
Structural Characterization and Molecular Dynamics Study of the REPI Fusion Protein from Papaver somniferum L.


Abstract
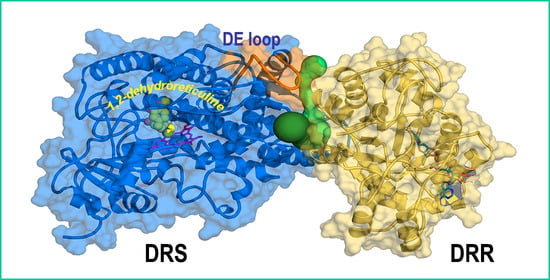
:
1. Introduction
2. Materials and Methods
2.1. Structures of Proteins, Cofactors, and Ligands
2.2. Structures of Protein–Cofactor–Ligand Complexes
2.3. All-Atom Molecular Dynamics (MD) Calculations
2.4. Structural Alignment, Assignment of Secondary Structure, Detection of Channels, and Molecular Graphics
2.5. Calculation of Poisson–Boltzmann (PB) Electrostatic Potentials (EPs)
3. Results
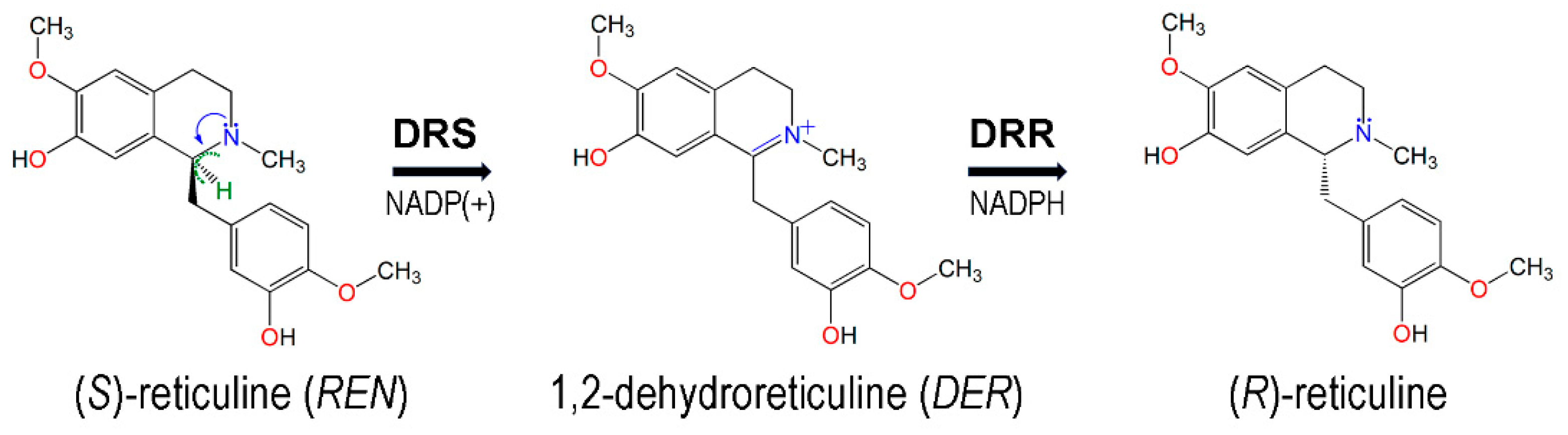
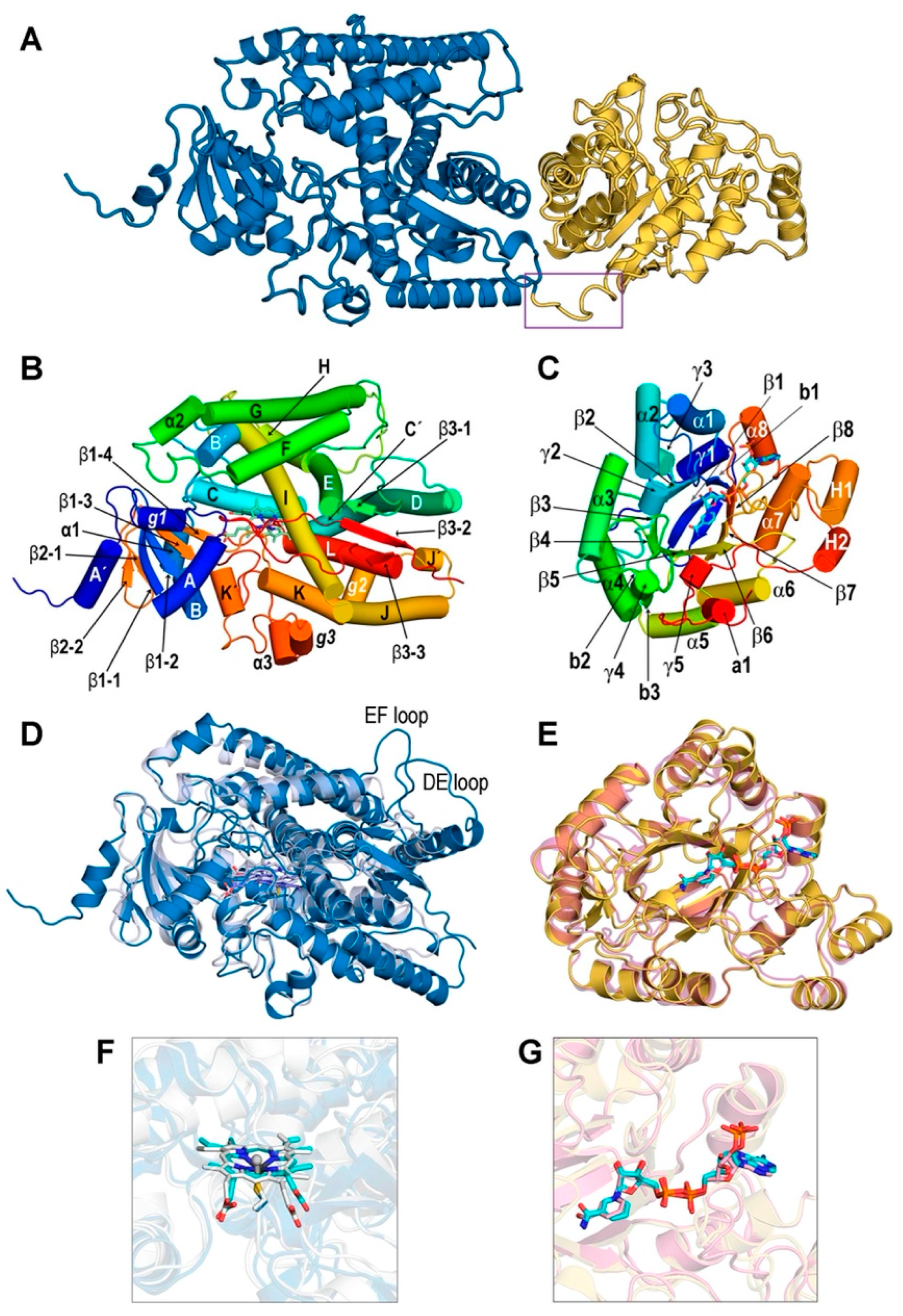
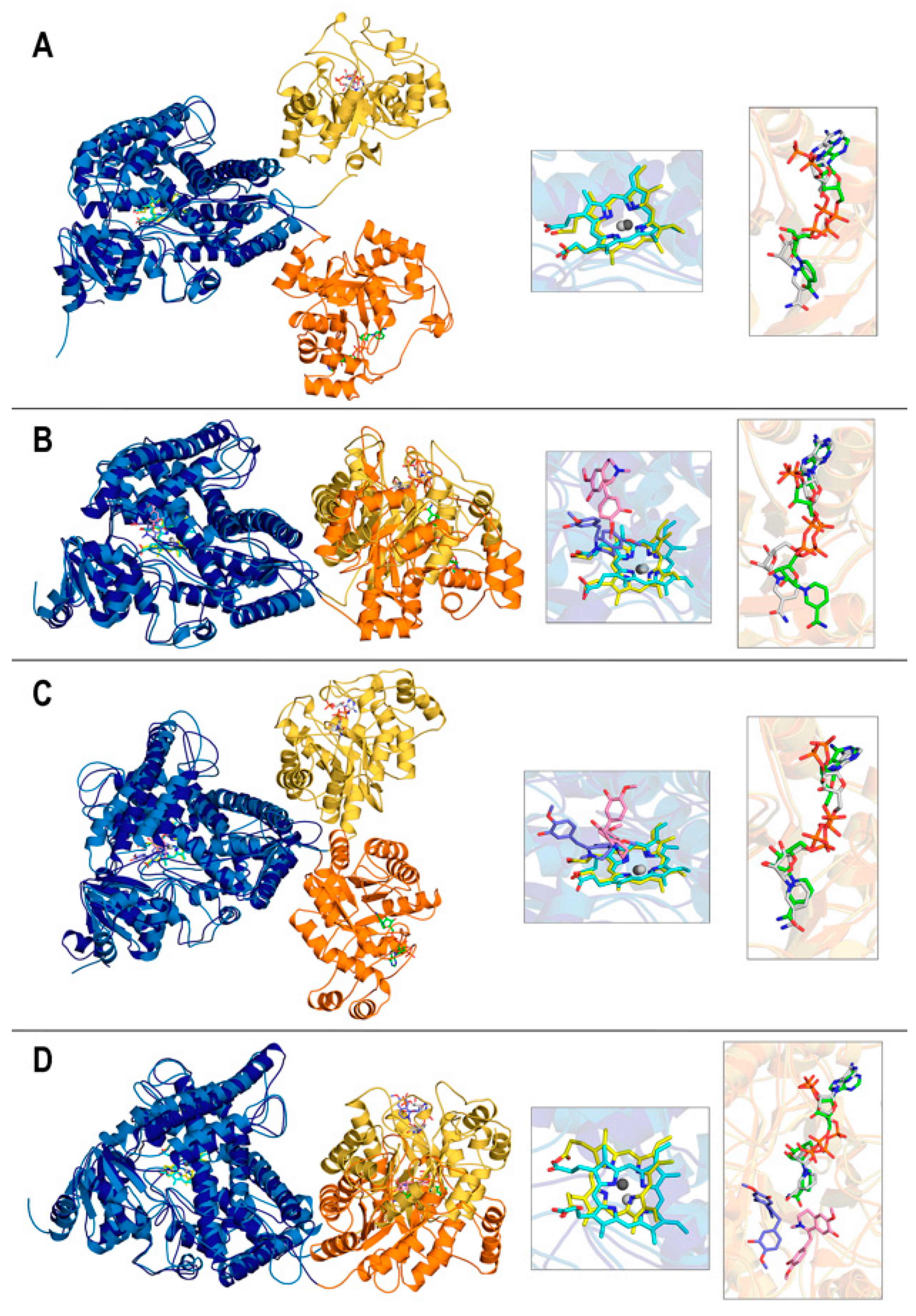
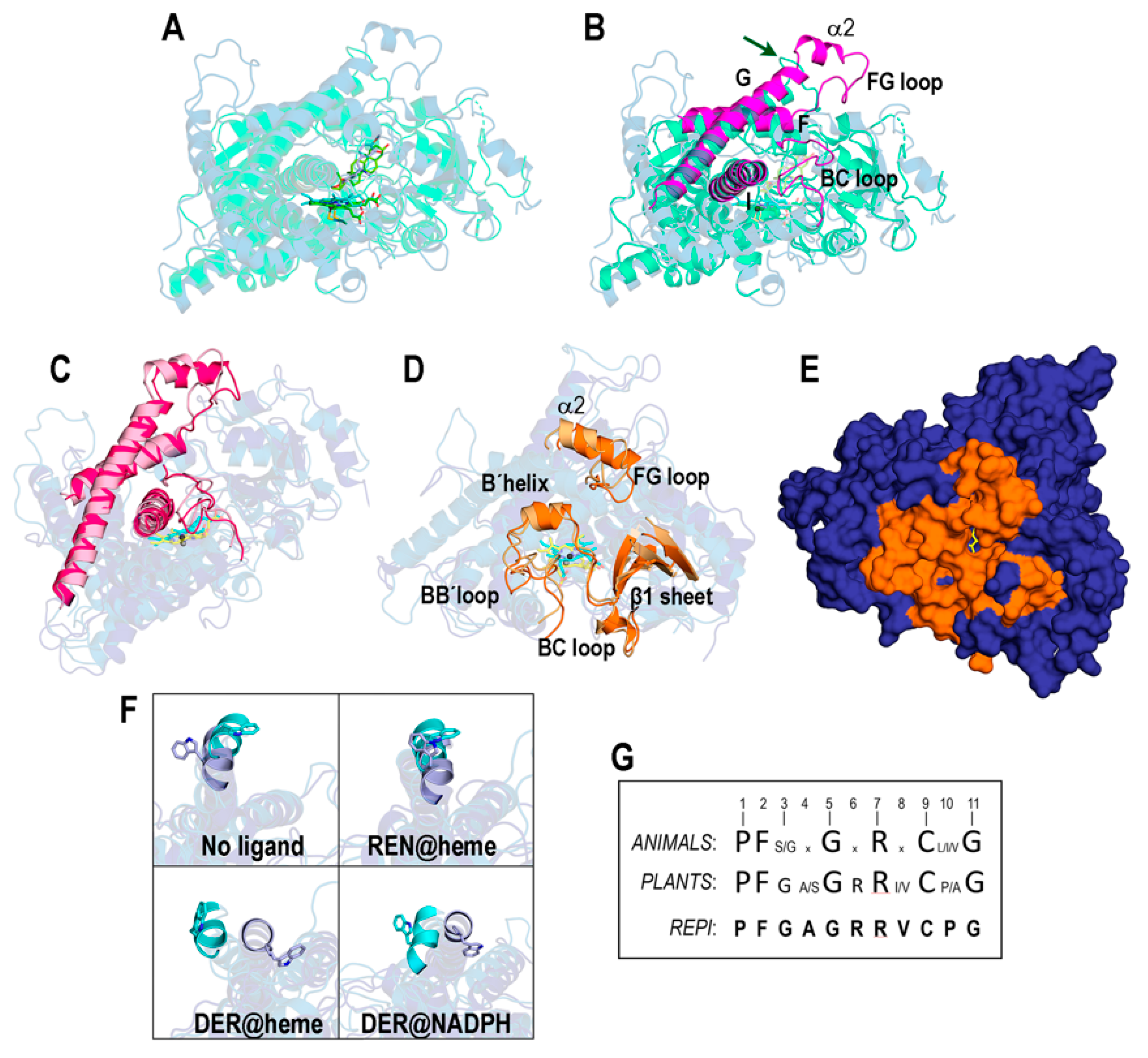
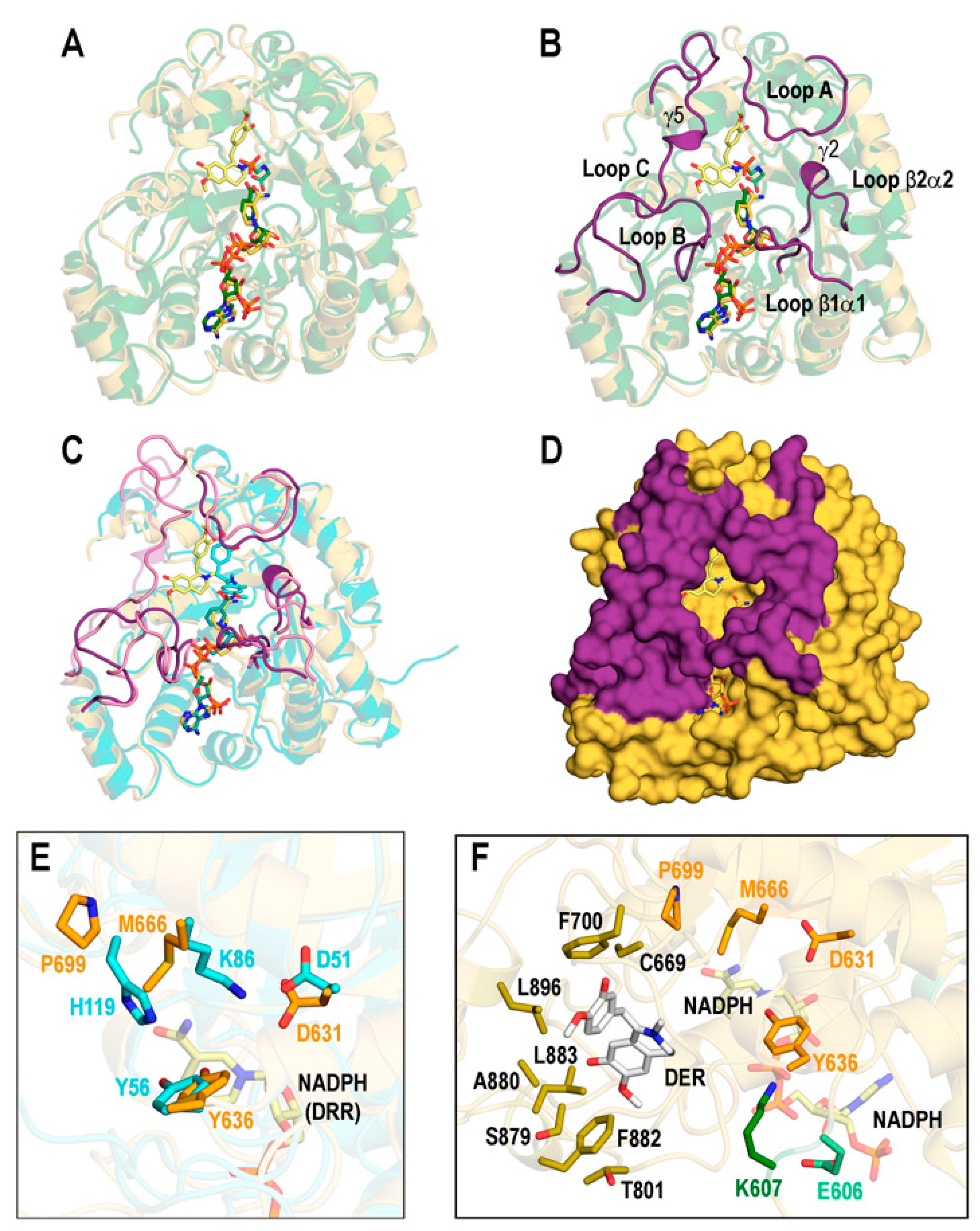
3.1. Structures of the REPI-Cofactor-Ligand Complexes
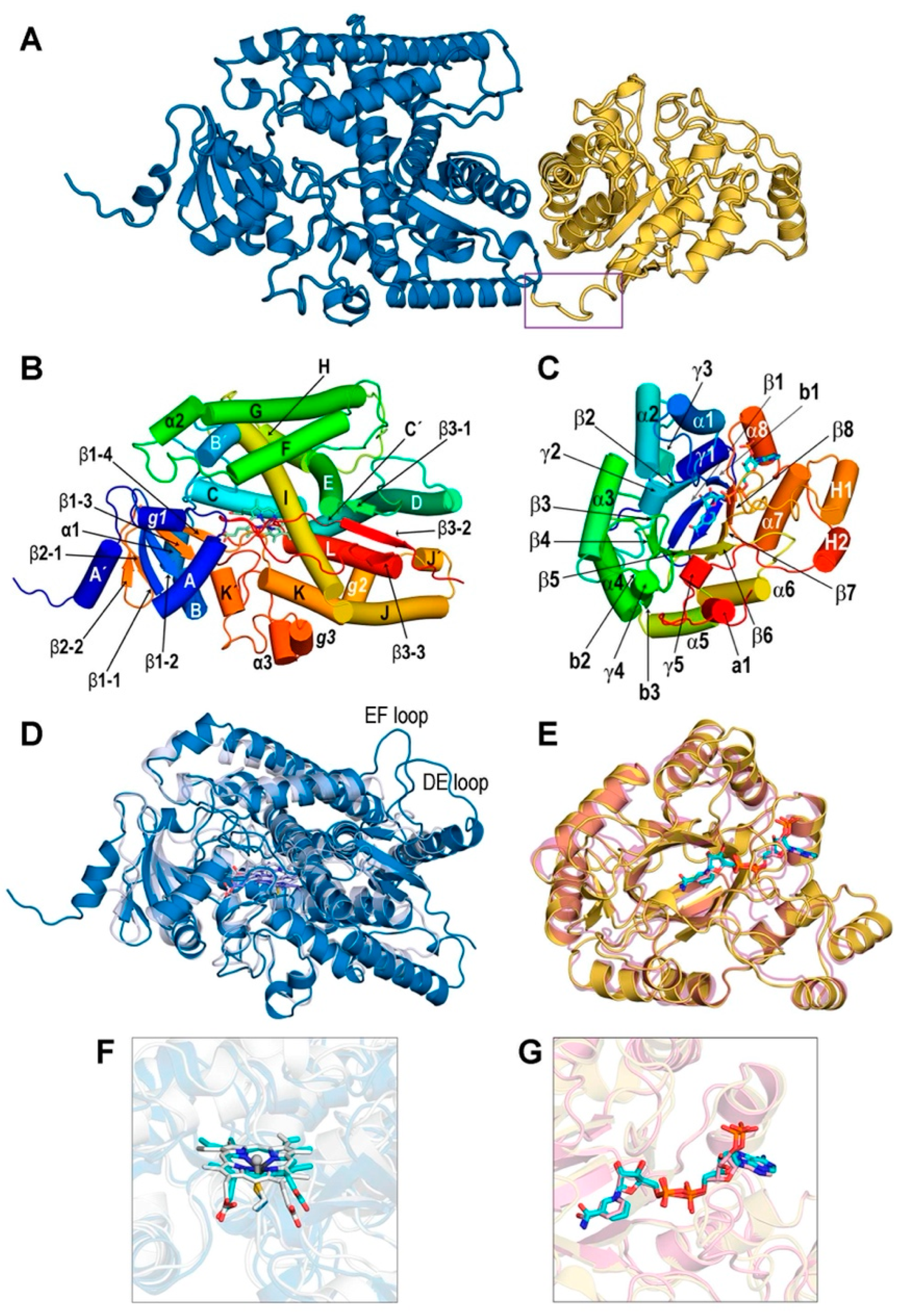
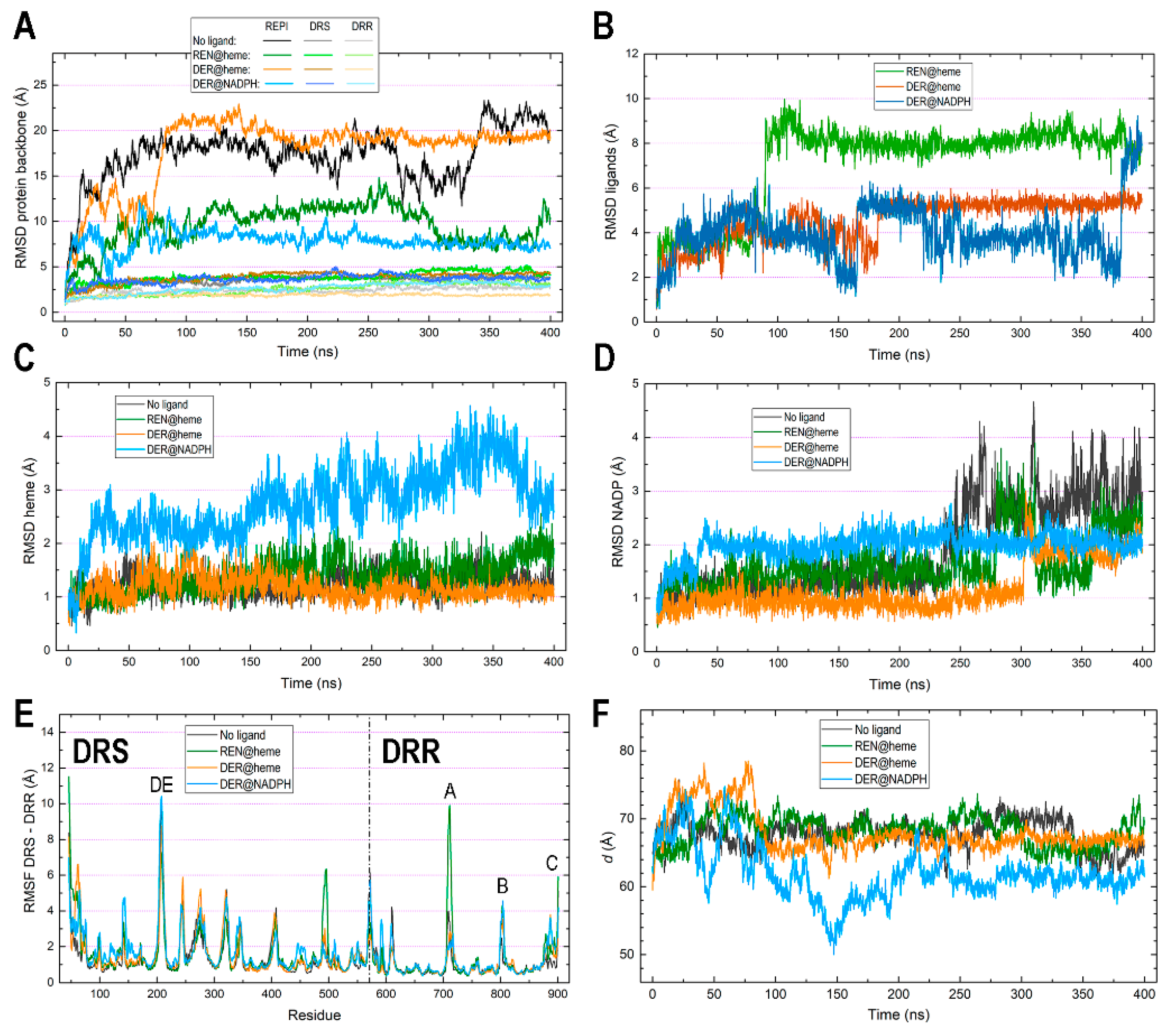
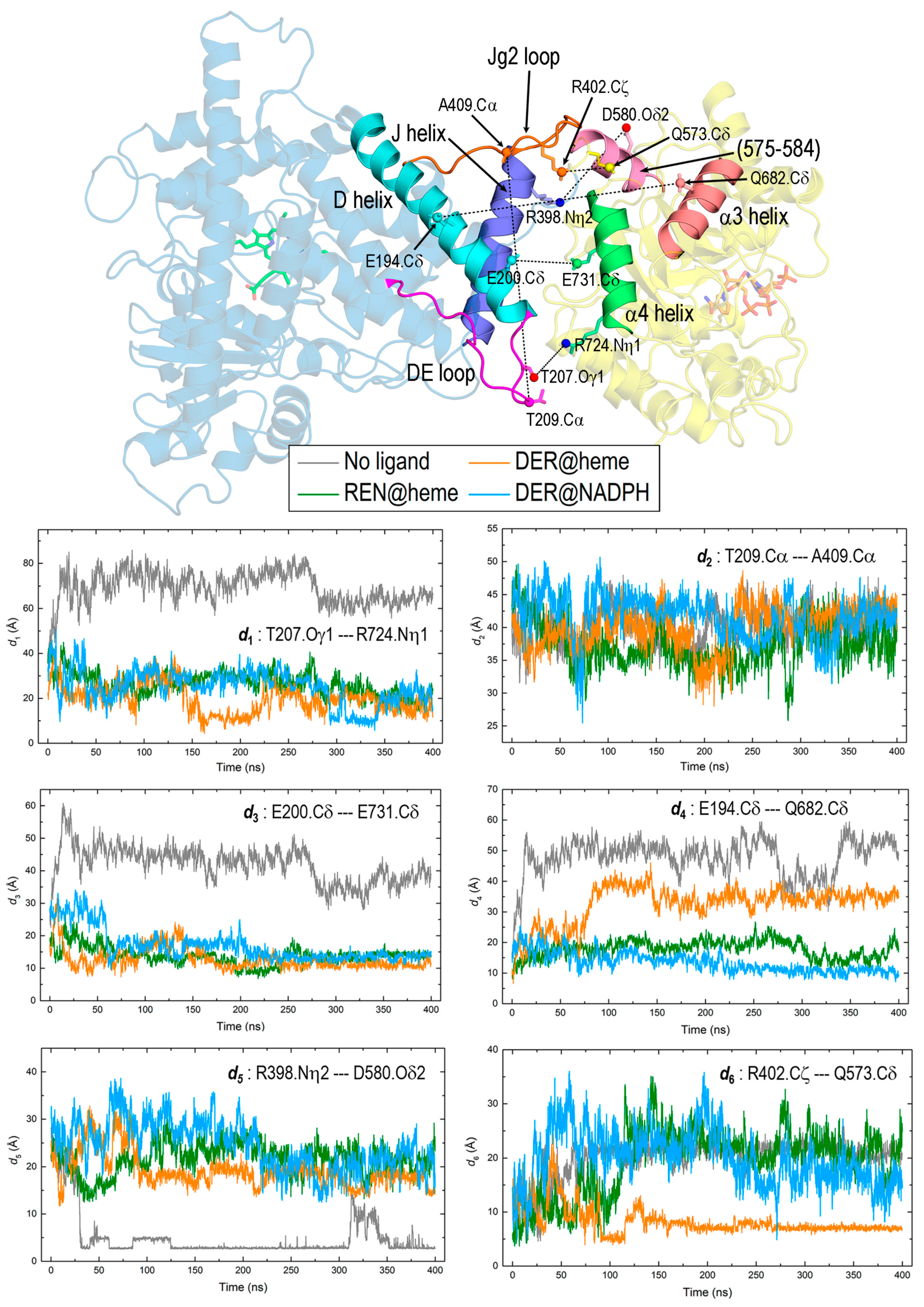
3.2. General Features of MD Simulations and Final Structures of REPI Complexes
3.3. Catalytic Sites of REPI
3.3.1. REPI-DRS

3.3.2. REPI-DRR
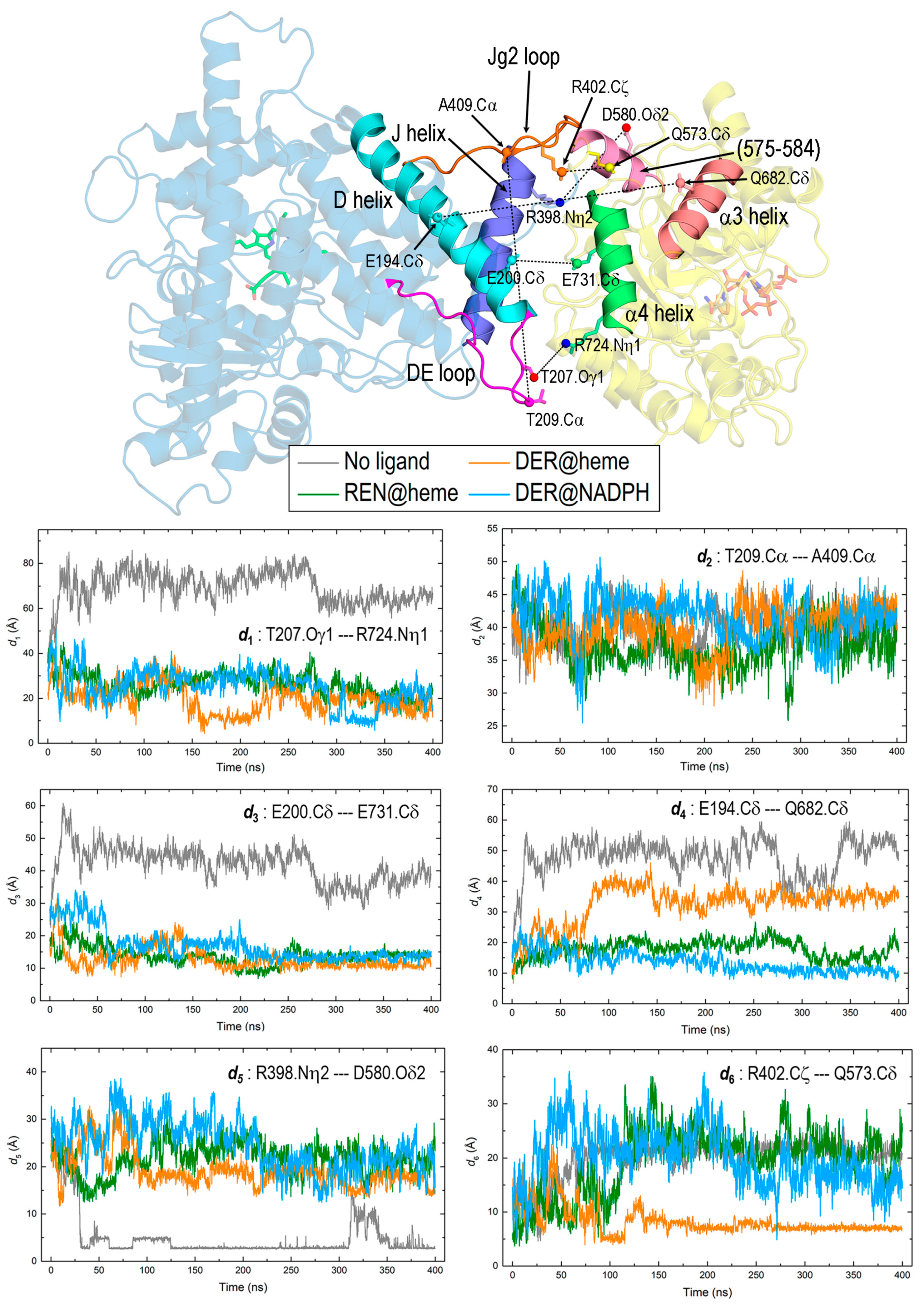
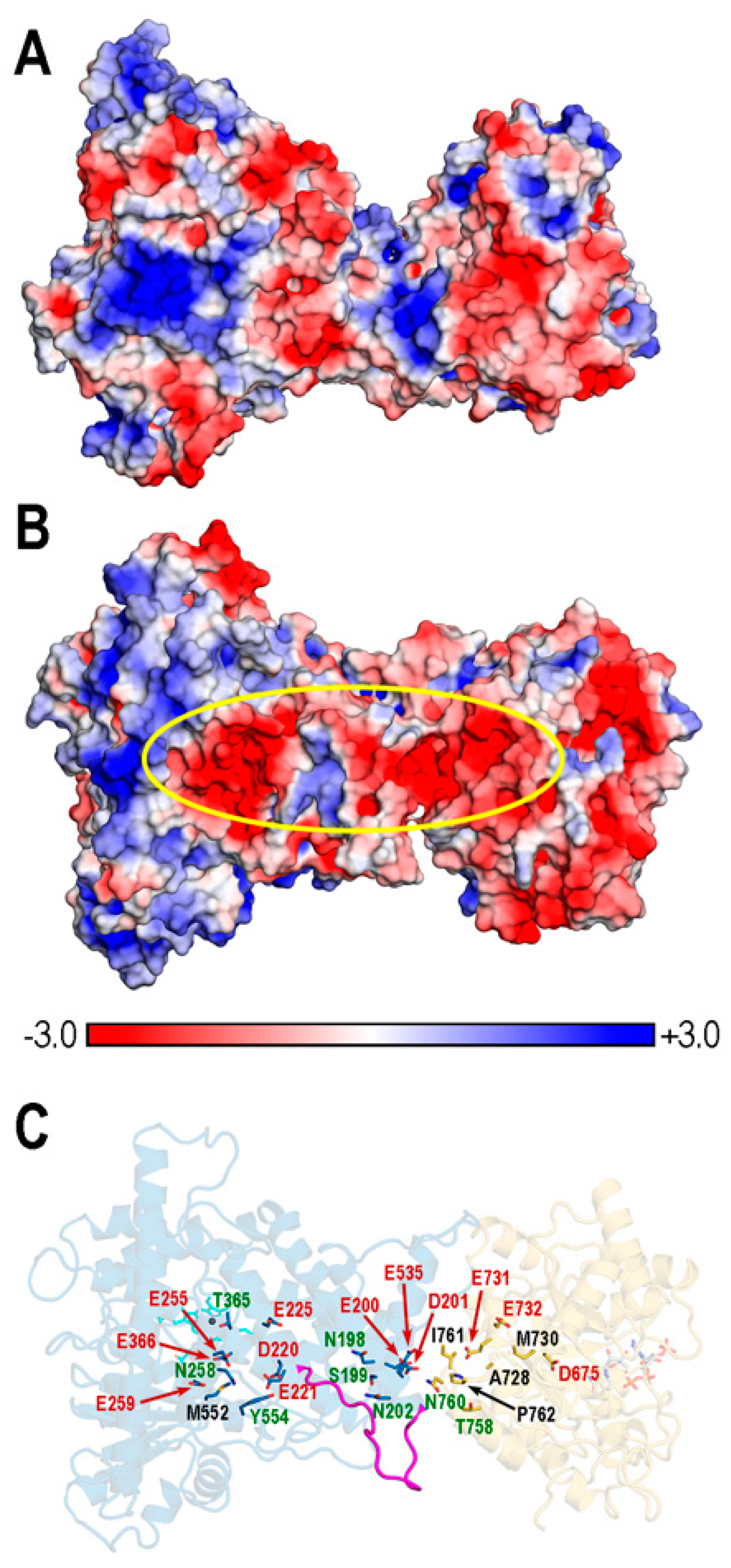
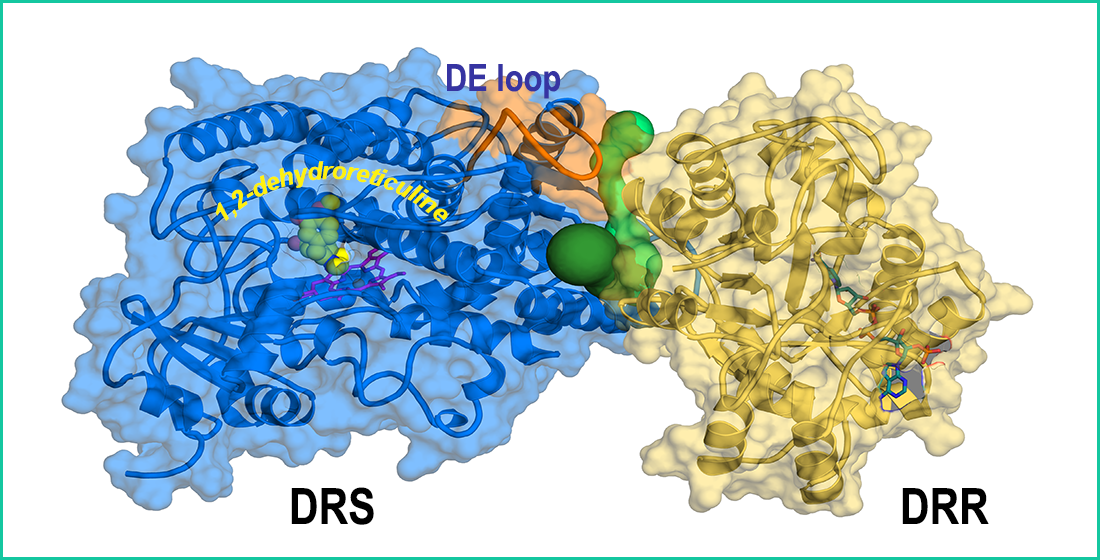
3.4. Interaction between DRS and DRR Domains
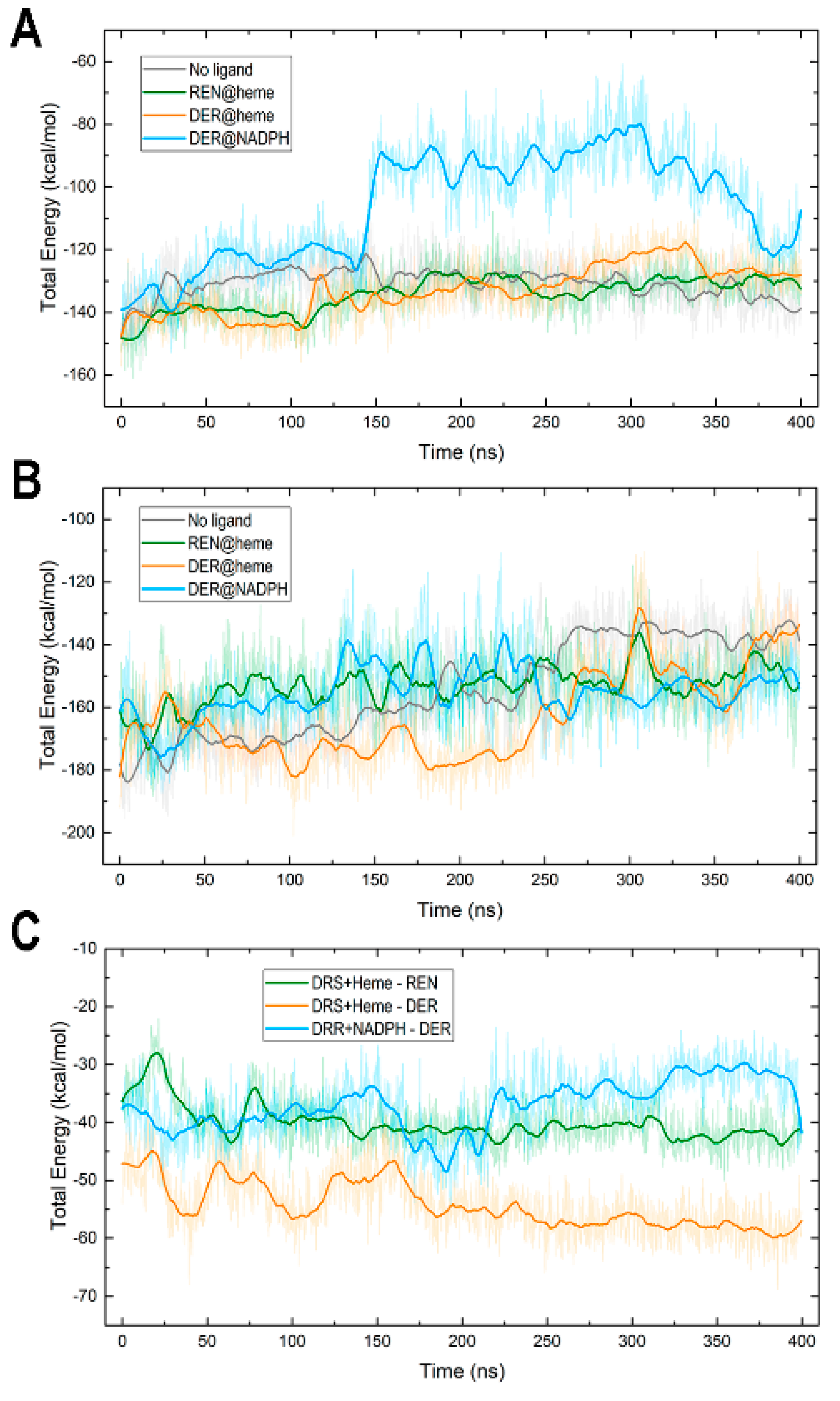
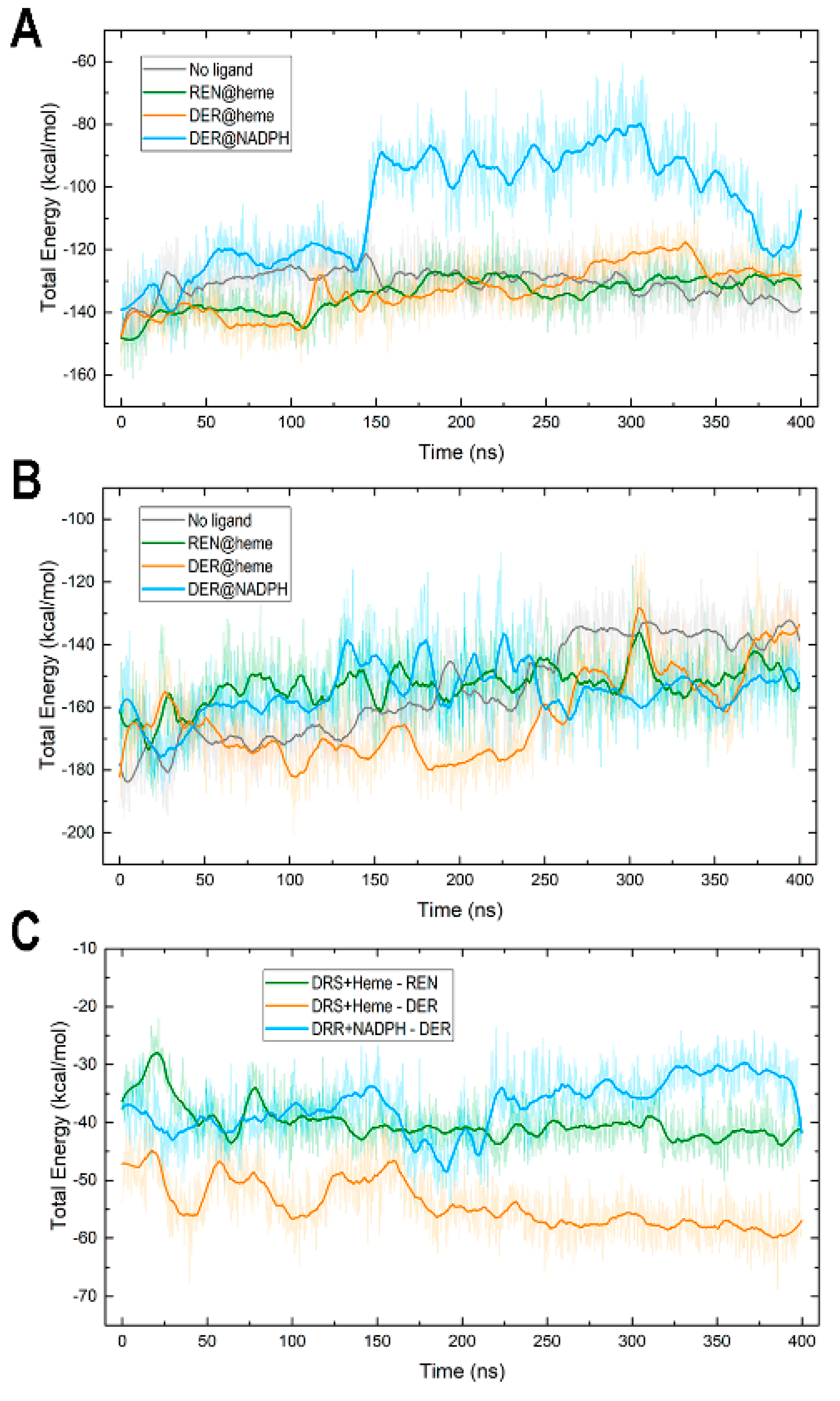
3.5. Protein–Cofactor and Protein–Ligand Interaction Energies and Water Effects
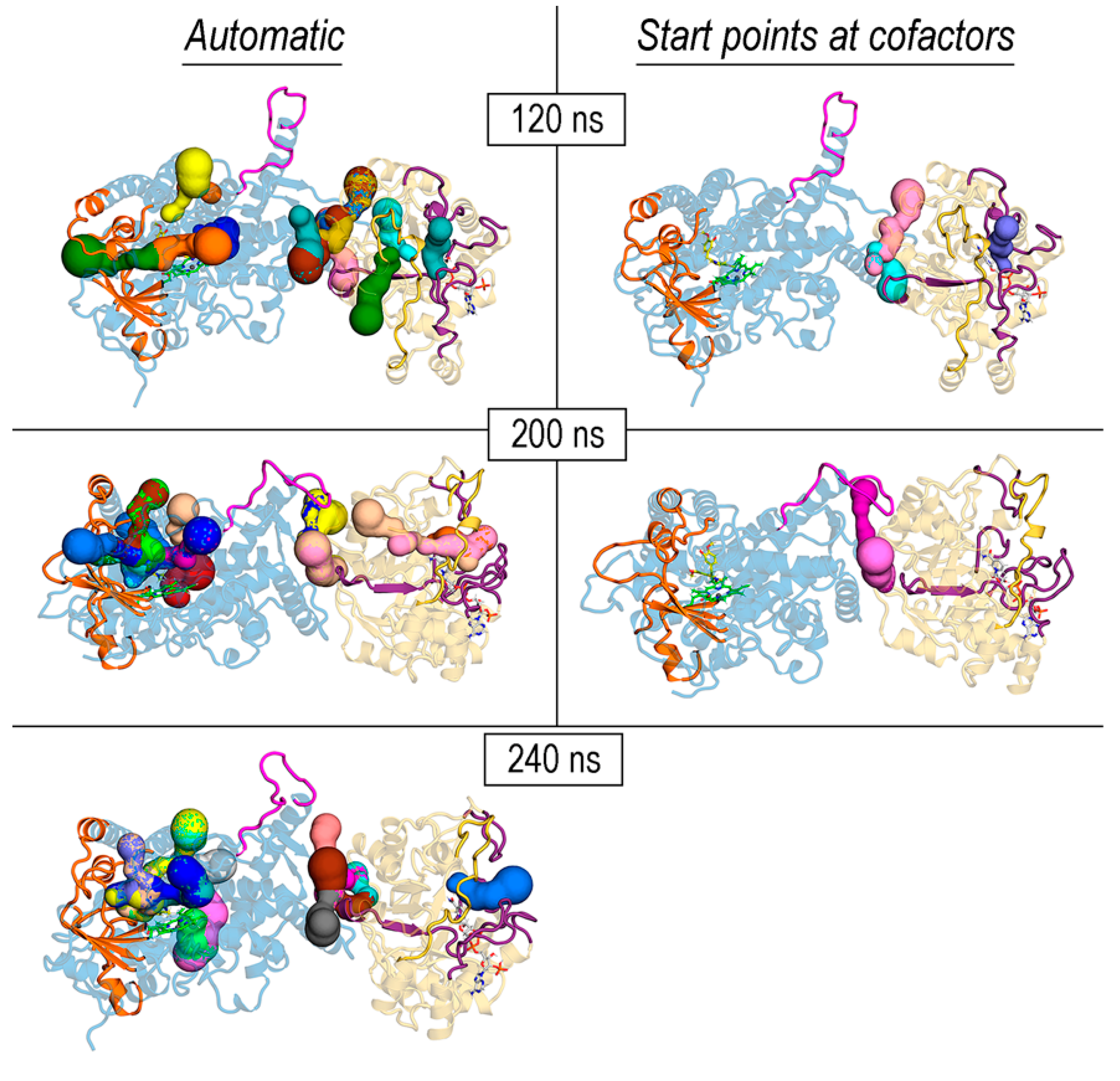
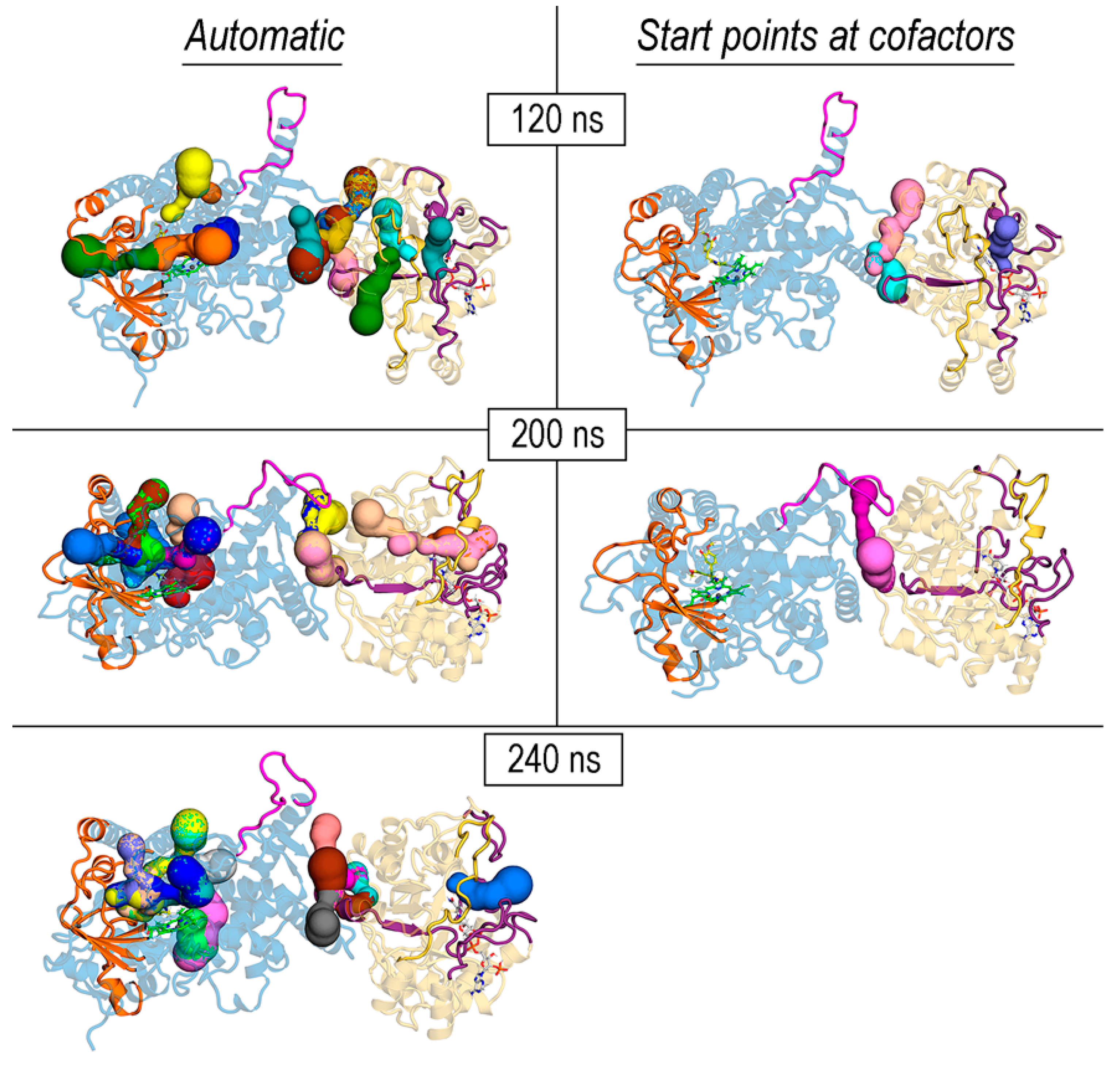
3.6. Exploration of Tunnels in REPI
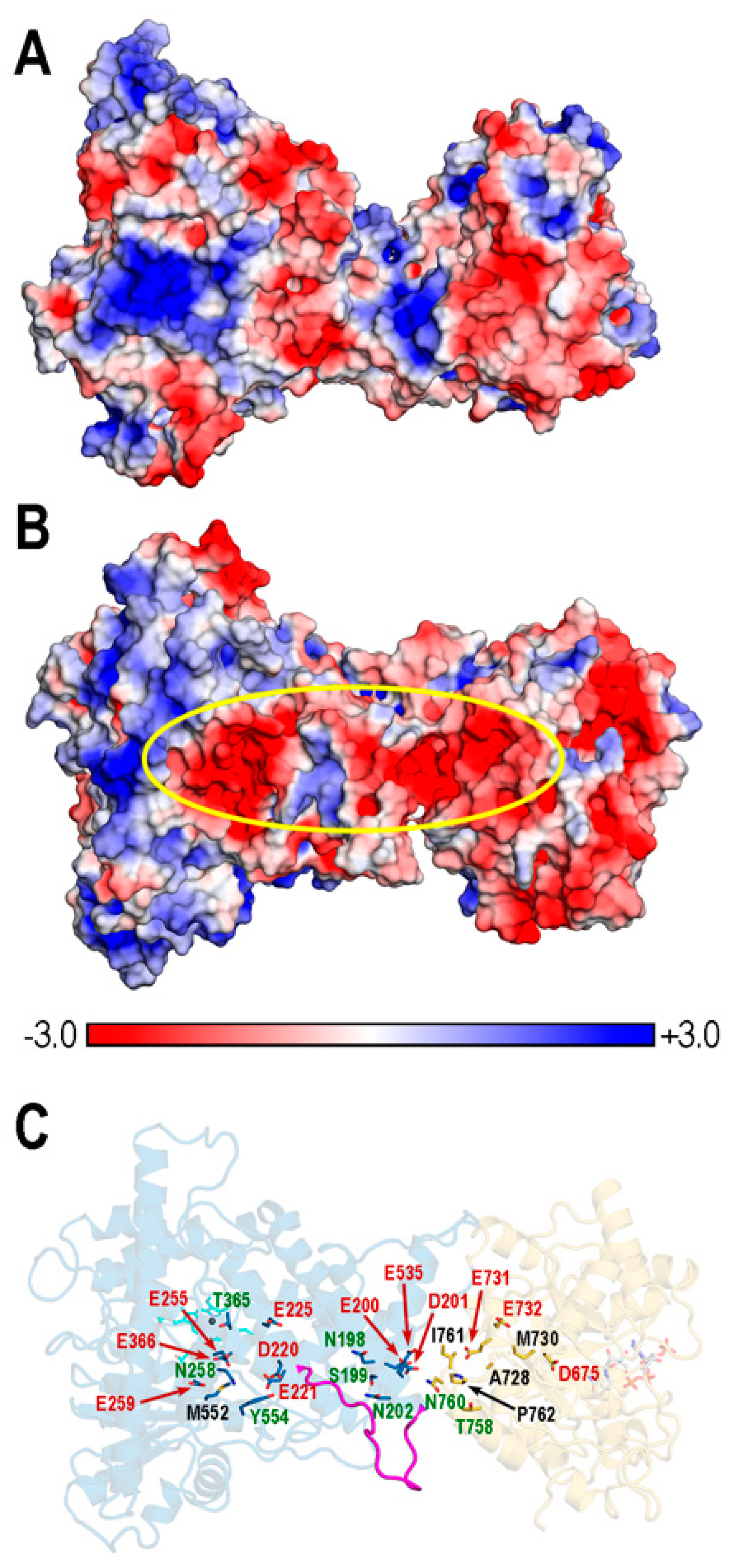
