

GDF-15 Inhibits ADP-Induced Human Platelet Aggregation through the GFRAL/RET Signaling Complex
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Human Platelets, Erythrocytes, and Leukocytes
2.2. Platelet Aggregation Assay
2.3. GDF-15-Related Receptors Microarray
2.4. Western Blot
2.5. Immunoprecipitation Assay
2.6. Statistical Analysis
3. Results
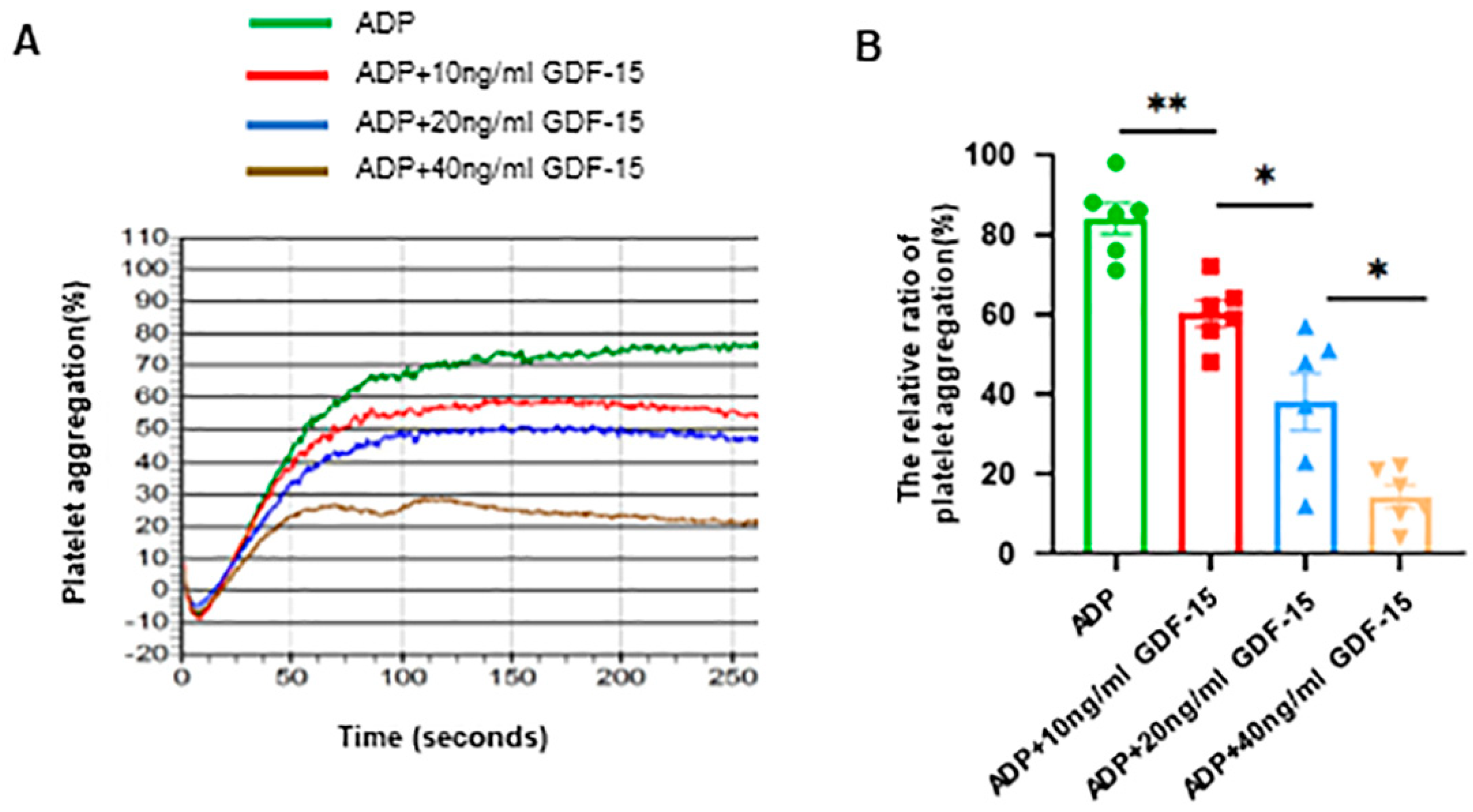
3.1. GDF-15 Inhibits Human Platelet Aggregation Triggered by ADP
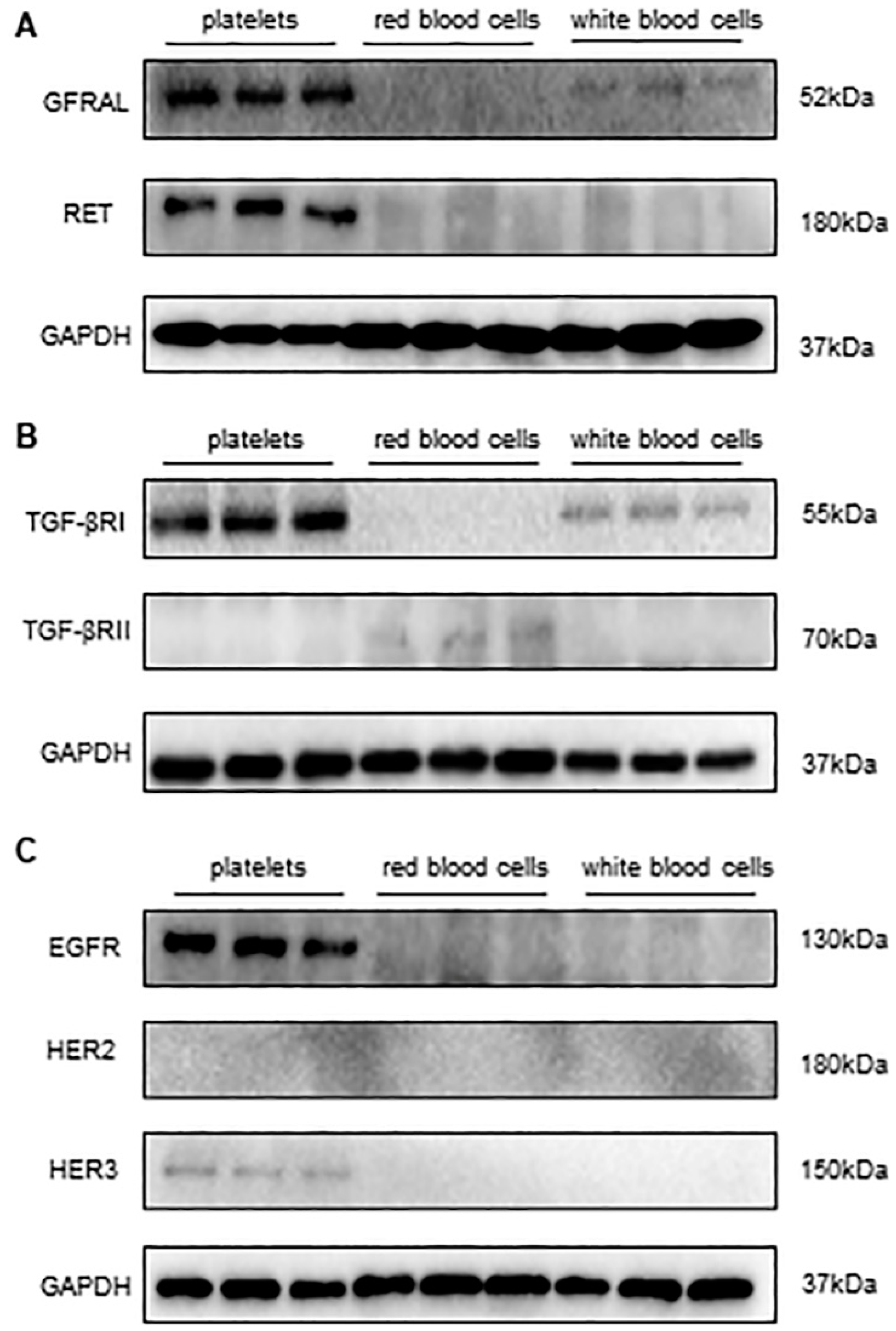
3.2. Detecting the Expression of Receptors of GDF-15 in Human Platelets
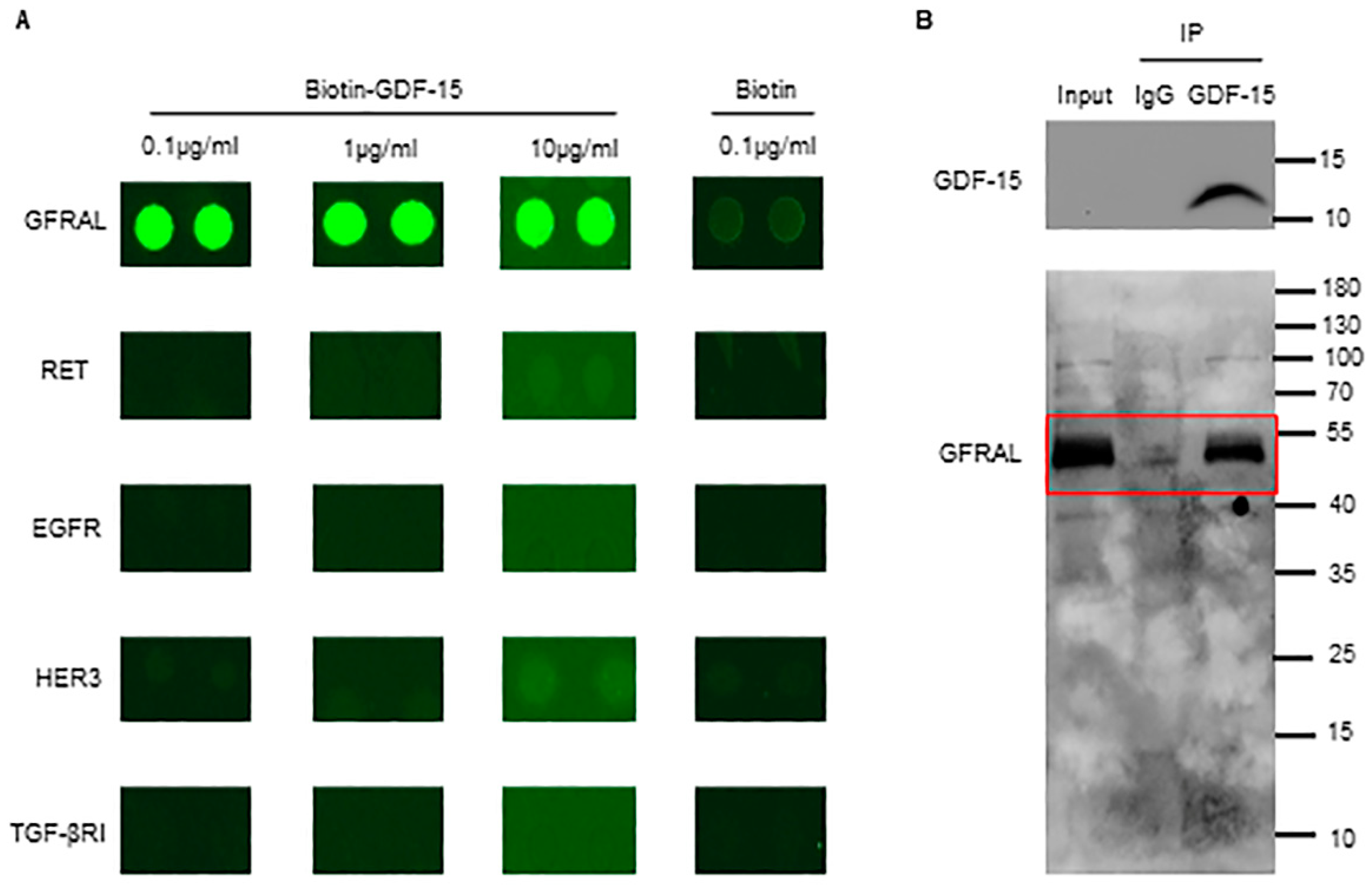
3.3. GDF-15 Interacts with GFRAL in Platelets
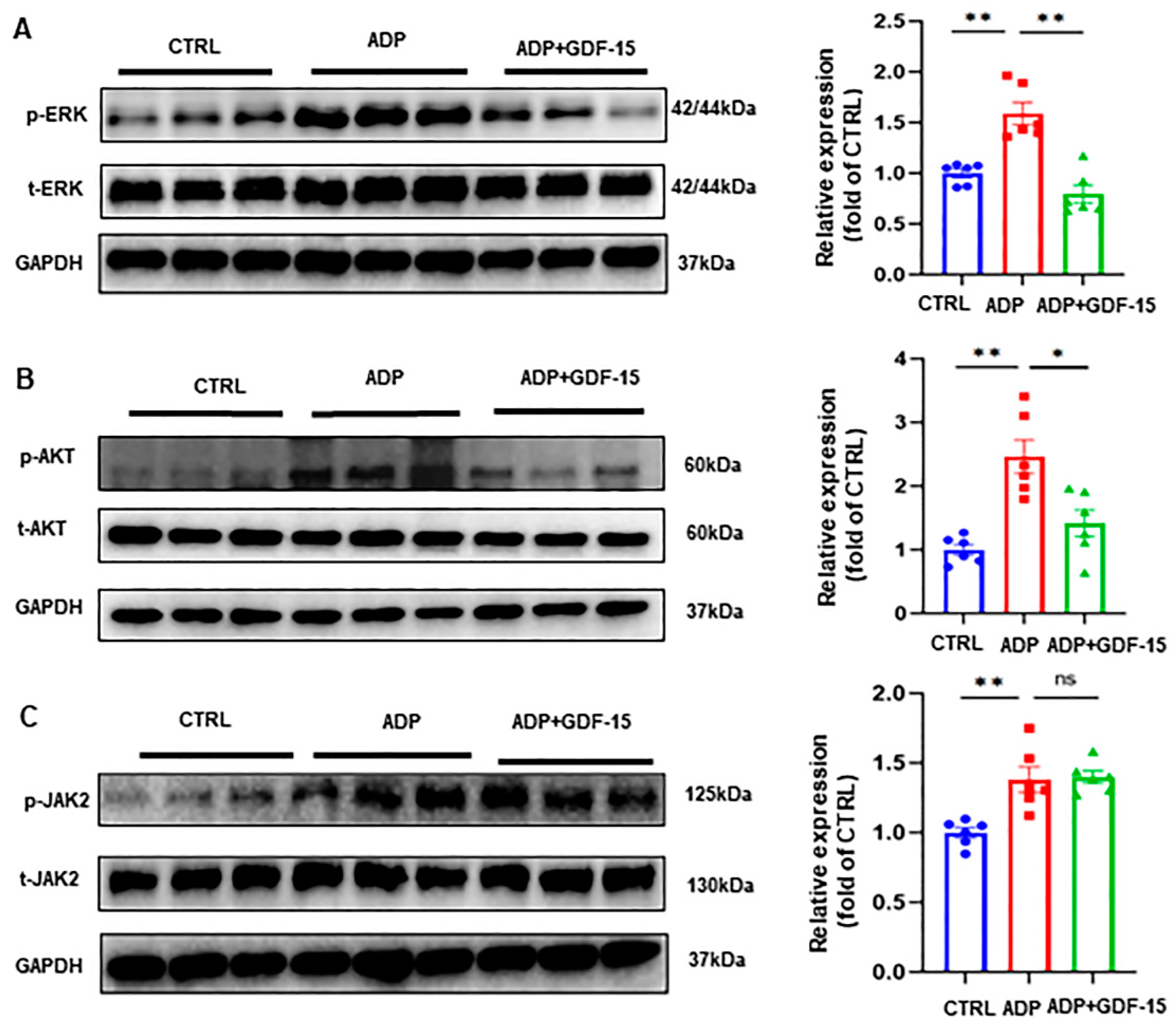
3.4. GDF-15 Inhibits ADP-Induced AKT and ERK Activation
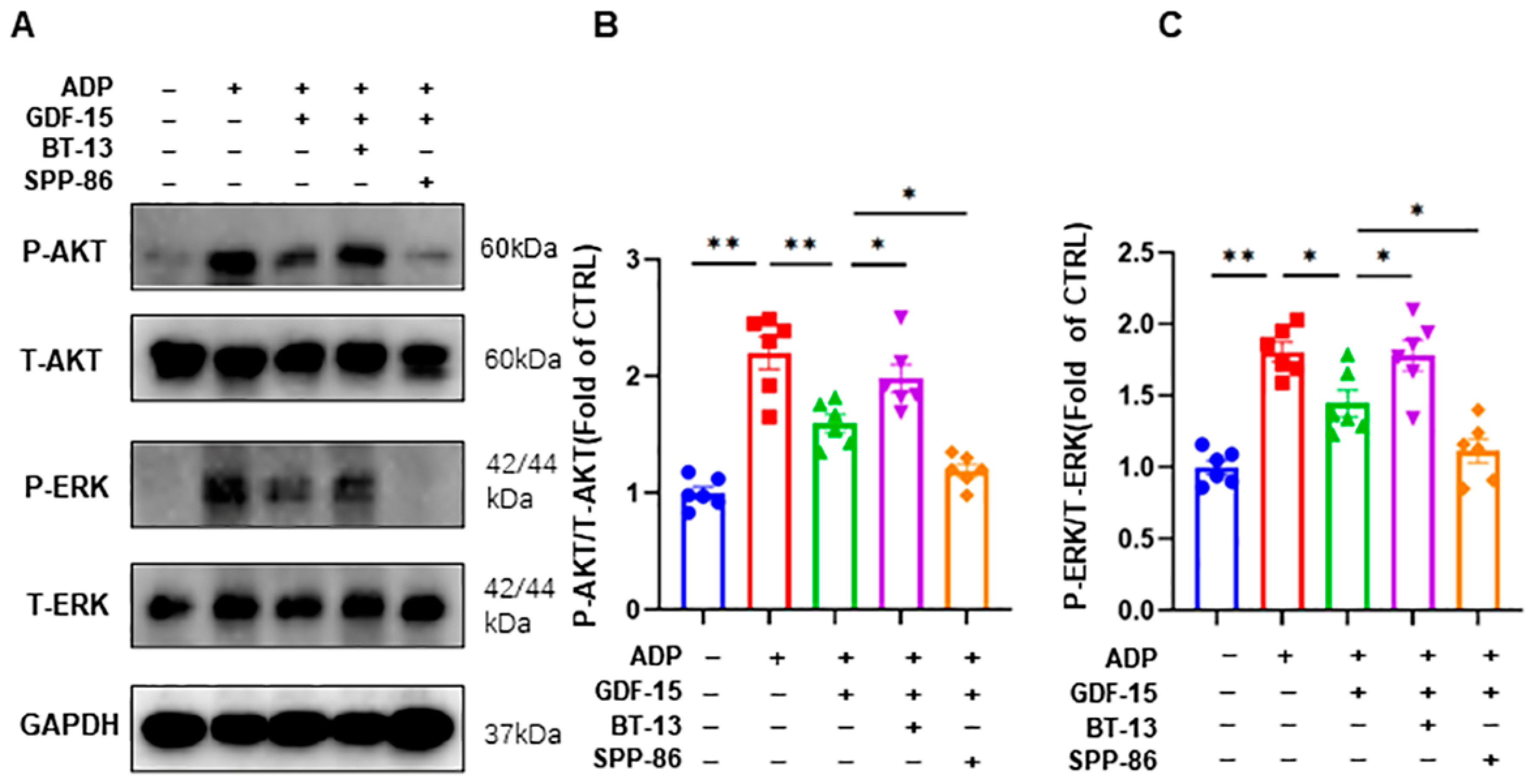
3.5. GDF-15 Inhibits ADP-Induced AKT and ERK Activation through GFRAL/RET Signaling Complex
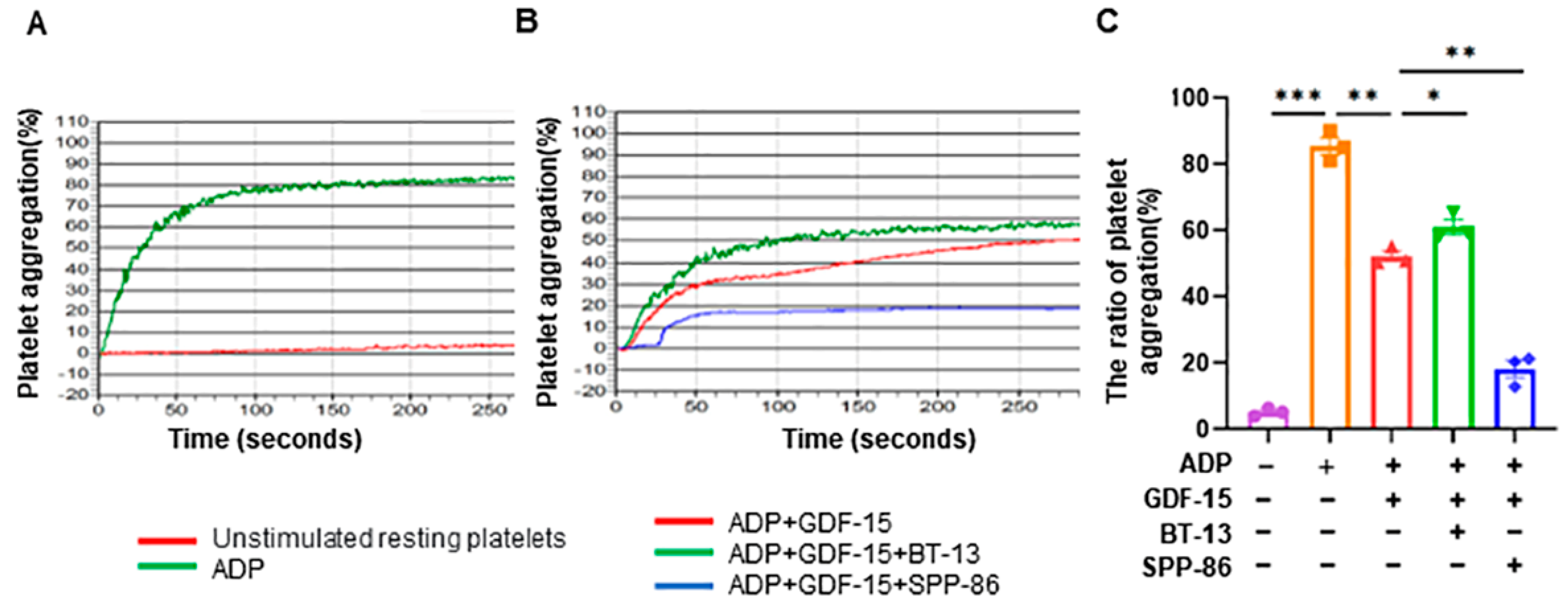
3.6. GDF-15 Inhibits ADP-Induced Human Platelet Aggregation through GFRAL/RET Signaling Complex
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vorchheimer, D.A.; Becker, R. Platelets in atherothrombosis. Mayo Clin. Proc. 2006, 81, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Kollnberger, M.; Wakefield, T.W.; Lammle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Collaboration, A.T. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002, 324, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Pulmonary Embolism Prevention (PEP) trial Collaborative Group. Prevention of pulmonary embolism and deep vein thrombosis with low dose aspirin: Pulmonary Embolism Prevention (PEP) trial. Lancet 2000, 355, 1295–1302. [Google Scholar] [CrossRef]
