
Poly(ADP-Ribose) Polymerase (PARP) Inhibitors for Cancer Therapy: Advances, Challenges, and Future Directions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
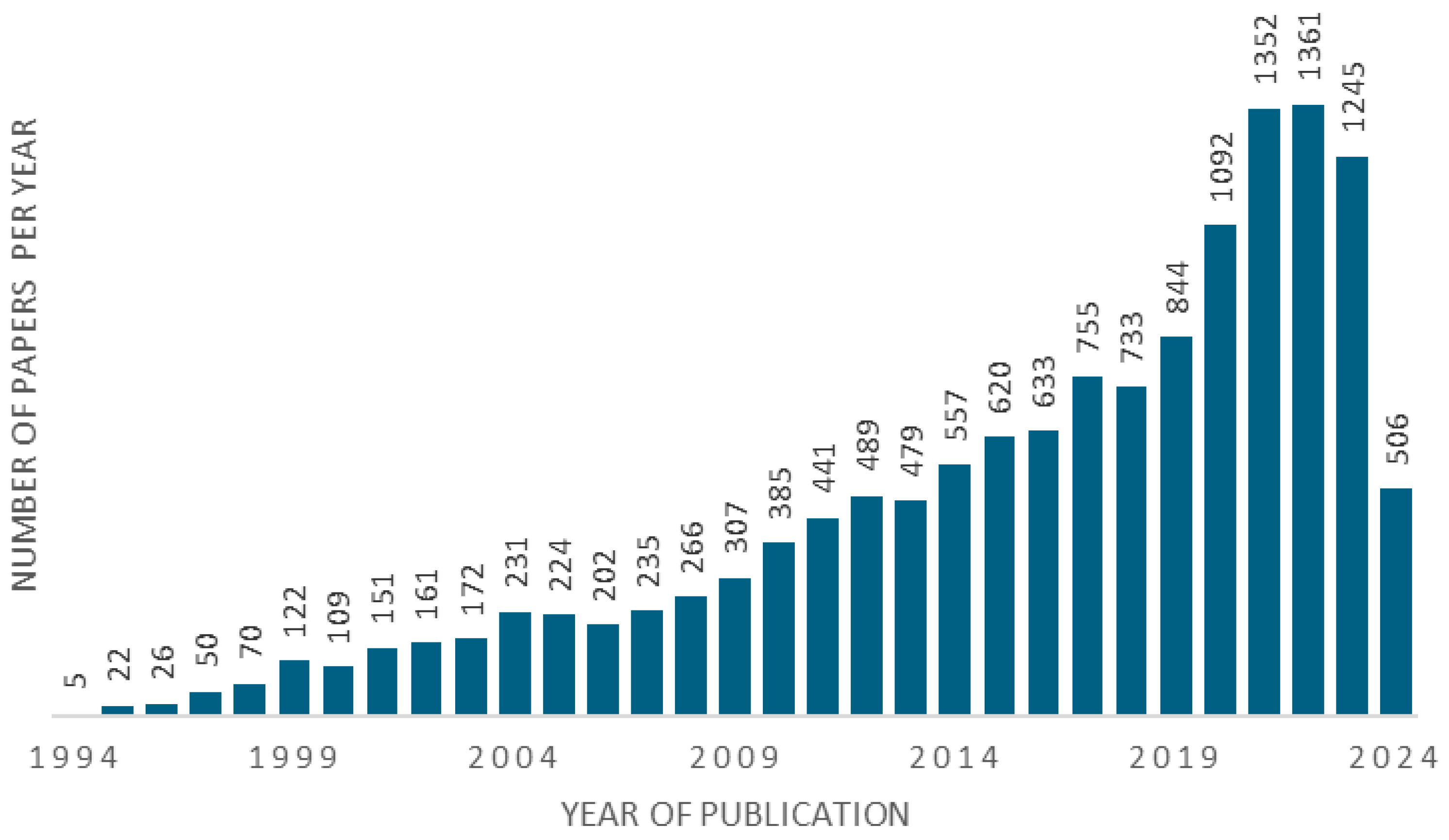
:1. Introduction
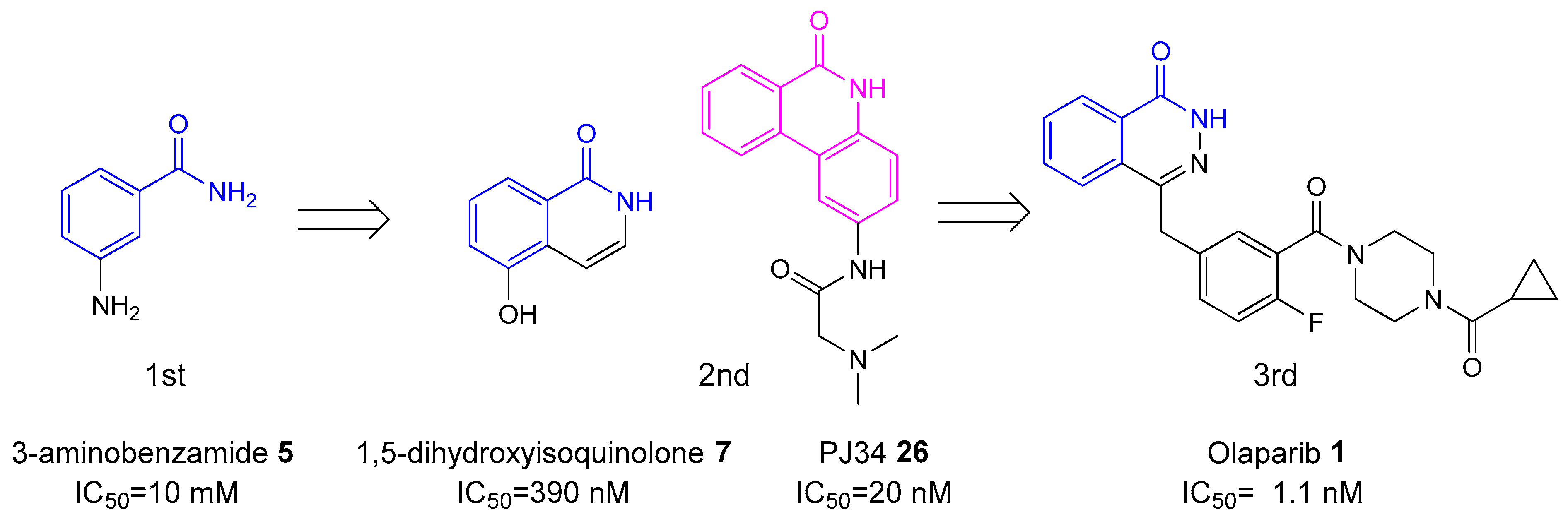
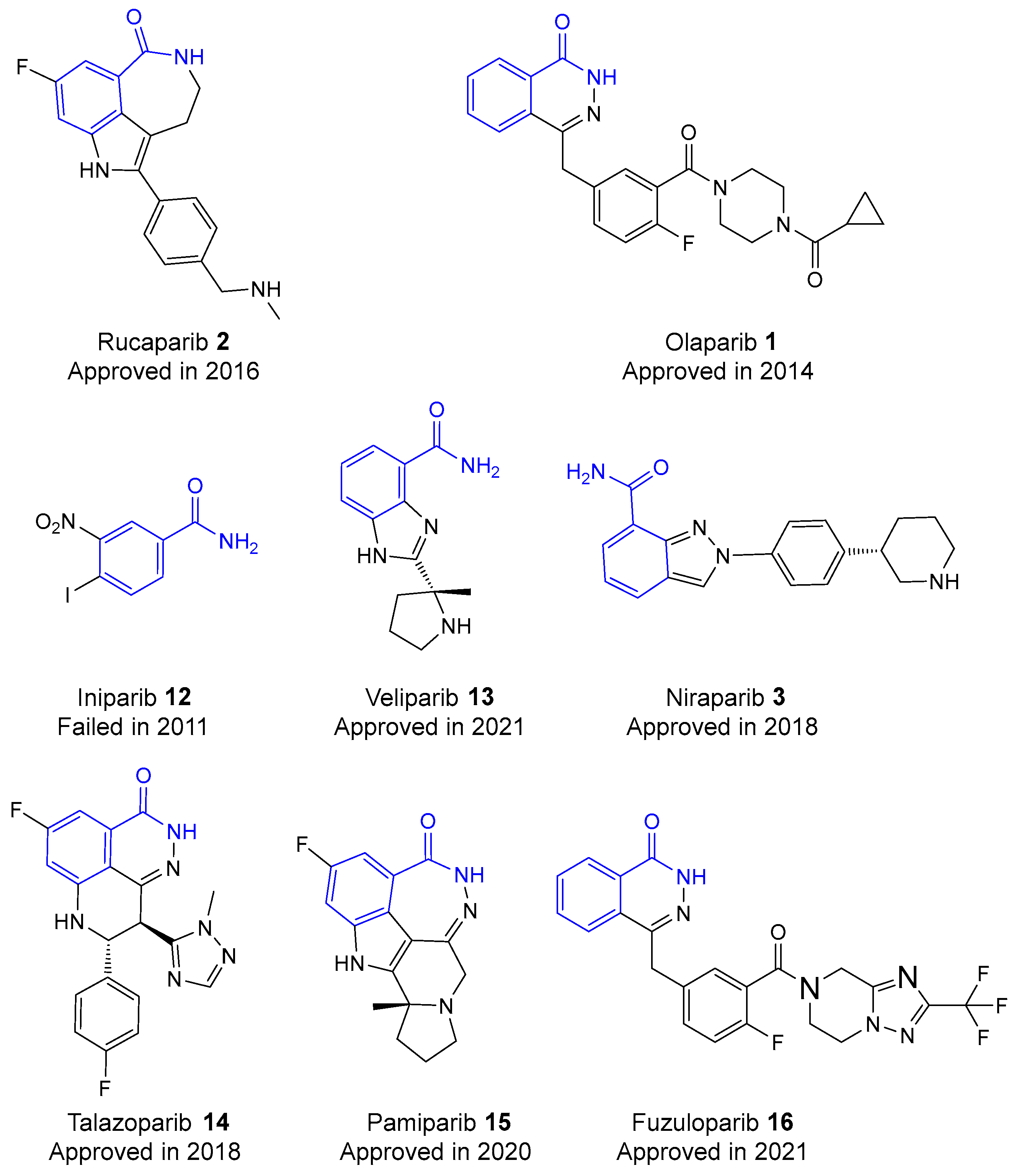
2. History of Development of PARP Inhibitors
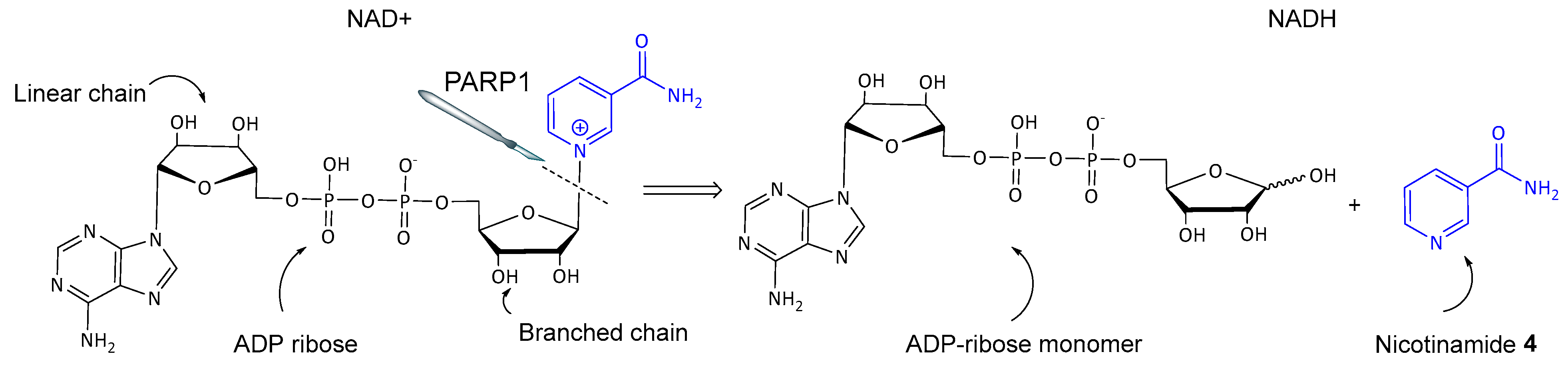
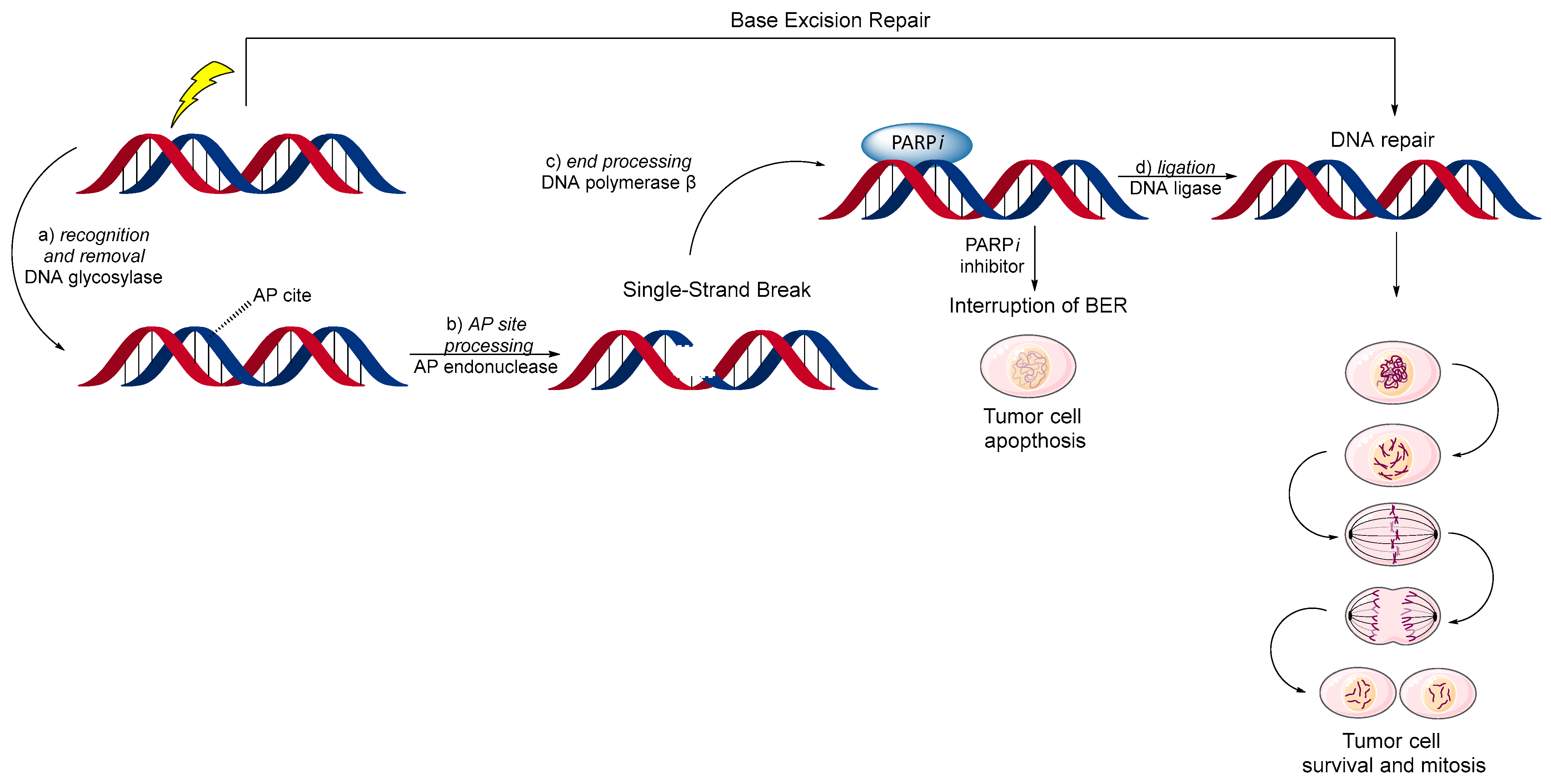