3.7. Poisson–Boltzmann (PB) Electrostatic Potential (EP) in REPI Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ziegler, J.; Facchini, P.J. Alkaloid biosynthesis: Metabolism and trafficking. Annu. Rev. Plant. Biol. 2008, 59, 735–769. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, J.; Diaz-Chávez, M.L.; Kramell, R.; Ammer, C.; Kutchan, T.M. Comparative macroarray analysis of morphine containing Papaver somniferum and eight morphine free Papaver species identifies an O-methyltransferase involved in benzylisoquinoline biosynthesis. Planta 2005, 222, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudian, M.; Rahimi-Moghaddam, P. The Anti-Cancer Activity of Noscapine: A Review. Recent Pat. Anticancer Drug Discov. 2009, 4, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, J.; Brandt, W.; Geißler, R.; Facchini, P.J. Removal of substrate inhibition and increase in maximal velocity in the short chain dehydrogenase/reductase salutaridine reductase involved in morphine biosynthesis. J. Biol. Chem. 2009, 284, 26758–26767. [Google Scholar] [CrossRef]
- Winzer, T.; Kern, M.; King, A.J.; Larson, T.R.; Teodor, R.I.; Donninger, S.L.; Li, Y.; Dowle, A.A.; Cartwright, J.; Bates, R.; et al. Morphinan biosynthesis in opium poppy requires a P450-oxidoreductase fusion protein. Science 2015, 349, 309–312. [Google Scholar] [CrossRef]
- Ziegler, J.; Voigtländer, S.; Schmidt, J.; Kramell, R.; Miersch, O.; Ammer, C.; Gesell, A.; Kutchan, T.M. Comparative transcript and alkaloid profiling in Papaver species identifies a short chain dehydrogenase/reductase involved in morphine biosynthesis. Plant J. 2006, 48, 177–192. [Google Scholar] [CrossRef]
- Catania, T.; Li, Y.; Winzer, T.; Harvey, D.; Meade, F.; Caridi, A.; Leech, A.; Larson, T.R.; Ning, Z.; Chang, J.; et al. A functionally conserved STORR gene fusion in Papaver species that diverged 16.8 million years ago. Nat. Commun. 2022, 13, 3150. [Google Scholar] [CrossRef]