- Luan, H.H.; Wang, A.; Hilliard, B.K.; Carvalho, F.; Rosen, C.E.; Ahasic, A.M.; Herzog, E.L.; Kang, I.; Pisani, M.A.; Yu, S.; et al. GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 2019, 178, 1231–1244 e1211. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Junker, M.; Tritschler, I.; Mittelbronn, M.; Dombrowski, Y.; Breit, S.N.; Tabatabai, G.; Wick, W.; Weller, M.; Wischhusen, J. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin. Cancer Res. 2010, 16, 3851–3859. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, Y.M.; Zheng, P.S.; Zhang, P. GDF15 promotes the proliferation of cervical cancer cells by phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J. Exp. Clin. Cancer Res. 2018, 37, 80. [Google Scholar] [CrossRef] [PubMed]
- Borner, T.; Shaulson, E.D.; Ghidewon, M.Y.; Barnett, A.B.; Horn, C.C.; Doyle, R.P.; Grill, H.J.; Hayes, M.R.; De Jonghe, B.C. GDF15 Induces Anorexia through Nausea and Emesis. Cell Metab. 2020, 31, 351–362.e5. [Google Scholar] [CrossRef]
- Kempf, T.; von Haehling, S.; Peter, T.; Allhoff, T.; Cicoira, M.; Doehner, W.; Ponikowski, P.; Filippatos, G.S.; Rozentryt, P.; Drexler, H.; et al. Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 50, 1054–1060. [Google Scholar] [CrossRef]
- Rohatgi, A.; Patel, P.; Das, S.R.; Ayers, C.R.; Khera, A.; Martinez-Rumayor, A.; Berry, J.D.; McGuire, D.K.; de Lemos, J.A. Association of growth differentiation factor-15 with coronary atherosclerosis and mortality in a young, multiethnic population: Observations from the Dallas Heart Study. Clin. Chem. 2012, 58, 172–182. [Google Scholar] [CrossRef]
