
An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Peptides
2.2. ThT Assay
2.3. SEM
2.4. Cell Culture and Viability
2.5. Phagocytosis Assay
2.6. Animals
2.7. Intraventricular Injection of Aβ25-35 and YS-RD11
2.8. Intranasal Injection of YS-RD11
2.9. Spontaneous Alternation in Y-Maze Test
2.10. Statistical Analysis
3. Results
3.1. Screening for Effective Peptides
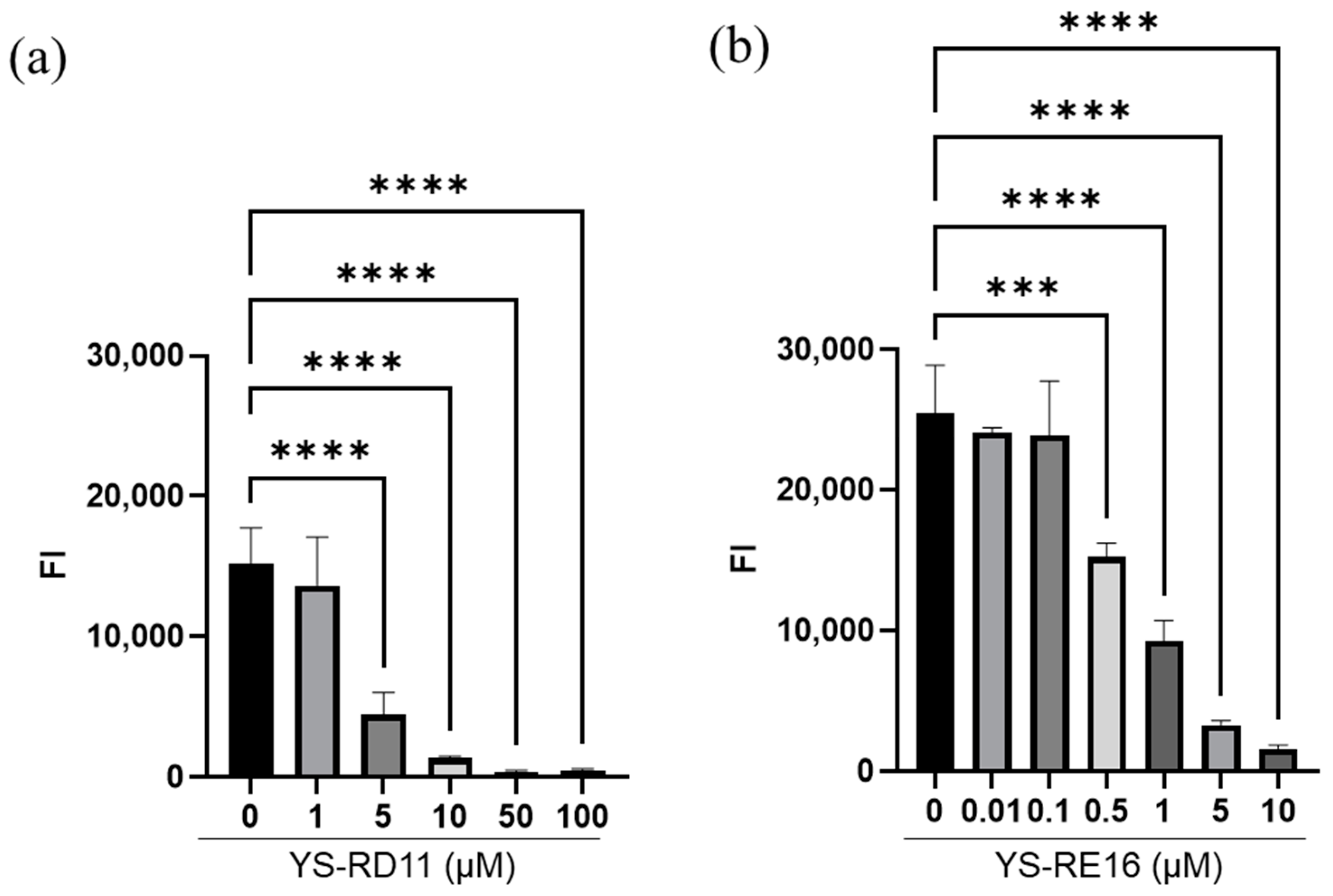
3.2. Inhibitory Effect of YS-RD11 and YS-RE16 on Aβ 25-35 Aggregation
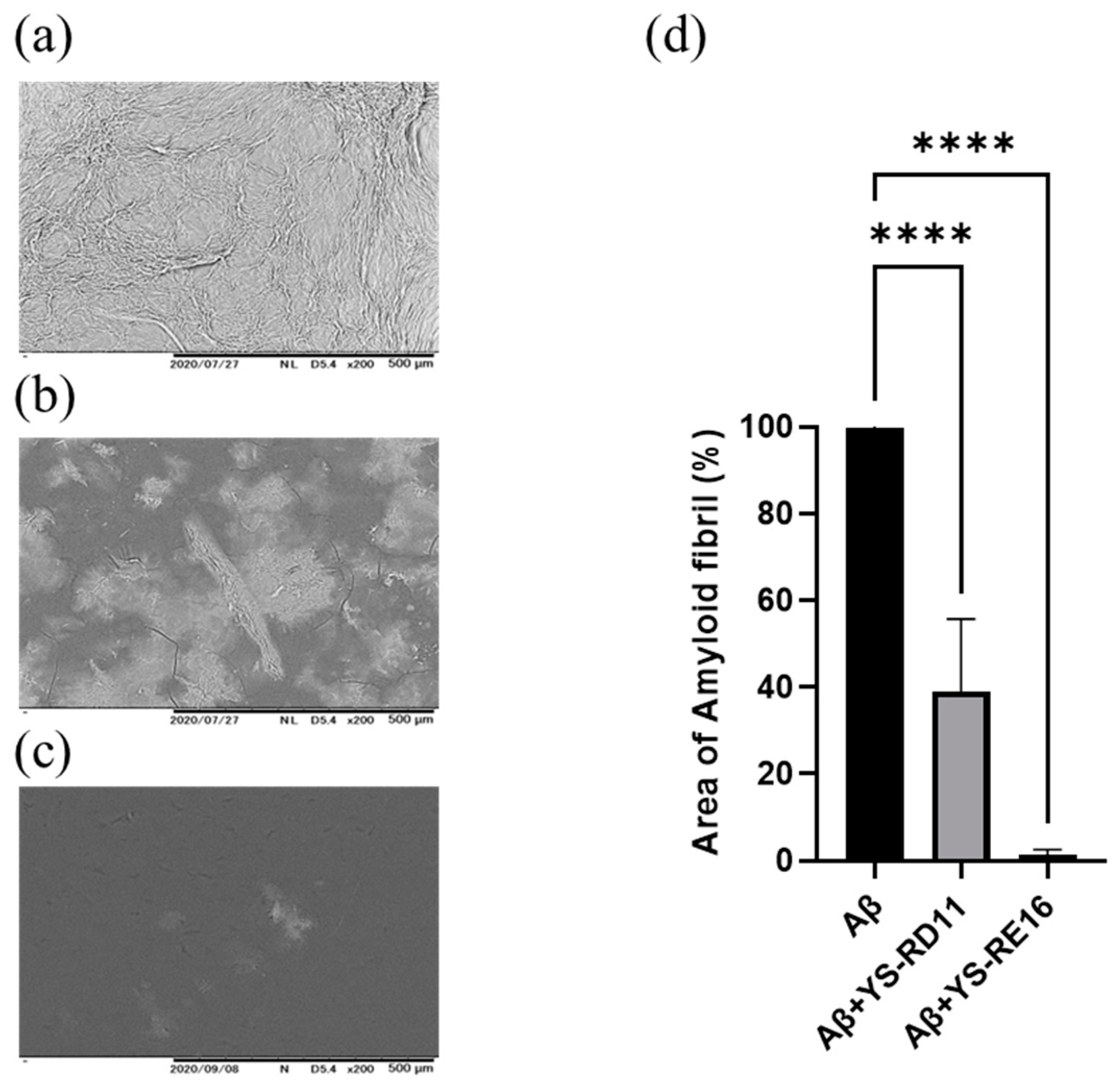
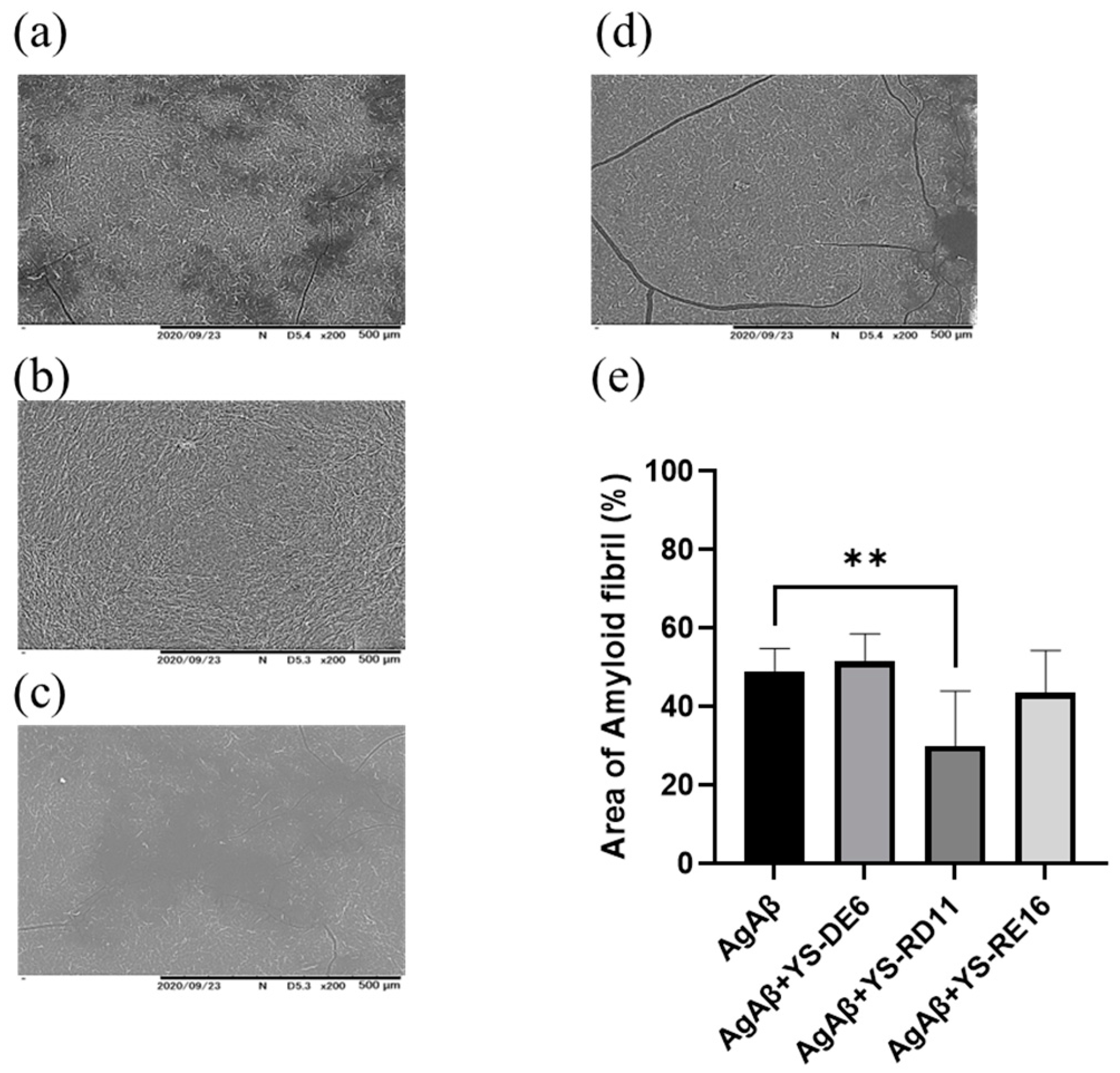
3.3. Resolving Effects on Aggregated Aβ 25-35
3.4. Cell Experiments
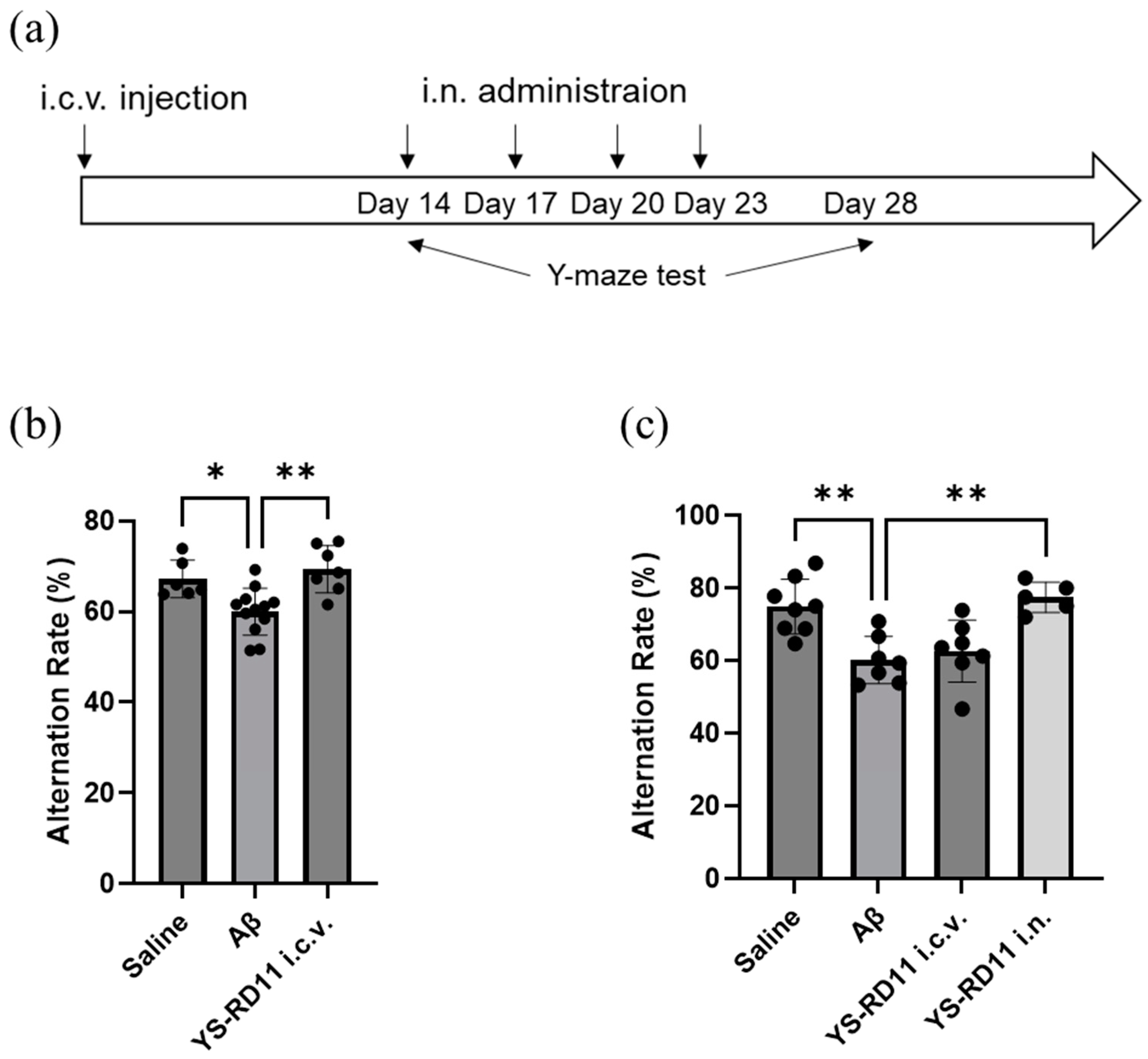
3.5. Animal Experiments
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aishwarya, R.; Abdullah, C.S.; Remex, N.S.; Bhuiyan, M.A.N.; Lu, X.H.; Dhanesha, N.; Stokes, K.Y.; Orr, A.W.; Kevil, C.G.; Bhuiyan, M.S. Diastolic dysfunction in Alzheimer’s disease model mice is associated with Aβ-amyloid aggregate formation and mitochondrial dysfunction. Sci. Rep. 2024, 14, 16715. [Google Scholar] [CrossRef] [PubMed]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I. Amyloid-β Pathology Is the Common Nominator Proteinopathy of the Primate Brain Aging. J. Alzheimers Dis. 2024, 100, S153–S164. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Deshmukh, R. Embelin Mitigates Amyloid-β-Induced Neurotoxicity and Cognitive Impairment in Rats: Potential Therapeutic Implications for Alzheimer’s Disease. Mol. Neurobiol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, K.N.; Manelli, A.M.; Blaine Stine, W.; Baker, L.K.; Krafft, G.A.; Ladu, M.J. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. New precision medicine avenues to the prevention of Alzheimer’s disease from insights into the structure and function of γ-secretases. EMBO J. 2024, 43, 887–903. [Google Scholar] [CrossRef]
- Dehury, B.; Kepp, K.P. Membrane dynamics of γ-secretase with the anterior pharynx-defective 1B subunit. J. Cell. Biochem. 2021, 122, 69–85. [Google Scholar] [CrossRef]
- Fuchino, K.; Mitsuoka, Y.; Masui, M.; Kurose, N.; Yoshida, S.; Komano, K.; Yamamoto, T.; Ogawa, M.; Unemura, C.; Hosono, M.; et al. Rational Design of Novel 1,3-Oxazine Based β-Secretase (BACE1) Inhibitors: Incorporation of a Double Bond To Reduce P-gp Efflux Leading to Robust Aβ Reduction in the Brain. J. Med. Chem. 2018, 61, 5122–5137. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J. Alzheimers Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Lopez, S.; Del Percio, C.; Forloni, G.; Frasca, A.; Drinkenburg, W.H.; Lizio, R.; Noce, G.; Ferri, R.; Soricelli, A.; Stocchi, F.; et al. Chronic BACE-1 Inhibitor Administration in TASTPM Mice (APP KM670/671NL and PSEN1 M146V Mutation): An EEG Study. Int. J. Mol. Sci. 2020, 21, 9072. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Green, B.D. Temporal Effects of Neuron-specific beta-secretase 1 (BACE1) Knock-in on the Mouse Brain Metabolome: Implications for Alzheimer’s Disease. Neuroscience 2019, 397, 138–146. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab—Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401—A clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. Correction: A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 2022, 14, 70. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic soluble amyloid oligomers drive alzheimer’s pathogenesis and represent a clinically validated target for slowing disease progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.X.; Wei, W.; Xu, S.Y. Protective effects of melatonin on cortico-hippocampal neurotoxicity induced by amyloid beta-peptide 25-35. Acta Pharmacol. Sin. 2002, 23, 71–76. [Google Scholar] [PubMed]
- Sun, M.K.; Alkon, D.L. Impairment of hippocampal CA1 heterosynaptic transformation and spatial memory by β-amyloid25–35. J. Neurophysiol. 2002, 87, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, T.; Li, G.; Zhou, L.; Sha, S.; Chen, L. Simvastatin prevents β-amyloid25-35-impaired neurogenesis in hippocampal dentate gyrus through α7nAChR-dependent cascading PI3K-Akt and increasing BDNF via reduction of farnesyl pyrophosphate. Neuropharmacology 2015, 97, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and Biological Activities of Amyloid Beta Peptide 25-35. Curr. Protein Pept. Sci. 2009, 999, 54–67. [Google Scholar] [CrossRef]
- Naldi, M.; Fiori, J.; Pistolozzi, M.; Drake, A.F.; Bertucci, C.; Wu, R.; Mlynarczyk, K.; Filipek, S.; De Simone, A.; Andrisano, V. Amyloid β-peptide 25-35 self-assembly and its inhibition: A model undecapeptide system to gain atomistic and secondary structure details of the Alzheimers disease process and treatment. ACS Chem. Neurosci. 2012, 3, 952–962. [Google Scholar] [CrossRef]