- Nakagawa, A.; Matsumura, E.; Koyanagi, T.; Katayama, T.; Kawano, N.; Yoshimatsu, K.; Yamamoto, K.; Kumagai, H.; Sato, F.; Minami, H. Total biosynthesis of opiates by stepwise fermentation using engineered Escherichia coli. Nat. Commun. 2016, 7, 10390. [Google Scholar] [CrossRef]
- Thodey, K.; Galanie, S.; Smolke, C.D. A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol. 2014, 10, 837–844. [Google Scholar] [CrossRef]
- Galanie, S.; Thodey, K.; Trenchard, I.J.; Interrante, M.F.; Smolke, C.D. Complete biosynthesis of opioids in yeast. Science 2015, 349, 1095–1100. [Google Scholar] [CrossRef]
- Lipp, A.; Ferenc, D.; Gütz, C.; Geffe, M.; Vierengel, N.; Schollmeyer, D.; Schafer, H.J.; Waldvogel, S.R.; Opatz, T. A Regio- and Diastereoselective Anodic Aryl–Aryl Coupling in the Biomimetic Total Synthesis of (−)-Thebaine. Angew. Chem. Int. Ed. 2018, 57, 11055–11059. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Winzer, T.; Yang, X.; Li, Y.; Ning, Z.; He, Z.; Teodor, R.; Lu, Y.; Bowser, T.A.; Graham, I.A.; et al. The opium poppy genome and morphinan production. Science 2018, 362, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gao, S.; Guo, L.; Wang, B.; Jia, Y.; Zhou, J.; Che, Y.; Jia, P.; Lin, J.; Xu, T.; et al. Three chromosome-scale Papaver genomes reveal punctuated patchwork evolution of the morphinan and noscapine biosynthesis pathway. Nat. Commun. 2021, 12, 6030. [Google Scholar] [CrossRef] [PubMed]
- Ozber, N.; Facchini, P.J. Phloem-specific localization of benzylisoquinoline alkaloid metabolism in opium poppy. J. Plant Physiol. 2022, 271, 153641. [Google Scholar] [CrossRef]