- Khan, S.Q.; Ng, K.; Dhillon, O.; Kelly, D.; Quinn, P.; Squire, I.B.; Davies, J.E.; Ng, L.L. Growth differentiation factor-15 as a prognostic marker in patients with acute myocardial infarction. Eur. Heart J. 2009, 30, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Wei, F.; Yang, C.; Xie, F.; Shuai, X.X.; Wang, M.; Yu, M. GDF-15 is associated with thrombus burden in patients with deep venous thrombosis. Thromb. Res 2020, 187, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Vestweber, D.; Zarbock, A. GDF-15 prevents platelet integrin activation and thrombus formation. J. Thromb. Haemost. 2013, 11, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Díez, J. Chronic heart failure as a state of reduced effectiveness of the natriuretic peptide system: Implications for therapy. Eur. J. Heart Fail. 2017, 19, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Schlittenhardt, D.; Schober, A.; Strelau, J.; Bonaterra, G.A.; Schmiedt, W.; Unsicker, K.; Metz, J.; Kinscherf, R. Involvement of growth differentiation factor-15/macrophage inhibitory cytokine-1 (GDF-15/MIC-1) in oxLDL-induced apoptosis of human macrophages in vitro and in arteriosclerotic lesions. Cell Tissue Res. 2004, 318, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, N.; Pfeffer, U.; Dell’Eva, R.; Ambrosini, C.; Noonan, D.M.; Albini, A. The transforming growth factor-beta family members bone morphogenetic protein-2 and macrophage inhibitory cytokine-1 as mediators of the antiangiogenic activity of N-(4-hydroxyphenyl)retinamide. Clin. Cancer Res. 2005, 11, 4610–4619. [Google Scholar] [CrossRef] [PubMed]
- Hechler, B.; Gachet, C. Purinergic Receptors in Thrombosis and Inflammation. Arter. Thromb. Vasc. Biol. 2015, 35, 2307–2315. [Google Scholar] [CrossRef]