- Nakamura, R.; Konishi, M.; Taniguchi, M.; Hatakawa, Y.; Akizawa, T. The discovery of shorter synthetic proteolytic peptides derived from Tob1 protein. Peptides 2019, 116, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hatakawa, Y.; Nakamura, R.; Konishi, M.; Sakane, T.; Saito, M.; Akizawa, T. Catalytides derived from the Box A region in the ANA/BTG3 protein cleave amyloid-β fragment peptide. Heliyon 2019, 5, e02454. [Google Scholar] [CrossRef]
- Hatakawa, Y.; Nakamura, R.; Konishi, M.; Sakane, T.; Tanaka, A.; Matsuda, A.; Saito, M.; Akizawa, T. Amyloid beta cleavage by ANA-TA9, a synthetic peptide from the ANA/BTG3 Box A region. Alzheimers Dement. Transl. Res. Clin. Interv. 2021, 7, e12146. [Google Scholar] [CrossRef]
- Nakamura, R.; Konishi, M.; Hatakawa, Y.; Saito, M.; Akizawa, T. The Novel Catalytic Peptide, A Synthetic Nona-Peptide (JAL-TA9) Derived from Tob1 Protein, Digests the Amyloid-β Peptide. J. R. Sci. 2019, 1, 30–35. [Google Scholar]
- Hatakawa, Y.; Tanaka, A.; Furubayashi, T.; Nakamura, R.; Konishi, M.; Akizawa, T.; Sakane, T. Direct Delivery of ANA-TA9, a Peptide Capable of Aβ Hydrolysis, to the Brain by Intranasal Administration. Pharmaceutics 2021, 13, 1673. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Higaki, K.; Ninomiya, H.; Luan, Z.; Iida, M.; Ogawa, S.; Suzuki, Y.; Ohno, K.; Nanba, E. Chemical chaperone therapy: Luciferase assay for screening of β-galactosidase mutations. Mol. Genet. Metab. 2010, 101, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Quan, S.; Bardwell, J.C. Chaperone discovery. Bioessays 2012, 34, 973–981. [Google Scholar] [CrossRef]