- Farrow, S.C.; Hagel, J.M.; Beaudoin, G.A.W.; Burns, D.C.; Facchini, P.J. Stereochemical inversion of (S)-reticuline by a cytochrome P450 fusion in opium poppy. Nat. Chem. Biol. 2015, 11, 728–732. [Google Scholar] [CrossRef]
- Allen, R.S.; Millgate, A.G.; Chitty, J.A.; Thisleton, J.; Miller, J.A.; Fist, A.J.; Gerlach, W.G.; Larkin, P.J. RNAi-mediated replacement of morphine with the nonnarcotic alkaloid reticuline in opium poppy. Nat. Biotechnol. 2004, 22, 1559–1566. [Google Scholar] [CrossRef]
- Ozber, N.; Carr, S.C.; Morris, J.S.; Liang, S.; Watkins, J.L.; Caldo, K.M.; Hagel, J.M.; Ng, K.K.S.; Facchini, P.J. Alkaloid binding to opium poppy major latex proteins triggers structural modification and functional aggregation. Nat. Commun. 2022, 13, 6768. [Google Scholar] [CrossRef]
- Chen, C.C.; Xue, J.; Peng, W.; Wang, B.; Zhang, L.; Liu, W.; Ko, T.; Huang, J.; Zhou, S.; Min, J.; et al. Structural insights into thebaine synthase 2 catalysis. Biochem. Biophys. Res. Commun. 2020, 529, 156–161. [Google Scholar] [CrossRef]
- Carr, S.C.; Torres, M.A.; Morris, J.S.; Facchini, P.J.; Ng, K.K.S. Structural studies of codeinone reductase reveal novel insights into aldo-keto reductase function in benzylisoquinoline alkaloid biosynthesis. J. Biol. Chem. 2021, 297, 101211. [Google Scholar] [CrossRef]
- Kluza, A.; Niedzialkowska, E.; Kurpiewska, K.; Wojdyla, Z.; Quesne, M.; Kot, E.; Porebski, P.J.; Borowski, T. Crystal structure of thebaine 6-O-demethylase from the morphine biosynthesis pathway. J. Struct. Biol. 2018, 202, 229–235. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucl. Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Srere, P.A. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 1987, 56, 89–124. [Google Scholar] [CrossRef] [PubMed]