- French, D.L.; Seligsohn, U. Platelet glycoprotein IIb/IIIa receptors and Glanzmann’s thrombasthenia. Arter. Thromb. Vasc. Biol. 2000, 20, 607–610. [Google Scholar] [CrossRef]
- Hodivala-Dilke, K.M.; McHugh, K.P.; Tsakiris, D.A.; Rayburn, H.; Crowley, D.; Ullman-Culleré, M.; Ross, F.P.; Coller, B.S.; Teitelbaum, S.; Hynes, R.O. Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J. Clin. Investig. 1999, 103, 229–238. [Google Scholar] [CrossRef]
- Feng, Y.; Yu, M.; Zhu, F.; Zhang, S.; Ding, P.; Wang, M. IL-9 Promotes the Development of Deep Venous Thrombosis by Facilitating Platelet Function. Thromb. Haemost. 2018, 118, 1885–1894. [Google Scholar] [CrossRef]
- Yuan, J.; Ding, P.W.; Yu, M.; Zhang, S.S.; Long, Q.; Cheng, X.; Liao, Y.H.; Wang, M. IL-17 Induces MPTP opening through ERK2 and P53 signaling pathway in human platelets. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Song, G.; Huang, W.; Liu, G.Z.; Deng, C.W.; Zeng, H.P.; Wang, L.; Zhang, F.C.; Zhang, X.; Jeong, J.S.; et al. Identification of new autoantigens for primary biliary cirrhosis using human proteome microarrays. Mol. Cell. Proteom. 2012, 11, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Hollopeter, G.; Jantzen, H.M.; Vincent, D.; Li, G.; England, L.; Ramakrishnan, V.; Yang, R.B.; Nurden, P.; Nurden, A.; Julius, D.; et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001, 409, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Garcia, C.; Prochnow, S.; Simeonova, I.K.; Strelau, J.; Holzl-Wenig, G.; Mandl, C.; Unsicker, K.; von Bohlen Und Halbach, O.; Ciccolini, F. Growth/differentiation factor 15 promotes EGFR signalling, and regulates proliferation and migration in the hippocampus of neonatal and