
Alternative Lengthening of Telomeres in Yeast: Old Questions and New Approaches
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
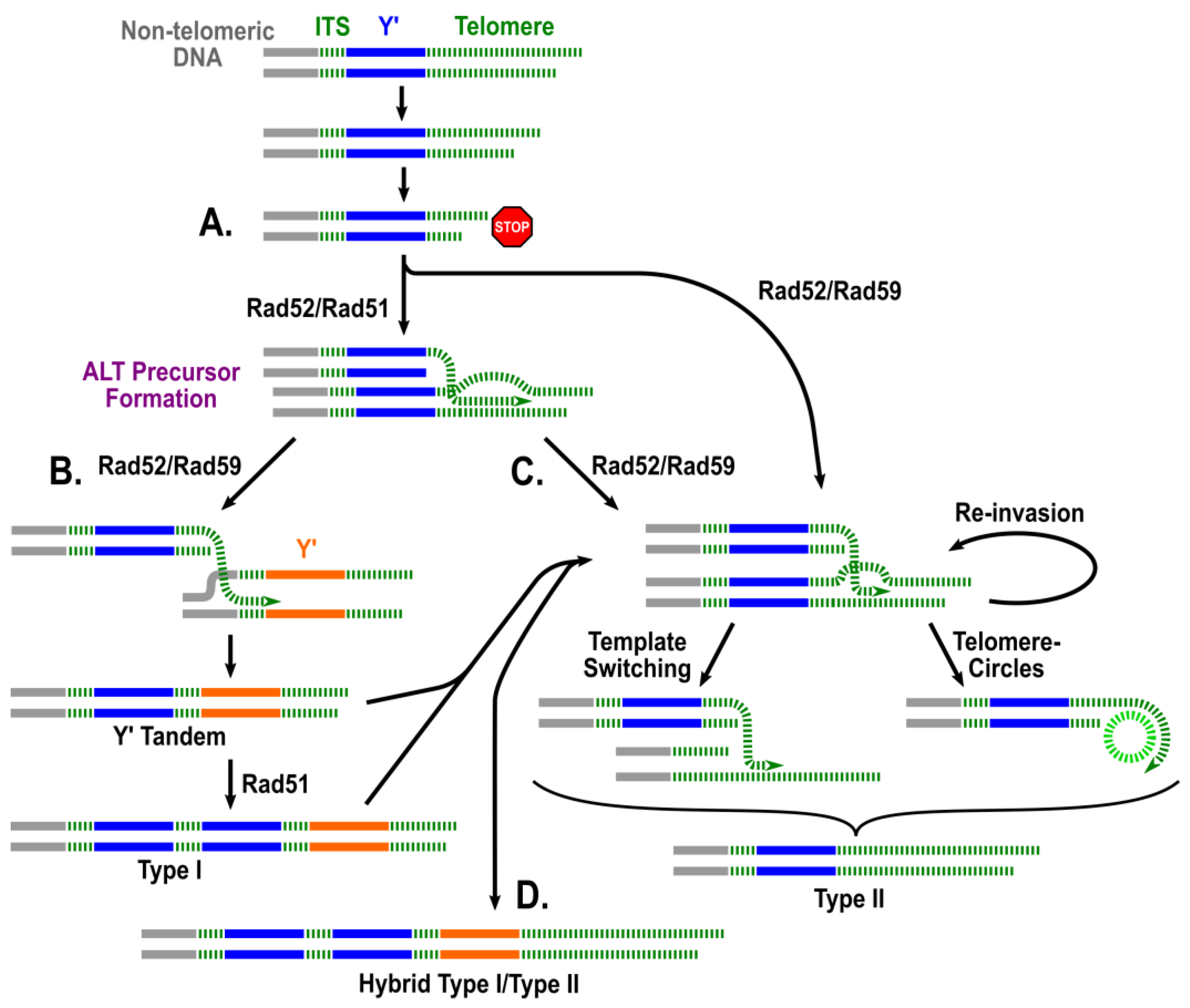
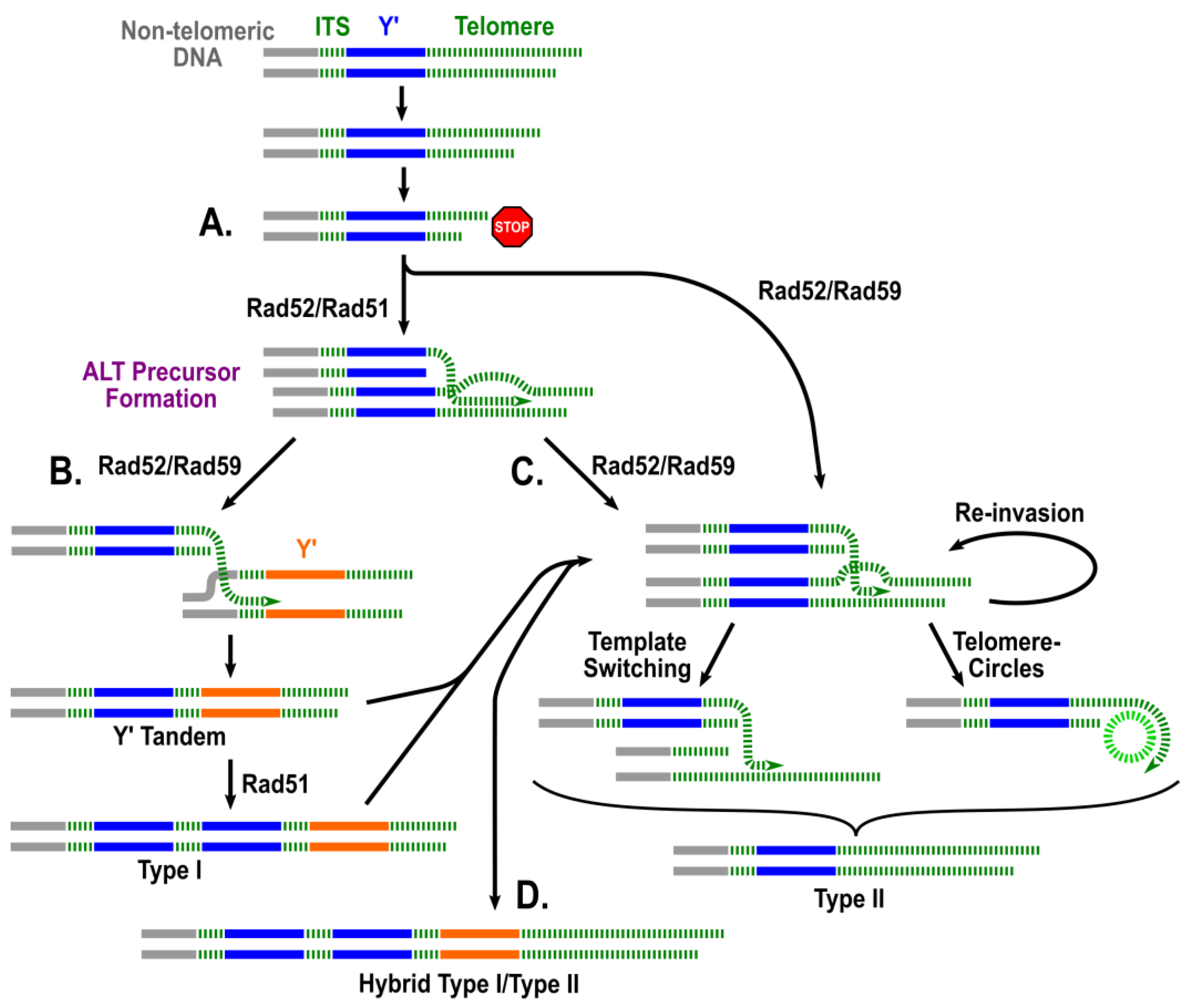
2. A Two-Pathway Model of ALT in Yeast: A Historical Perspective
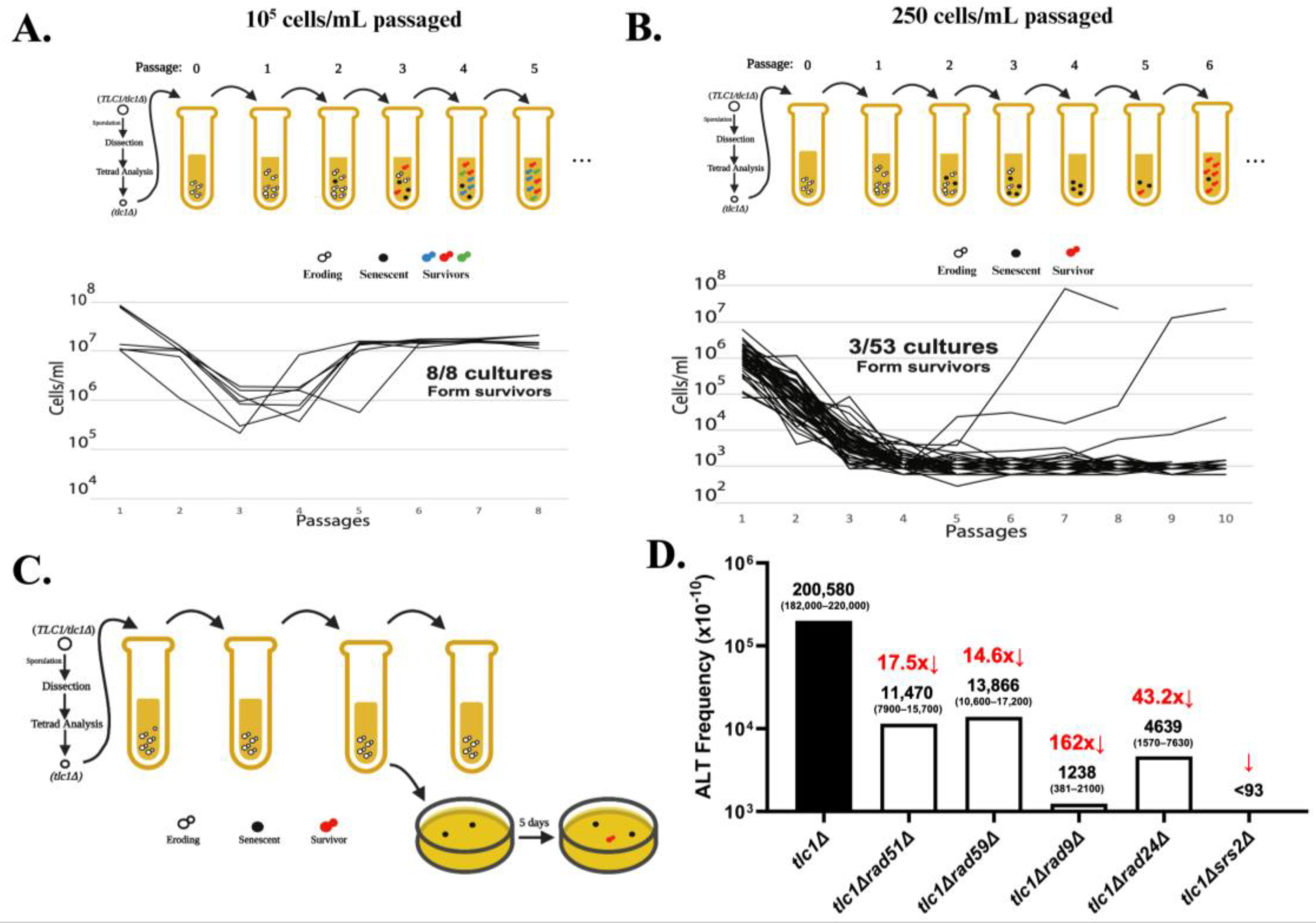
3. Investigation of the Process of ALT Establishment
4. Determining the Frequency and the Precise Structure of ALT Survivors
5. Determining the Mechanisms Governing the Development of ALT Survivors
6. Summary and Future Prospects
- The newly developed precise methods that allow for the calculation of the frequency of ALT and for the determination of the structure of individual chromosomal ends can be applied to determine the consequences of any genetic, structural, or environmental change on the frequency and telomere structures of ALT survivors. This will allow us to determine the mechanism of ALT through the identification of the specific roles that are played by various genetic factors in ALT.