- Ovadi, J. Physiological Significance of Metabolic Channelling. J. Theor. Biol. 1991, 152, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Miles, E.W.; Rhee, S.; Davies, D.R. The molecular basis of substrate channeling. J. Biol. Chem. 1999, 274, 12193–12196. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Holden, H.M.; Raushel, F.M. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem. 2001, 70, 149–180. [Google Scholar] [CrossRef] [PubMed]
- Winkel, B.S.J. Metabolic channeling in plants. Annu. Rev. Plant Biol. 2004, 55, 85–107. [Google Scholar] [CrossRef]
- Jørgensen, K.; Rasmussen, A.V.; Morant, M.; Nielsen, A.H.; Bjarnholt, N.; Zagrobelny, M.; Bak, S.; Møller, B.L. Metabolon formation and metabolic channeling in the biosynthesis of plant natural products. Curr. Opin. Plant Biol. 2005, 8, 280–291. [Google Scholar] [CrossRef]
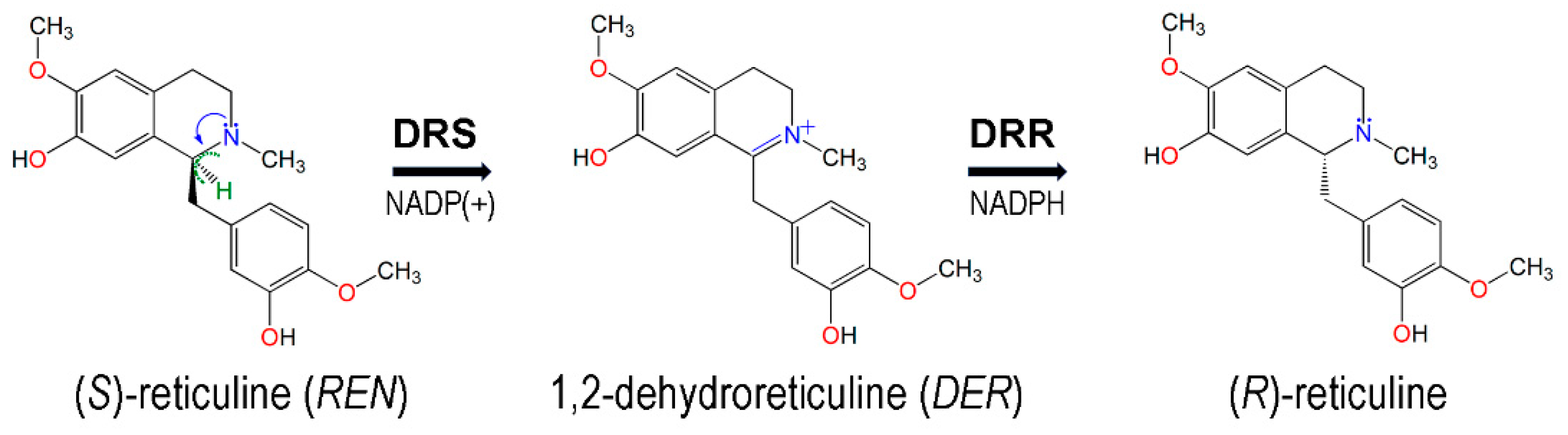
- He, X.S.; Brossi, A. 1,2-dehydroreticuline: Conversion of iminium salts into enamines. J. Nat. Prod. 1993, 56, 973–975. [Google Scholar] [CrossRef]
- Arroyo-Mañez, P.; Bikiel, D.E.; Boechi, L.; Capece, L.; Di Lella, S.; Estrin, D.A.; Martí, M.A.; Moreno, D.M.; Nadra, A.D.; Petruk, A.A. Protein dynamics and ligand migration interplay as studied by computer simulation. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 1054–1064. [Google Scholar] [CrossRef]
- Amaro, R.; Luthey-Schulten, Z. Molecular dynamics simulations of substrate channeling through an a-b barrel protein. Chem. Phys. 2004, 307, 147–155. [Google Scholar] [CrossRef]
- Smith, N.E.; Vrielink, A.; Attwood, P.V.; Corry, B. Binding and channeling of alternative substrates in the enzyme DmpFG: A molecular dynamics study. Biophys. J. 2014, 106, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Brezovsky, J.; Chovancova, E.; Gora, A.; Pavelka, A.; Biedermannova, L.; Damborsky, J. Software