young adult mice. Development 2014, 141, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Cang, W.; Li, Q.; Liao, X.; Zhan, M.; Deng, H.; Li, S.; Jin, W.; Pang, Z.; Qiu, X.; et al. Erlotinib overcomes paclitaxel-resistant cancer stem cells by blocking the EGFR-CREB/GRβ-IL-6 axis in MUC1-positive cer-vical cancer. Oncogenesis 2019, 8, 70. [Google Scholar] [CrossRef]
- Okamoto, M.; Koma, Y.I.; Kodama, T.; Nishio, M.; Shigeoka, M.; Yokozaki, H. Growth Differentiation Factor 15 Promotes Progression of Esophageal Squamous Cell Carcinoma via TGF-β Type II Receptor Activation. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2020, 87, 100–113. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, G.; Liu, Y.; Chen, R.; Zhao, D.; McAlister, V.; Mele, T.; Liu, K.; Zheng, X. GDF15 Regulates Malat-1 Circular RNA and Inactivates NFkappaB Signaling Leading to Immune Tolerogenic DCs for Preventing Alloimmune Rejection in Heart Transplantation. Front. Immunol. 2018, 9, 2407. [Google Scholar] [CrossRef]
- Artz, A.; Butz, S.; Vestweber, D. GDF-15 inhibits integrin activation and mouse neutrophil recruitment through the ALK-5/TGF-betaRII heterodimer. Blood 2016, 128, 529–541. [Google Scholar] [CrossRef]
- Xu, X.Y.; Nie, Y.; Wang, F.F.; Bai, Y.; Lv, Z.Z.; Zhang, Y.Y.; Li, Z.J.; Gao, W. Growth differentiation factor (GDF)-15 blocks norepinephrine-induced myocardial hypertrophy via a novel pathway involving inhibition of epidermal growth factor receptor transactivation. J. Biol. Chem. 2014, 289, 10084–10094. [Google Scholar] [CrossRef]
- Park, Y.J.; Lee, H.; Lee, J.H. Macrophage inhibitory cytokine-1 transactivates ErbB family receptors via the activation of Src in SK-BR-3 human breast cancer cells. BMB Rep. 2010, 43, 91–96. [Google Scholar] [CrossRef]
