | 25 patients | Antibody titre, 6MWT, 10 m walk, timed up-and-go test, four-stair climb, MFM-20, MRC, HHD, ACTIVLIM, SF-36, iNQOL, muscle MRI (fat fraction) |
| Alandy-Dy J. (2019) [44] | Variable clinical features and genotype-phenotype correlations in 18 patients with late-onset Pompe disease | Cohort study | 18 patients *** | Ambulatory status (type of assistive device being used), 6MWT, MRC scale and dynamometry, respiratory status (type of ventilatory support required), pulmonary function, FVC upright and supine, MIP, SNIP |
| Diaz-Manera J. (2021) [5] | Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial | Randomised controlled trial | 49 patients **** | FVC, MIP, MEP, upper extremity muscle strength (HHD), QMFT, SF-12, PCS and MCS, gait, stairs, Gower’s Manoeuvre, chair composite score, GMFM-88, EQ-5D, Pediatric Quality of Life Inventory, anti-alglucosidase alfa antibodies and neutralising IgG antibodies, urinary hexose tetrasaccharide, serum creatine kinase, alanine aminotransferase, aspartate aminotransferase, treatment-emergent adverse events, infusion-associated reactions, Rasch-built-Pompe-specific activity scale, patient global impression of change, Pompe disease symptom scale, Pompe disease impact scale |
| Winkler, M. (2022) [45] | Long-term effects of enzyme replacement therapy in an elderly cohort of late-onset Pompe disease | Retrospective, cohort study | 6 patients | 6MWT, MRC sum score, QMFT, FVC sitting and supine, CK, anti-rhGAA IgG antibody titres, muscle biopsy |
| Author (Year) [Ref. #] | Patients (Patients with Antibody Titres Measured) | Sex (M/F) | Age (Mean/Median) | Age Onset (Mean/Median) | Age at Diagnosis (Mean/Median) | Dosing | Treatment Duration | % of Patients with High Antibody Titre | Antibody Titre Range | Neutralising Effects Antibodies | Definition High Antibody Titre HSAT | % of Patients with IARs | Treatment Response |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Regnery, C. (2012) [3] | 1–38 (38) | 20 M, 18 F | 53.11 years average (range 27–73) | 36.2 years ± 10.7 years | 41.6 years (mean) | 20 mg/kg/eow | 36 months | 5% | 1:600–1:819 200 | Yes, in one patient (2.6%) | Not defined | 18% (7 patients) | Stable disease or improvement (Walton Gardner Medwin Score, MRC score); improved, unchanged, declined disease (timed function tests), or stable disease (SF-36); significant mean decrease in CK (improvement). Overall conclusion: stabilises natural disease course. One female discontinued ERT as a result of declining in her neuromuscular state, based on development of high anti-GAA antibody titres (1:819,000). |
| Diaz-Manera, J. (2021) [5] | 1–49 *** (48) | 25 M, 24 F | 20-78 (mean 50.3 SD ± 13.7) years | 6.1–73.2 (mean ± SD 37.7 ± 15.7) years | 17.1–76.7 (mean ± SD 48.2 ± 14.6) years | 20 mg/kg/eow | 49 weeks | 33% (high and persistent) | 1:100–1:409,600 | Yes, in four patients (8%) | ≥12,800, sustained not defined | 33% (16 patients) | Changes from baseline to week 49 in least-squares mean (SE). Improvements in upright FVC (% predicted) (0.46% (SE 0.93)); 6MWT (2.19 m (10.40)); 6MWT (% predicted) (0.31 (1.62)); MIP (% predicted) (4.29 (2.19)); MEP (% predicted) (8.38 (2.96)); HHD, lower extremity (153.72 (48.54)); HHD, upper extremity (109.67 (38.98)); QMFT total score (1.89 (0.69)); SF-12 PCS score (1.60 (1.07)); and SF-12 MCS (0.76 (1.32)). Effect of antibody titres on treatment outcome not assessed. |