tools for identification, visualization and analysis of protein tunnels and channels. Biotechnol. Adv. 2013, 31, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Petřek, M.; Otyepka, M.; Banáš, P.; Košinová, P.; Koča, J.; Damborský, J. CAVER: A new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinform. 2006, 7, 316. [Google Scholar] [CrossRef] [PubMed]
- Petrek, M.; Kosinova, P.; Koca, J.; Otyepka, M. MOLE: A Voronoi diagram-based explorer of molecular channels, pores, and tunnels. Structure 2007, 15, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, E.; Fishelovitch, D.; Wolfson, H.J.; Halperin, D.; Nussinov, R. MolAxis: A server for identification of channels in macromolecules. Nucleic Acids Res. 2008, 36, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Svobodová Vařeková, R.; Berka, K.; Pravda, L.; Navrátilová, V.; Banáš, P.; Ionescu, C.; Otyepka, M.; Koča, J. MOLE 2.0: Advanced approach for analysis of biomacromolecular channels. J. Cheminform. 2013, 5, 39. [Google Scholar] [CrossRef]
- Pravda, L.; Berka, K.; Varekova, R.S.; Sehnal, D.; Banas, P.; Laskowski, R.A.; Koca, J.; Otyepka, M. Anatomy of enzyme channels. BMC Bioinform. 2014, 15, 379. [Google Scholar] [CrossRef]
- Cojocaru, V.; Winn, P.J.; Wade, R.C. The ins and outs of cytochrome P450s. Biochim. Biophys. Acta 2007, 1770, 390–401. [Google Scholar] [CrossRef]
- Dubey, K.D.; Shaik, S. Cytochrome P450—The Wonderful Nanomachine Revealed through Dynamic Simulations of the Catalytic Cycle. Acc. Chem. Res. 2019, 52, 389–399. [Google Scholar] [CrossRef]