3. Base Excision Repair (BER) Mechanism
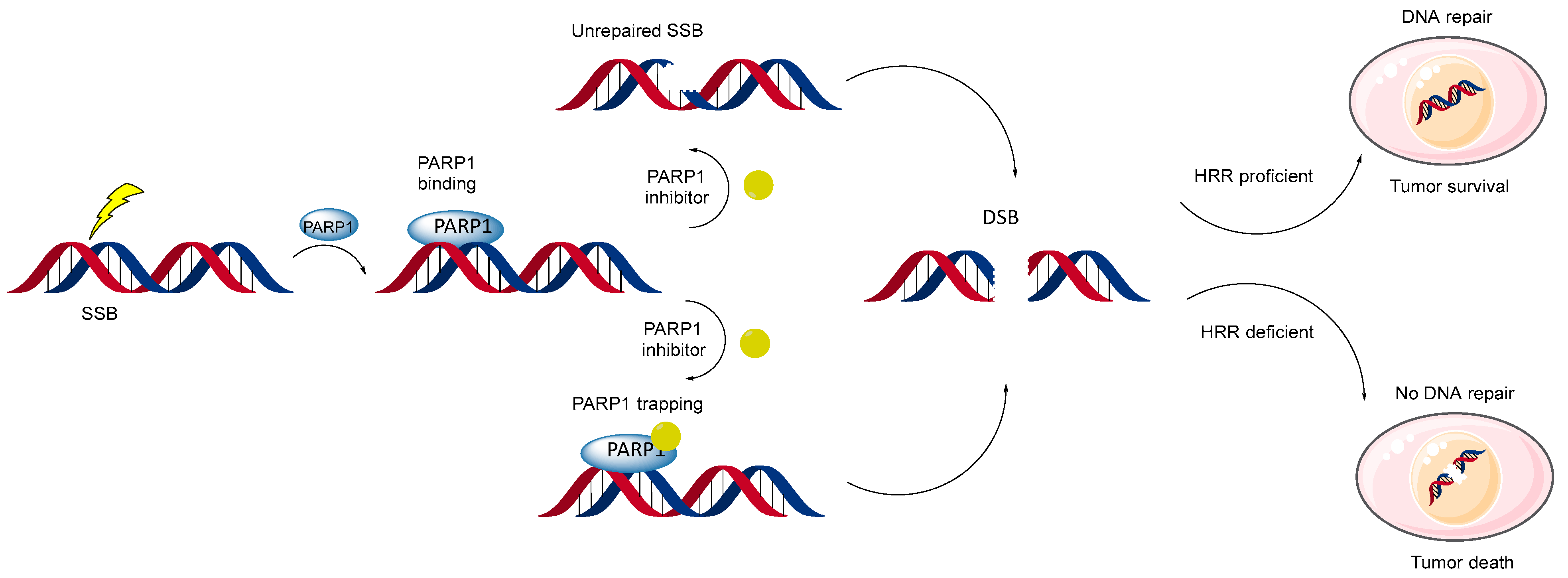
4. Two Models of Action of PARP Inhibitors
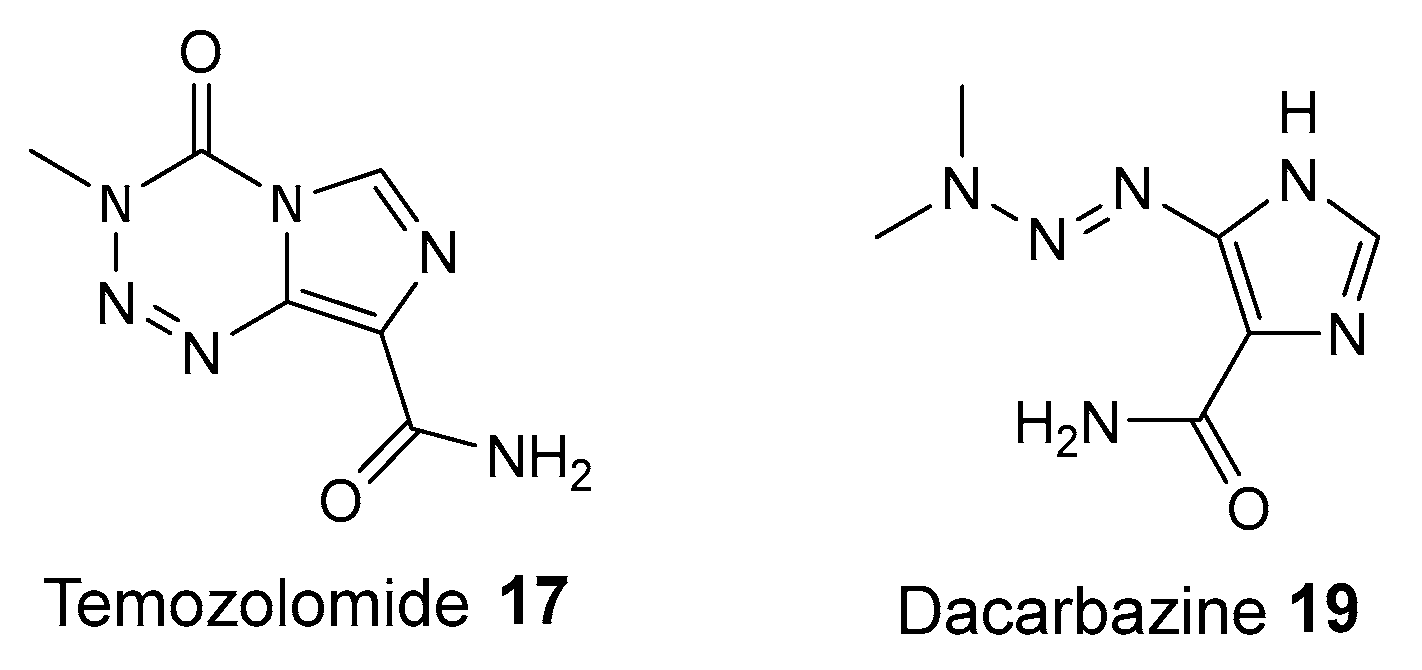
5. Synergy of PARP1 Inhibitors and DNA Methylating Agents
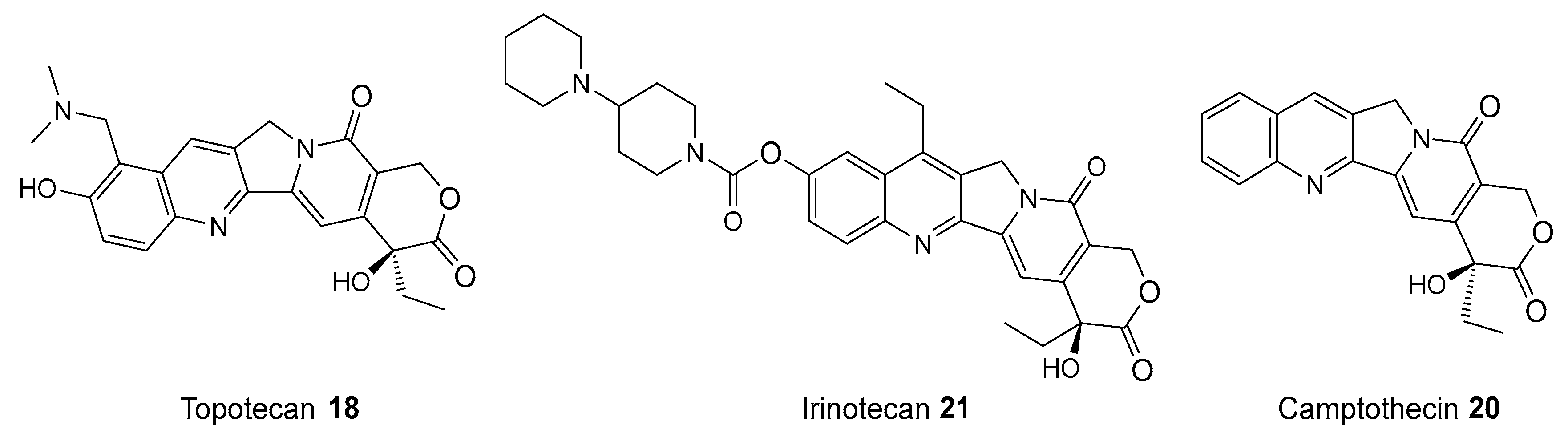
6. Synergy of PARP1 Inhibitors and Topoisomerase I (TOP1) Inhibitors
7. Synergy of PARP1 Inhibitors and Radiotherapy
8. PARP1 Inhibitors with Cytostatic Drugs