| Van Der Ploeg, A.T. (2010) [7] | 1–60 (59) | 34 M, 26 F | NR | 30.3 years (mean) ± 12.3, range 5.3–58.6 | NR | 20 mg/kg/eow | 78 weeks | 24% (supplementary data > 1:25,600) | 1:200–1:819,200 | 18 (31%) tested positive for inhibition of enzyme uptake (out of 59) | 1:25,600–819,200, Q4 titre, sustained not defined | 28% (17 patients) | Significantly increased 6MWT, % pred FVC. Increase in quantitative muscle testing, MIP, MEP, and SF-36 on a group level. No consistent association was found between the serum IgG antibody titre and the coprimary efficacy end points. |
| Patel, T.T. (2012) [35] | 1–60 (60) | NR | NR | NR | NR | 20 mg/kg/eow | NR | Periodically high titres unknown, 10% out of all 60 patients had high sustained antibody titres | NR | NR | ≥1:51,200 on two or more occasions at or beyond 6 months on ERT | At least 3% of the 60 patients had IARs (two out of three reported in detail) | Not reported on a group level. Effect of antibody titres on treatment outcome not assessed on a group level. |
| Case, L.E. (2015) [38] | 1–4 (4) | 2 M, 2 F | NR | NR | NR | 20 mg/kg/week or 40 mg/kg/eow | 52 weeks | 0% | NR | None | ≥1:51,200 on more than 2 occasions at or beyond 6 months on ERT | 25% (1 patient) | 2 patients’ motor skills improved on ERT, one patient’s motor skills maintained baseline status, one patient experienced a decline in respiratory status. Effect of antibody titres on treatment outcome not assessed. |
| Masat, E. (2016) [40] | 1–28 (24) | 13 M, 15 F | Average age 58.2 years, SD 2.4 years; median 57.5 years | NR | NR | 20 mg/kg/eow | 78 months on average (SD 28.8; median 84) | 21% (out of 24 patients with more than 3 years on ERT) | <1:6400–≥1:25,000 | Subset had elevated levels of anti-rhGAA IgG1 and IgG4 antibodies. No inhibitory activity was measured | Not defined | NR | Initial improvement or stabilisation of 6MWT and FVC measurements followed by a downward trend. No correlation was found between the evolution of FVC and 6MWT and the measurements of immune responses to rhGAA after long-term ERT |
| De Vries, J.M. (2017) [41] | 1–73 (73) | 37 M, 36 F | NR | 32.1 years (1.4–62.2) | 41.3 (1.4–72.7) years | 20 mg/kg/eow | 40 months (range 24–42 months) | 22% | 0 to 1:156,250 | A total of 16 patients with high titres. A 15–85% neutralising effect in 8 patients with maximum titre ≥1:156,250.A 0–15% neutralising effect in patients with maximum titre 1:31,250. Only one patient evidenced interference of high sustained anti-rhGAA antibodies with ERT | ≥1:31,250, sustained not defined | 18% (13 patients) | In all three titre groups (none-or-low, intermediate, high), patients with improving, stable, and declining clinical courses were present. The occurrence of IARs positively correlated with the height of the titre (p = 0.001). The total number of IARs that a patient experienced during the study period increased with higher antibody titres (ρ = 0.46, p < 0.001). |
| Filosto, M. (2019) [42] | 1–64 (64) | 29 M, 35 F | median 46.45 ± 17.64 years | NR | NR | 20 mg/kg/eow | 4–136 months ERT | 3.1% | 0 to 1:31,250 | NR | >1:31,250, sustained not defined | NR | Increased, stable, and decreased clinical functioning. No statistical significance was found in relating the T0–T1 delta differences and antibody titres, except for MRC sum score values in a subgroup of patients treated for <36 months, in which those with a null antibody titre showed a greater clinical improvement than patients with a positive titre. |