- Combining computational modeling with ultra-long (for example, ONT) sequencing will allow for the tracking of structural changes of individual chromosomes during the entire process of ALT: from the beginning of telomere erosion through the formation of ALT survivors and their maintenance. Such analysis is expected to identify the discrete steps of ALT development and to characterize their structural parameters and genetic control. Specifically, it will be very important to establish connections between early ALT precursors that are formed at the beginning of the process and the final ALT survivors. It will be also important to identify the mechanisms and the main components conducive to the formation of ALT precursors including delineating relevant chromosome structures, determining the events that trigger ALT, and identifying the proteins and signaling pathways involved.
- It has been proposed that extrachromosomal telomeric circles may serve as templates for telomeric extension during the formation of Type II and possibly “hybrid” type survivors. To test this experimentally, telomeric circles could be introduced exogenously (by transformation) into telomerase deficient yeast. We would predict that if indeed extrachromosomal telomeric circles can serve as templates for telomere extension, and not merely represent a byproduct of survivor formation, providing the circles exogenously would (1) increase the frequency of survivor formation; (2) result in the earlier formation of survivors from precursors; (3) result in a greater pro-portion of survivors having a type II or hybrid end structure, and (4) possibly compensate for the absence of some proteins that were proposed to be specifically involved in Type II formation (e.g., Rad59, Sgs1, etc.).
- We believe that the results of research focused on ALT establishment in yeast will establish a roadmap to guide future studies of ALT in human models. For example, based on recent success in converting ALT-negative into ALT-positive cells (for example, via infection with the KSHV virus [62]), we envision that the methods and approaches developed in yeast will soon be able to be translated into detecting and characterizing the individual steps of ALT establishment in human cells. Specifically, the population genetics approach to determine ALT frequency can be used to test the current hypothesis that several genetically independent ALT pathways operate in humans (e.g., RAD52-dependent and -independent ALT pathways [63,64]) or, conversely, whether these pathways intermingle into a more complex unified pathway. Also, future studies of human ALT will greatly benefit from the molecular genetics and genomic approaches to the structural analysis of chromosome ends in ALT precursor cell populations, as well as in ALT outcomes. These approaches hold potential translational significance. They offer the ability to provide insights into specific molecular signatures of pre-senescent cells at different steps during ALT establishment, potentially serving as diagnostic biomarkers for the early detection of ALT-mediated aging/carcinogenic events. Additionally, we expect that the research in yeast will identify the primary proteins driving ALT, which would inform the study of human homologs of these yeast ALT genes and the development of new targeted, mechanism-based, anti-ALT therapeutics.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wellinger, R.J.; Zakian, V.A. Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: Beginning to end. Genetics 2012, 191, 1073–1105. [Google Scholar] [CrossRef]
- Doksani, Y.; de Lange, T. The role of double-strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb. Perspect. Biol. 2014, 6, a016576. [Google Scholar] [CrossRef]
- Teixeira, M.T. Saccharomyces cerevisiae as a Model to Study Replicative Senescence Triggered by Telomere Shortening. Front. Oncol. 2013, 3, 101. [Google Scholar] [CrossRef]