- Sharma, L.; Sharma, A. Influence of cyclodextrin ring substituents on folding-related aggregation of bovine carbonic anhydrase. Eur. J. Biochem. 2001, 268, 2456–2463. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Kuboi, R. Oxidative refolding of Denatured/Reduced lysozyme utilizing the chaperone-like function of liposomes and immobilized liposome chromatography. Biotechnol. Prog. 1999, 15, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Khanna, R.; Schilling, A.; Flanagan, J.J.; Pellegrino, L.J.; Brignol, N.; Lun, Y.; Guillen, D.; Ranes, B.E.; Frascella, M.; et al. Co-administration with the pharmacological chaperone AT1001 increases recombinant human α-galactosidase A tissue uptake and improves substrate reduction in Fabry mice. Mol. Ther. 2012, 20, 717–726. [Google Scholar] [CrossRef]
- Mogk, A.; Tomoyasu, T.; Goloubinoff, P.; Rüdiger, S.; Röder, D.; Langen, H.; Bukau, B. Identification of thermolabile Escherichia coli proteins: Prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 1999, 18, 6934–6949. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Ishikawa, T. A nuclear localization signal is essential for stress-induced dimer-to-trimer transition of heat shock transcription factor 3. J. Biol. Chem. 2000, 275, 34665–34671. [Google Scholar] [CrossRef]
- Pike, C.J.; Walencewicz-Wasserman, A.J.; Kosmoski, J.; Cribbs, D.H.; Glabe, C.G.; Cotman, C.W. Structure-activity analyses of beta-amyloid peptides: Contributions of the beta 25-35 region to aggregation and neurotoxicity. J. Neurochem. 1995, 64, 253–265. [Google Scholar] [CrossRef]