- The UniProt Consortium, UniProt: The Universal Protein Knowledgebase in 2023. Nucl. Acids Res. 2023, 51, D523–D531. [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Dali server: Structural unification of protein families. Nucl. Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on TM-score. Nucl. Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Chen, Z.X.; He, K.; Shi, C.; Huang, L.X.; Xiao, C.L.; Chang, Z.Z. The crystal structure and molecular docking of CYP76AH3 in the Tanshinone Biosynthesis Pathway. Chin. J. Biochem. Mol. Biol. 2022, 38, 488–494. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Luthra, A.; Denisov, I.G.; Sligar, S.G. Spectroscopic features of cytochrome P450 reaction intermediates. Arch. Biochem. Biophys. 2011, 507, 26–35. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hu, Y.; Chen, C.C.; Huang, J.W.; Min, J.; Dai, L.; Guo, R.T. Structural analysis and engineering of aldo-keto reductase from glyphosate-resistant Echinochloa colona. J. Hazard. Mater. 2022, 436, 129191. [Google Scholar] [CrossRef]
- Winkler, A.; Lyskowski, A.; Riedl, S.; Puhl, M.; Kutchan, T.M.; Macheroux, P.; Gruber, K. A concerted mechanism for berberine bridge enzyme. Nat. Chem. Biol. 2008, 4, 739–741. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; Zaslavsky, L.; et al. PubChem 2023 update. Nucl. Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kern, N.; Brown, T.; Lee, J.; Im, W. CHARMM-GUI PDB Manipulator: Various PDB structural modifications for biomolecular modeling and simulation. J. Mol. Biol. 2023, 435, 167995. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Chulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar] [CrossRef] [PubMed]
- Shindyalov, I.N.; Bourne, P.E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998, 11, 739–747. [Google Scholar] [CrossRef] [PubMed]
- PyMOL Molecular Graphics System; Version 2.5; Schrödinger LLC: New York, NY, USA, 2021.