- Yang, L.; Chang, C.C.; Sun, Z.; Madsen, D.; Zhu, H.; Padkjaer, S.B.; Wu, X.; Huang, T.; Hultman, K.; Paulsen, S.J.; et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017, 23, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, B.; Wu, X.; Cheng, S.Y.; Paraoan, L.; Zhou, J. Identification, expression and functional characterization of the GRAL gene. J. Neurochem. 2005, 95, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, J.; Yin, L.; Yang, J.; Zheng, Y.; Zhang, M.; Ni, B.; Wang, H. Upregulated GDF-15 expression facilitates pancreatic ductal adenocarcinoma progression through orphan receptor GFRAL. Aging 2020, 12, 22564–22581. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Narita, S.; Koizumi, A.; Nara, T.; Numakura, K.; Satoh, S.; Nanjo, H.; Habuchi, T. Macrophage inhibitory cytokine-1 induced by a high-fat diet promotes prostate cancer progression by stimulating tumor-promoting cytokine production from tumor stromal cells. Cancer Commun. 2021, 41, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jin, Y.J.; Lee, M.S.; Kim, Y.M.; Lee, H. Macrophage inhibitory cytokine-1 promotes angiogenesis by eliciting the GFRAL-mediated endothelial cell signaling. J. Cell. Physiol. 2021, 236, 4008–4023. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Crawley, S.; Chen, M.; Ayupova, D.A.; Lindhout, D.A.; Higbee, J.; Kutach, A.; Joo, W.; Gao, Z.; Fu, D.; et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 2017, 550, 255–259. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Reséndiz, J.C.; Kroll, M.H.; Lassila, R. Protease-activated receptor-induced Akt activation--regulation and possible function. J. Thromb. Haemost. 2007, 5, 2484–2493. [Google Scholar] [CrossRef]