| Alandy-Dy, J. (2019) [44] | 1–18 * (NR, at least 2) | 14 M, 4 F | 22-74 (mean ± SD 53.72 ± 14.09/median 56.50) years | 5–58 (mean ± SD 29.56 ± 15.80/median 30.00) years | 11–65 (mean ± SD 43.61 ± 15.82/median 44.00) years | 20 mg/kg/eow ** | 2–11 years (up to 10.7 years after treatment initiation) | 11.1% | NR | NR | Not defined | 5.6% (1 patient) | Upright FVC; significant improvement in the patients’ decline after starting ERT (−0.17 per year (p < 0.0001)). Supine FVC; patients declined −0.55 per year after starting ERT (p = 0.047). Post ERT, MIP declined at 0.92 per year (p = 0.0169) and SNIP declined at 1.93 per year (p = 0.0226). Post ERT 6MWT was declining by 11.6 m per year (p < 0.0001). Effect of antibody titres on treatment outcome not assessed. |
| Winkler, M. (2022) [45] | 1–6 (6) | 3 M, 3 F | 59–80 (median 72.5) years | 40–64 (49) years | Diagnostic gap of 5–26 years (median 7.5) Age at beginning of ERT of 52–69 (median 63) years | 20 mg/kg/eow | 7–12 years (median 8.5) | 16.6% | 0 to 1:102,400 **** | NR | ≥1:31,250, sustained not defined | 0% (0 patients) | 6MWT improved in 4/6, and 2/6 each showed an improvement or stabilisation in muscle strength and FVC supine. FVC showed a decline in all patients in a sitting position, and QMFT worsened in 5/6. CK levels decreased in all patients. Antibody titres were not associated with treatment effects. Highest titres were present in best responders who were female, still ambulatory, and without ventilatory support at follow-up. |
| Author (Year) [Ref. #] | Patient No. | Sex | Age | Allele 1, Protein Change | Allele 2, Protein Change | Age at Onset | Age at Diagnosis | Age at Start of Treatment | Dosing | Treatment Duration | Highest Antibody Titre (Neutralising Effect) | Definition High Antibody Titre and High Sustained Antibody Titre (HSAT) | IARs | Treatment Response |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| De Vries, J.M. (2010) [29] | 1 # | M | NR | c. -32-13T>G p.[=,0] * | c.1548G>A * p.Trp516X | 29 years | 39 years | 50 years | 20 mg/kg/eow | 35 months | 1:800,000 (42% of alglucosidase alfa captured by antibodies, uptake inhibited) | Not defined | ≥1 | Poor; declining clinical parameters on ERT |
| Papadopoulos, C (2012) [34] | 1 | F | 37 | c.-32-13T>G p.[=,0] * | c.2066_2070dup p.(Ala691Serfs*7) * | NR | NR | 37 years | 20 mg/kg/eow *** | 24 weeks | 1:102,400 (no neutralising antibodies) | Not defined | None mentioned | NR |
| Patel, T.T. (2012) [35] | 1 | M | NR | c.-32-13T>G p.[=,0] * | c.2238G>A p.(Trp746*) * | 32 years | 28 years | 37 years | 20 mg/kg/eow | 255 weeks | 1:102,400 (NR) | ≥1:51,200 on two or more occasions at or beyond 6 months on ERT | No significant ones | Clinical decline |
| Patel, T.T. (2012) [35] | 2 | F | NR | c.-32-13T>G p.[=,0] * | c.1075G>A p.[(Gly359Arg) (Val358Aspfs*33)] * | 41 years | 49 years | 56 years | 20 mg/kg/eow | 202 weeks | 1:204,800 (NR) | ≥1 | Initial stable disease; clinical decline after 54 weeks | |
| Patel, T.T. (2012) [35] | 3 | F | NR | c.-32-13T>G p.[=,0] * | c.1076-22T>G p.? * | 25 years | 41 years | 57 years | 20 mg/kg/eow | 68 weeks | 1:819,200 (NR) | ≥1 | Initial improvement or stable disease until week 32; rapid clinical decline at 60 weeks | |
| De Vries, J.M. (2017) [41] | 1 # | NR | NR | NR | NR | NR | NR | NR | 20 mg/kg/eow | NR | 1:3,906,250 (strong neutralising effects) | ≥1:31,250, sustained not defined | NR | Clear interference of ERT by anti-rhGAA antibodies: decline in MRC and FVC scores |
| De Vries, J.M. (2017) [41] | 2 | NR | NR | NR | NR | NR | NR | NR | 20 mg/kg/eow | NR | ≥156,250 (strong neutralising effects) | NR | Unclear due to high, stable MRC scores and FVC | |