- Nakamura, R.; Konishi, M.; Higashi, Y.; Saito, M.; Akizawa, T. Five-mer peptides prevent short-term spatial memory deficits in Aβ25-35-induced Alzheimer’s model mouse by suppressing Aβ25-35 aggregation and resolving its aggregate form. Alzheimers Res. Ther. 2023, 15, 83. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Akizawa, T.; Konishi, M. Structure–Activity Relationship of 5-mer Catalytides, GSGYR and RYGSG. Biomolecules 2022, 12, 1766. [Google Scholar] [CrossRef]
- Hatakawa, Y.; Nakamura, R.; Akizawa, T.; Konishi, M.; Matsuda, A.; Oe, T.; Saito, M.; Ito, F. SKGQA, a Peptide Derived from the ANA/BTG3 Protein, Cleaves Amyloid-β with Proteolytic Activity. Biomolecules 2024, 14, 586. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kamatsuka, Y.; Ohishi, A.; Nishida, K.; Nagasawa, K. P2X7 receptors regulate engulfing activity of non-stimulated resting astrocytes. Biochem. Biophys. Res. Commun. 2013, 439, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Aratake, T.; Shimizu, S.; Shimizu, T.; Nakamura, K.; Tsuda, M.; Yawata, T.; Ueba, T.; Saito, M. Influence of extracellular zinc on M1 microglial activation. Sci. Rep. 2017, 7, 43778. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-Β25-35-induced learning and memory impairment in mice, an experimental model of alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Namsechi, R.; Sim, V.L. Structure-based peptide design to modulate amyloid beta aggregation and reduce cytotoxicity. PLoS ONE 2015, 10, e0129087. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.K.; Samuel, D.; Jayaraman, G.; Srimathi, T.; Yu, C. The role of proline in the prevention of aggregation during protein folding in vitro. Biochem. Mol. Biol. Int. 1998, 46, 509–517. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Li, Q.; Wu, Y.; Chen, J.; Xuan, A.; Wang, X. Microglia and immunotherapy in Alzheimer’s disease. Acta Neurol. Scand. 2022, 145, 273–278. [Google Scholar] [CrossRef]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Colonna, M. Microglia in Alzheimer’s disease: A target for immunotherapy. J. Leukoc. Biol. 2019, 106, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takayama, K.; Furubayashi, T.; Mori, K.; Takemura, Y.; Amano, M.; Maeda, C.; Inoue, D.; Kimura, S.; Kiriyama, A.; et al. Transnasal Delivery of the Peptide Agonist Specific to Neuromedin-U Receptor 2 to the Brain for the Treatment of Obesity. Mol. Pharm. 2020, 17, 32–39. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | |
|---|---|
| YS-YE20 | YKNMRETLVYLTHLDYDDTE |
| YS-YR5 | YKNMR |
| YS-DE6 | DYDDTE |
| YS-RD11 | RETLVYLTHLD |
| YS-YD15 | YKNMRETLVYLTHLD |
| YS-RE16 | RETLVYLTHLDYDDTE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, R.; Matsuda, A.; Higashi, Y.; Hayashi, Y.; Konishi, M.; Saito, M.; Akizawa, T. An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model. Biomolecules 2024, 14, 1234. https://doi.org/10.3390/biom14101234
Nakamura R, Matsuda A, Higashi Y, Hayashi Y, Konishi M, Saito M, Akizawa T. An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model. Biomolecules. 2024; 14(10):1234. https://doi.org/10.3390/biom14101234
Chicago/Turabian StyleNakamura, Rina, Akira Matsuda, Youichirou Higashi, Yoshihiro Hayashi, Motomi Konishi, Motoaki Saito, and Toshifumi Akizawa. 2024. "An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model" Biomolecules 14, no. 10: 1234. https://doi.org/10.3390/biom14101234