- Adam, F.; Kauskot, A.; Rosa, J.P.; Bryckaert, M. Mitogen-activated protein kinases in hemostasis and thrombosis. J. Thromb. Haemost. 2008, 6, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lin, K.C.; Huang, S.Y.; Thomas, P.A.; Wu, Y.H.; Wu, H.C.; Lin, K.H.; Sheu, J.R. Role of a Janus kinase 2-dependent signaling pathway in platelet activation. Thromb. Res. 2014, 133, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; He, L.; Li, W.; Xu, C.; Zhang, J.; Wang, D.; Dou, K.; Zhuang, R.; Jin, B.; Zhang, W.; et al. GDF15 induces immunosuppression via CD48 on regulatory T cells in hepatocellular carcinoma. J. Immuno Ther. Cancer 2021, 9, e002787. [Google Scholar] [CrossRef] [PubMed]
- Tarfiei, G.A.; Shadboorestan, A.; Montazeri, H.; Rahmanian, N.; Tavosi, G.; Ghahremani, M.H. GDF15 induced apoptosis and cytotoxicity in A549 cells depends on TGFBR2 expression. Cell Biochem. Funct. 2019, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Hosen, N.; Ichihara, H.; Mugitani, A.; Aoyama, Y.; Fukuda, Y.; Kishida, S.; Matsuoka, Y.; Nakajima, H.; Kawakami, M.; Yamagami, T.; et al. CD48 as a novel molecular target for antibody therapy in multiple myeloma. Br. J. Haematol. 2012, 156, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Zhang, W.; Colman, R.W. Thrombin regulates intracellular cyclic AMP concentration in human platelets through phosphorylation/activation of phosphodiesterase 3A. Blood 2007, 110, 1475–1482. [Google Scholar] [CrossRef]
- Li, D.; August, S.; Woulfe, D.S. GSK3beta is a negative regulator of platelet function and thrombosis. Blood 2008, 111, 3522–3530. [Google Scholar] [CrossRef]