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Hasemann, C.A.; Kurumbail, R.G.; Boddupalli, S.S.; Peterson, J.A.; Deisenhofer, J. Structure and function of cytochromes P450: A comparative analysis of three crystal structures. Structure 1995, 2, 41–62. [Google Scholar] [CrossRef]
- Holm, L. DALI and the persistence of protein shape. Protein Sci. 2020, 29, 128–140. [Google Scholar] [CrossRef]
- Guo, J.; Ma, X.; Cai, Y.; Ma, Y.; Zhan, Z.; Zhou, Y.J.; Liu, W.; Guan, M.; Yang, J.; Cui, G.; et al. Cytochrome P450 promiscuity leads to a bifurcating biosynthetic pathway for tanshinones. New Phytol. 2016, 210, 525–534. [Google Scholar] [CrossRef]
- Wang, Y.; Yi, H.; Wang, M.; Yu, O.; Jez, J.M. Structural and kinetic analysis of the unnatural fusion protein 4-coumaroyl-CoA ligase: Stilbene synthase. J. Am. Chem. Soc. 2011, 133, 20684–20687. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, K.; Hino, T.; Kanadani, M.; Watanabe, B.; Lee, H.J.; Mizutani, M.; Nagano, S. Structural insights into a key step of brassinosteroid biosynthesis and its inhibition. Nat. Plants 2019, 5, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Otyepka, M.; Skopalik, J.; Anzenbacherova, E.; Anzenbacher, P. What common structural features and variations of mammalian P450s are known to date? Biochim. Biophys. Acta 2007, 1770, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Urban, P.; Lautier, T.; Pompon, D.; Truan, G. Ligand access channels in cytochrome P450 enzymes: A review. Int. J. Mol. Sci. 2018, 19, 1617. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. The uniqueness of tryptophan in biology: Properties, metabolism, interactions and localizations in proteins. Int. J. Mol. Sci. 2020, 21, 8776. [Google Scholar] [CrossRef] [PubMed]
- Khemaissa, S.; Sagan, S.; Walrant, A. Tryptophan, an amino acid endowed with unique properties and its many roles in membrane proteins. Crystals 2021, 11, 1032. [Google Scholar] [CrossRef]
- Lakshikanth, G.S.; Krishnamoorthy, G. Solvent-exposed tryptophans probe the dynamics at protein surfaces. Biophys. J. 1999, 77, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Campanini, B.; Raboni, S.; Vaccari, S.; Zhang, L.; Cook, P.F.; Hazlett, T.L.; Mozzarelli, A.; Bettati, S. Surface-exposed tryptophan residues are essential for O-acetylserine sulfhydrylase structure, function, and stability. J. Biol. Chem. 2003, 278, 17511–37519. [Google Scholar] [CrossRef]
- Cuevas-Zuviría, B.; Mínguez-Toral, M.; Díaz-Perales, A.; Garrido-Arandia, M.; Pacios, L.F. Structural dynamics of the lipid antigen-binding site of CD1d protein. Biomolecules 2020, 10, 532. [Google Scholar] [CrossRef]
- Rathod, D.C.; Vaidya, S.M.; Hopp, M.T.; Kuhl, T.; Imhof, D. Shapes and patterns of heme-binding motifs in mammalian heme-binding proteins. Biomolecules 2023, 13, 1031. [Google Scholar] [CrossRef]
- Saxena, A.; Singh, P.; Yadav, D.K.; Sharma, P.; Alam, S.; Khan, F.; Thul, S.T.; Shukla, R.K.; Gupta, V.; Sangwan, N.S. Identification of cytochrome P450 heme motif in plants proteome. Plant Omics J. 2013, 6, 1–12. [Google Scholar]
- Niu, G.; Guo, Q.; Wang, J.; Liu, L. Structural basis for plant lutein biosynthesis from a-carotene. Proc. Natl. Acad. Sci. USA 2020, 117, 14150–14157. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.S. Fundamental mechanisms of substrate channeling. Methods Enzymol. 1999, 308, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.J.; Lee, Y.S.; Yuan, M.; Alkotaini, B.; Zhao, J.; Blumenthal, E.; Minteer, S.D. Substrate channeling by a rationally designed fusion protein in a biocatalytic cascade. J. Am. Chem. Soc. Au 2021, 1, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Stroud, R.M. An electrostatic highway. Nat. Struct. Biol. 1994, 1, 131–134. [Google Scholar] [CrossRef]
- Elcock, A.H.; Potter, M.J.; Matthews, D.A.; Knighton, D.R.; McCammon, J.A. Electrostatic channeling in the bifunctional enzyme dihydrofolate reductase-thymidylate synthase. J. Mol. Biol. 1996, 262, 370–374. [Google Scholar] [CrossRef]
- Jamil, O.K.; Cravens, A.; Payne, J.T.; Kim, C.Y.; Smolke, C.D. Biosynthesis of tetrahydropapaverine and semisynthesis of papaverine in yeast. Proc. Natl. Acad. Sci. USA 2022, 119, 33. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DRS-Heme | DRR-NADP | Enzyme-Substrate | ||||
|---|---|---|---|---|---|---|
| Complex | Average | Std. Dev. | Average | Std. Dev. | Average | Std. Dev. |
| No ligand | −130.7 | 4.1 | −153.1 | 6.0 | - | - |
| REN@heme | −134.2 | 5.3 | −154.4 | 15.3 | −40.1 | 3.0 |
| DER@heme | −132.9 | 7.4 | −159.9 | 13.2 | −54.3 | 3.9 |
| DER@NADPH | −106.4 | 17.2 | −155.2 | 7.7 | −37.0 | 4.3 |
| #HBs Heme–Water | #HBs NADP–Water | #HBs Ligand–Water | ||||
|---|---|---|---|---|---|---|
| Complex | Average | Std. Dev. | Average | Std. Dev. | Average | Std. Dev. |
| No ligand | 3.2 | 0.5 | 5.7 | 1.0 | - | - |
| REN@heme | 2.3 | 0.4 | 6.3 | 0.9 | 1.0 | 0.2 |
| DER@heme | 2.7 | 0.9 | 5.6 | 1.7 | 0.5 | 0.3 |
| DER@NADPH | 4.5 | 1.5 | 7.1 | 0.8 | 0.7 | 0.3 |
| #waters-heme | #waters-NADP | |||||
| No ligand | 19.2 | 3.0 | 46.1 | 8.0 | ||
| REN@heme | 24.1 | 4.9 | 50.4 | 5.5 | ||
| DER@heme | 15.8 | 3.3 | 42.9 | 6.0 | ||
| DER@NADPH | 36.6 | 7.7 | 46.2 | 5.1 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Bárcena, A.; Fernandez-Pacios, L.; Giraldo, P. Structural Characterization and Molecular Dynamics Study of the REPI Fusion Protein from Papaver somniferum L. Biomolecules 2024, 14, 2. https://doi.org/10.3390/biom14010002
Diaz-Bárcena A, Fernandez-Pacios L, Giraldo P. Structural Characterization and Molecular Dynamics Study of the REPI Fusion Protein from Papaver somniferum L. Biomolecules. 2024; 14(1):2. https://doi.org/10.3390/biom14010002
Chicago/Turabian StyleDiaz-Bárcena, Alba, Luis Fernandez-Pacios, and Patricia Giraldo. 2024. "Structural Characterization and Molecular Dynamics Study of the REPI Fusion Protein from Papaver somniferum L." Biomolecules 14, no. 1: 2. https://doi.org/10.3390/biom14010002
APA StyleDiaz-Bárcena, A., Fernandez-Pacios, L., & Giraldo, P. (2024). Structural Characterization and Molecular Dynamics Study of the REPI Fusion Protein from Papaver somniferum L. Biomolecules, 14(1), 2. https://doi.org/10.3390/biom14010002