- Bonnell, E.; Pasquier, E.; Wellinger, R.J. Telomere Replication: Solving Multiple End Replication Problems. Front. Cell Dev. Biol. 2021, 9, 668171. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V. Telomere maintenance without telomerase. Oncogene 2002, 21, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.T.; Gilson, E. Telomere maintenance, function and evolution: The yeast paradigm. Chromosome Res. 2005, 13, 535–548. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef]
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef]
- Lundblad, V.; Szostak, J.W. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 1989, 57, 633–643. [Google Scholar] [CrossRef]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.A.; Reddel, R.R. Telomere maintenance and cancer—Look, no telomerase. Nat. Rev. Cancer 2002, 2, 879–884. [Google Scholar] [CrossRef]
- Teng, S.C.; Chang, J.; McCowan, B.; Zakian, V.A. Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol. Cell 2000, 6, 947–952. [Google Scholar] [CrossRef]
- Teng, S.C.; Zakian, V.A. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 8083–8093. [Google Scholar] [CrossRef] [PubMed]
- Lendvay, T.S.; Morris, D.K.; Sah, J.; Balasubramanian, B.; Lundblad, V. Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics 1996, 144, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ijpma, A.; Greider, C.W. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol. Cell. Biol. 2001, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Le, S.; Moore, J.K.; Haber, J.E.; Greider, C.W. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 1999, 152, 143–152. [Google Scholar] [CrossRef]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-strand break repair in the absence of RAD51 in yeast: A possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef]
- Malkova, A.; Naylor, M.L.; Yamaguchi, M.; Ira, G.; Haber, J.E. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol. Cell. Biol. 2005, 25, 933–944. [Google Scholar] [CrossRef]
- Bosco, G.; Haber, J.E. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 1998, 150, 1037–1047. [Google Scholar] [CrossRef]
- Davis, A.P.; Symington, L.S. RAD51-dependent break-induced replication in yeast. Mol. Cell. Biol. 2004, 24, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Lydeard, J.R.; Jain, S.; Yamaguchi, M.; Haber, J.E. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007, 448, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Harari, Y.; Kupiec, M. Mec1(ATR) is needed for extensive telomere elongation in response to ethanol in yeast. Curr. Genet. 2018, 64, 223–234. [Google Scholar] [CrossRef]
- Hu, Y.; Tang, H.B.; Liu, N.N.; Tong, X.J.; Dang, W.; Duan, Y.M.; Fu, X.H.; Zhang, Y.; Peng, J.; Meng, F.L.; et al. Telomerase-null survivor screening identifies novel telomere recombination regulators. PLoS Genet. 2013, 9, e1003208. [Google Scholar] [CrossRef]
- Huang, P.; Pryde, F.E.; Lester, D.; Maddison, R.L.; Borts, R.H.; Hickson, I.D.; Louis, E.J. SGS1 is required for telomere elongation in the absence of telomerase. Curr. Biol. 2001, 11, 125–129. [Google Scholar] [CrossRef]
- Johnson, F.B.; Marciniak, R.A.; McVey, M.; Stewart, S.A.; Hahn, W.C.; Guarente, L. The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J. 2001, 20, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Tseng, S.F.; Chang, S.H.; Lin, C.C.; Teng, S.C. Involvement of replicative polymerases, Tel1p, Mec1p, Cdc13p, and the Ku complex in telomere-telomere recombination. Mol. Cell. Biol. 2002, 22, 5679–5687. [Google Scholar] [CrossRef]
- Bertuch, A.A.; Lundblad, V. EXO1 contributes to telomere maintenance in both telomerase-proficient and telomerase-deficient Saccharomyces cerevisiae. Genetics 2004, 166, 1651–1659. [Google Scholar] [CrossRef]
- Beenstock, J.; Sicheri, F. The structural and functional workings of KEOPS. Nucleic Acids Res. 2021, 49, 10818–10834. [Google Scholar] [CrossRef]