- Lin, L.L.; Wartmann, M.; Lin, A.Y.; Knopf, J.L.; Seth, A.; Davis, R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell 1993, 72, 269–278. [Google Scholar] [CrossRef]
- Bledzka, K.; Smyth, S.S.; Plow, E.F. Integrin αIIbβ3: From discovery to efficacious therapeutic target. Circ. Res. 2013, 112, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.M.; Tolley, N.D.; Bunting, M.; Schwertz, H.; Jiang, H.; Lindemann, S.; Yost, C.C.; Rubner, F.J.; Albertine, K.H.; Swoboda, K.J.; et al. Escaping the nuclear confines: Signal-dependent pre-mRNA splicing in anucleate platelets. Cell 2005, 122, 379–391. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, B.; Tang, W.; Wen, S.; Chen, F.; Yang, C.; Wang, M.; Yang, Y.; Liang, W. GDF-15 Inhibits ADP-Induced Human Platelet Aggregation through the GFRAL/RET Signaling Complex. Biomolecules 2024, 14, 38. https://doi.org/10.3390/biom14010038
Xie B, Tang W, Wen S, Chen F, Yang C, Wang M, Yang Y, Liang W. GDF-15 Inhibits ADP-Induced Human Platelet Aggregation through the GFRAL/RET Signaling Complex. Biomolecules. 2024; 14(1):38. https://doi.org/10.3390/biom14010038
Chicago/Turabian StyleXie, Baikang, Wenjing Tang, Shuang Wen, Fen Chen, Chao Yang, Min Wang, Yong Yang, and Wei Liang. 2024. "GDF-15 Inhibits ADP-Induced Human Platelet Aggregation through the GFRAL/RET Signaling Complex" Biomolecules 14, no. 1: 38. https://doi.org/10.3390/biom14010038
APA StyleXie, B., Tang, W., Wen, S., Chen, F., Yang, C., Wang, M., Yang, Y., & Liang, W. (2024). GDF-15 Inhibits ADP-Induced Human Platelet Aggregation through the GFRAL/RET Signaling Complex. Biomolecules, 14(1), 38. https://doi.org/10.3390/biom14010038