| De Vries, J.M. (2017) [41] | 3 | NR | NR | NR | NR | NR | NR | NR | 20 mg/kg/eow | NR | ≥156,250 (strong neutralising effects) | NR | Unclear due to high, stable MRC scores and FVC | |
| De Vries, J.M. (2017) [41] | 4 | NR | NR | NR | NR | NR | NR | NR | 20 mg/kg/eow | NR | ≥156,250 (strong neutralising effects) | NR | Unclear due to high, stable MRC scores and FVC | |
| De Vries, J.M. (2017) [41] | 5 | NR | NR | N | NR | NR | NR | NR | 20 mg/kg/eow | NR | ≥156,250 (temporary neutralising effects) | NR | No effect of antibodies | |
| De Vries, J.M. (2017) [41] | 6 | NR | NR | NR | NR | NR | NR | NR | 20 mg/kg/eow | NR | ≥156,250 (temporary neutralising effects) | NR | No effect of antibodies | |
| De Vries, J.M. (2017) [41] | 7 | NR | NR | NR | NR | NR | NR | NR | 20 mg/kg/eow | NR | ≥156,250 (temporary neutralising effects) | NR | No effect of antibodies | |
| De Vries, J.M. (2017) [41] | 8 | NR | NR | NR | NR | NR | NR | NR | 20 mg/kg/eow | NR | ≥156,250 (no neutralising effects) | NR | No effect of antibodies | |
| Fernandez-Simon, E. (2019) [43] | 15 | F | 65 | c.1781G>A p.(Arg594His) * | c. 1194+5G>A p.? * | NR | NR | 64 ** | 20 mg/kg/eow | NR | 1:51,200 (NR) | >1:31,200, sustained not defined | NR | Results of muscle function tests and spirometry remained stable after one year of progression in this patient, and there were not significant differences in mean thighs fat fraction |
| Alandy-Dy, J. (2019) [44] | 9 | F | 74 | c.-32-13T>G p.[=,0] * | c.2655_2656delCG p.(Val886Glufs*2) * | 30s | 62 | 63 | 20 mg/kg/eow *** | 11 years ** | 1:12,800 (NR) | Not defined | ≥1 | NR |
| Alandy-Dy, J. (2019) [44] | 10 | M | 70 | c.-32-13T>G p.[=,0] * | c.2655_2656delCG p.(Val886Glufs*2) * | 58 | 65 | 66 | 20 mg/kg/eow *** | 4 years ** | High antibody titre (NR) | Not defined | None | NR |
| Winkler, M. (2022) [45] | 6 | F | NR | c.-32-13T>G p.[=,0] * | NR | Not reported | Not reported | Not reported | 20 mg/kg/eow | Not reported | 1:102,400 (NR) **** | ≥1:31,250, sustained not defined | None | MRC improvement followed by stabilisation, QMFT improvement, 6MWT improvement, FVC sitting stabilisation followed by decline, FVC supine improvement |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ditters, I.A.M.; van Kooten, H.A.; van der Beek, N.A.M.E.; van der Ploeg, A.T.; Huidekoper, H.H.; van den Hout, J.M.P. Are Anti-rhGAA Antibodies a Determinant of Treatment Outcome in Adults with Late-Onset Pompe Disease? A Systematic Review. Biomolecules 2023, 13, 1414. https://doi.org/10.3390/biom13091414
Ditters IAM, van Kooten HA, van der Beek NAME, van der Ploeg AT, Huidekoper HH, van den Hout JMP. Are Anti-rhGAA Antibodies a Determinant of Treatment Outcome in Adults with Late-Onset Pompe Disease? A Systematic Review. Biomolecules. 2023; 13(9):1414. https://doi.org/10.3390/biom13091414
Chicago/Turabian StyleDitters, Imke A. M., Harmke A. van Kooten, Nadine A. M. E. van der Beek, Ans T. van der Ploeg, Hidde H. Huidekoper, and Johanna M. P. van den Hout. 2023. "Are Anti-rhGAA Antibodies a Determinant of Treatment Outcome in Adults with Late-Onset Pompe Disease? A Systematic Review" Biomolecules 13, no. 9: 1414. https://doi.org/10.3390/biom13091414
APA StyleDitters, I. A. M., van Kooten, H. A., van der Beek, N. A. M. E., van der Ploeg, A. T., Huidekoper, H. H., & van den Hout, J. M. P. (2023). Are Anti-rhGAA Antibodies a Determinant of Treatment Outcome in Adults with Late-Onset Pompe Disease? A Systematic Review. Biomolecules, 13(9), 1414. https://doi.org/10.3390/biom13091414