- He, M.H.; Liu, J.C.; Lu, Y.S.; Wu, Z.J.; Liu, Y.Y.; Wu, Z.; Peng, J.; Zhou, J.Q. KEOPS complex promotes homologous recombination via DNA resection. Nucleic Acids Res. 2019, 47, 5684–5697. [Google Scholar] [CrossRef]
- Downey, M.; Houlsworth, R.; Maringele, L.; Rollie, A.; Brehme, M.; Galicia, S.; Guillard, S.; Partington, M.; Zubko, M.K.; Krogan, N.J.; et al. A genome-wide screen identifies the evolutionarily conserved KEOPS complex as a telomere regulator. Cell 2006, 124, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Rizki, A.; Lundblad, V. Defects in mismatch repair promote telomerase-independent proliferation. Nature 2001, 411, 713–716. [Google Scholar] [CrossRef] [PubMed]
- McEachern, M.J.; Haber, J.E. Break-induced replication and recombinational telomere elongation in yeast. Annu. Rev. Biochem. 2006, 75, 111–135. [Google Scholar] [CrossRef] [PubMed]
- McEachern, M.J.; Iyer, S. Short telomeres in yeast are highly recombinogenic. Mol. Cell 2001, 7, 695–704. [Google Scholar] [CrossRef]
- Natarajan, S.; Groff-Vindman, C.; McEachern, M.J. Factors influencing the recombinational expansion and spread of telomeric tandem arrays in Kluyveromyces lactis. Eukaryot. Cell 2003, 2, 1115–1127. [Google Scholar] [CrossRef]
- Natarajan, S.; McEachern, M.J. Recombinational telomere elongation promoted by DNA circles. Mol. Cell. Biol. 2002, 22, 4512–4521. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, P.; Dubarry, M.; Hardy, J.; Lisby, M.; Simon, M.N.; Geli, V. Telomeric C-circles localize at nuclear pore complexes in Saccharomyces cerevisiae. EMBO J. 2022, 41, e108736. [Google Scholar] [CrossRef]
- Larrivee, M.; Wellinger, R.J. Telomerase- and capping-independent yeast survivors with alternate telomere states. Nat. Cell Biol. 2006, 8, 741–747. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chang, H.H.; Wu, K.J.; Tseng, S.F.; Lin, C.C.; Lin, C.P.; Teng, S.C. Extrachromosomal telomeric circles contribute to Rad52-, Rad50-, and polymerase delta-mediated telomere-telomere recombination in Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 327–336. [Google Scholar] [CrossRef]
- Horowitz, H.; Haber, J.E. Identification of autonomously replicating circular subtelomeric Y’ elements in Saccharomyces cerevisiae. Mol. Cell. Biol. 1985, 5, 2369–2380. [Google Scholar] [CrossRef]
- Chang, M.; Dittmar, J.C.; Rothstein, R. Long telomeres are preferentially extended during recombination-mediated telomere maintenance. Nat. Struct. Mol. Biol. 2011, 18, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.H.; Duan, Y.M.; Liu, Y.T.; Cai, C.; Meng, F.L.; Zhou, J.Q. Telomere recombination preferentially occurs at short telomeres in telomerase-null type II survivors. PLoS ONE 2014, 9, e90644. [Google Scholar] [CrossRef] [PubMed]
- Churikov, D.; Charifi, F.; Simon, M.N.; Geli, V. Rad59-facilitated acquisition of Y’ elements by short telomeres delays the onset of senescence. PLoS Genet. 2014, 10, e1004736. [Google Scholar] [CrossRef]
- Abdallah, P.; Luciano, P.; Runge, K.W.; Lisby, M.; Geli, V.; Gilson, E.; Teixeira, M.T. A two-step model for senescence triggered by a single critically short telomere. Nat. Cell Biol. 2009, 11, 988–993. [Google Scholar] [CrossRef]
- Coutelier, H.; Xu, Z.; Morisse, M.C.; Lhuillier-Akakpo, M.; Pelet, S.; Charvin, G.; Dubrana, K.; Teixeira, M.T. Adaptation to DNA damage checkpoint in senescent telomerase-negative cells promotes genome instability. Genes Dev. 2018, 32, 1499–1513. [Google Scholar] [CrossRef]
- Fallet, E.; Jolivet, P.; Soudet, J.; Lisby, M.; Gilson, E.; Teixeira, M.T. Length-dependent processing of telomeres in the absence of telomerase. Nucleic Acids Res. 2014, 42, 3648–3665. [Google Scholar] [CrossRef] [PubMed]
- Khadaroo, B.; Teixeira, M.T.; Luciano, P.; Eckert-Boulet, N.; Germann, S.M.; Simon, M.N.; Gallina, I.; Abdallah, P.; Gilson, E.; Geli, V.; et al. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat. Cell Biol. 2009, 11, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, S.; Glowczewski, L.; Berman, J. MEC3, MEC1, and DDC2 are essential components of a telomere checkpoint pathway required for cell cycle arrest during senescence in Saccharomyces cerevisiae. Mol. Biol. Cell 2002, 13, 2626–2638. [Google Scholar] [CrossRef]
- Ijpma, A.S.; Greider, C.W. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 987–1001. [Google Scholar] [CrossRef]
- Grandin, N.; Charbonneau, M. Control of the yeast telomeric senescence survival pathways of recombination by the Mec1 and Mec3 DNA damage sensors and RPA. Nucleic Acids Res. 2007, 35, 822–838. [Google Scholar] [CrossRef]
- Xu, Z.; Fallet, E.; Paoletti, C.; Fehrmann, S.; Charvin, G.; Teixeira, M.T. Two routes to senescence revealed by real-time analysis of telomerase-negative single lineages. Nat. Commun. 2015, 6, 7680. [Google Scholar] [CrossRef] [PubMed]
- Kockler, Z.W.; Comeron, J.M.; Malkova, A. A unified alternative telomere-lengthening pathway in yeast survivor cells. Mol. Cell 2021, 81, 1816–1829.e5. [Google Scholar] [CrossRef] [PubMed]
- Elango, R.; Sheng, Z.; Jackson, J.; DeCata, J.; Ibrahim, Y.; Pham, N.T.; Liang, D.H.; Sakofsky, C.J.; Vindigni, A.; Lobachev, K.S.; et al. Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat. Commun. 2017, 8, 1790. [Google Scholar] [CrossRef]
- Niu, H.; Klein, H.L. Multifunctional roles of Saccharomyces cerevisiae Srs2 protein in replication, recombination and repair. FEMS Yeast Res. 2017, 17, fow111. [Google Scholar] [CrossRef] [PubMed]
- Marcand, S.; Brevet, V.; Gilson, E. Progressive cis-inhibition of telomerase upon telomere elongation. EMBO J. 1999, 18, 3509–3519. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.S.; Gottschling, D.E. TLC1: Template RNA component of Saccharomyces cerevisiae telomerase. Science 1994, 266, 404–409. [Google Scholar] [CrossRef]
- Ira, G.; Haber, J.E. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol. Cell. Biol. 2002, 22, 6384–6392. [Google Scholar] [CrossRef]
- Tsaponina, O.; Haber, J.E. Frequent Interchromosomal Template Switches during Gene Conversion in S. cerevisiae. Mol. Cell 2014, 55, 615–625. [Google Scholar] [CrossRef]
- Anand, R.P.; Tsaponina, O.; Greenwell, P.W.; Lee, C.S.; Du, W.; Petes, T.D.; Haber, J.E. Chromosome rearrangements via template switching between diverged repeated sequences. Gene Dev. 2014, 28, 2394–2406. [Google Scholar] [CrossRef]
- Sugawara, N.; Ira, G.; Haber, J.E. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol. Cell. Biol. 2000, 20, 5300–5309. [Google Scholar] [CrossRef]
- Shampay, J.; Szostak, J.W.; Blackburn, E.H. DNA sequences of telomeres maintained in yeast. Nature 1984, 310, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.P.; Marzec, P.; Idilli, A.I.; Sarek, G.; Vancevska, A.; Bower, M.; Farrell, P.J.; Ojala, P.M.; Feldhahn, N.; Boulton, S.J. Oncogenic herpesvirus KSHV triggers hallmarks of alternative lengthening of telomeres. Nat. Commun. 2021, 12, 512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Epum, E.A.; Haber, J.E. DNA replication: The recombination connection. Trends Cell Biol. 2022, 32, 45–57. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musmaker, K.; Wells, J.; Tsai, M.-C.; Comeron, J.M.; Malkova, A. Alternative Lengthening of Telomeres in Yeast: Old Questions and New Approaches. Biomolecules 2024, 14, 113. https://doi.org/10.3390/biom14010113
Musmaker K, Wells J, Tsai M-C, Comeron JM, Malkova A. Alternative Lengthening of Telomeres in Yeast: Old Questions and New Approaches. Biomolecules. 2024; 14(1):113. https://doi.org/10.3390/biom14010113
Chicago/Turabian StyleMusmaker, Kendra, Jacob Wells, Meng-Chia Tsai, Josep M. Comeron, and Anna Malkova. 2024. "Alternative Lengthening of Telomeres in Yeast: Old Questions and New Approaches" Biomolecules 14, no. 1: 113. https://doi.org/10.3390/biom14010113
APA StyleMusmaker, K., Wells, J., Tsai, M.-C., Comeron, J. M., & Malkova, A. (2024). Alternative Lengthening of Telomeres in Yeast: Old Questions and New Approaches. Biomolecules, 14(1), 113. https://doi.org/10.3390/biom14010113