9. Dual PARP and HDAC (Histone Deacetylases) Inhibitors
10. Problems of Using Existing PARP1 Inhibitors
11. Perspectives in Greener Synthesis of PARP Inhibitors
12. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Sun, Y.; Li, C.; Xue, Y.; Ba, X. Targeting the DNA Damage Response for Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 15907. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Deng, W.; Lin, S.H. Poly (ADP-ribose) polymerases (PARPs) and PARP inhibitor-targeted therapeutics. Anticancer Agents Med. Chem. 2019, 19, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.Y.; Jie, Y.E.; Cheng, S.W.; Ling, G.L.; Ming, H.V.Y. PARP Inhibitors in Breast and Ovarian Cancer. Cancers 2023, 15, 2357. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pu, W.; Zheng, Q.; Ai, M.; Chen, S.; Peng, Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol. Cancer 2022, 21, 99. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Hou, J.; He, Z.; Liu, T.; Chen, D.; Wang, B.; Wen, Q.; Zheng, X. Evolution of Molecular Targeted Cancer Therapy: Mechanisms of Drug Resistance and Novel Opportunities Identified by CRISPR-Cas9 Screening. Front. Oncol. 2022, 12, 755053. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. Int. J. Mol. Sci. 2022, 23, 8412. [Google Scholar] [CrossRef]
- Alcaraz, M.J.; Megías, J.; García-Arnandis, I.; Clérigues, V.; Guillén, M.I. New molecular targets for the treatment of osteoarthritis. Biochem. Pharmacol. 2010, 80, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Agu, P.C.; Afiukwa, C.A.; Orji, O.U.; Ezeh, E.M.; Ofoke, I.H.; Ogbu, C.O.; Ugwuja, E.I.; Aja, P.M. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 2023, 13, 13398. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Löser, D.A.; Shibata, A.; Shibata, A.K.; Woodbine, L.J.; Jeggo, P.A.; Chalmers, A.J. Sensitization to Radiation and Alkylating Agents by Inhibitors of Poly(ADP-ribose) Polymerase Is Enhanced in Cells Deficient in DNA Double-Strand Break Repair. Mol. Cancer Ther. 2010, 9, 1775–1787. [Google Scholar] [CrossRef]
- Ali, A.A.E.; Timinszky, G.; Arribas-Bosacoma, R.; Kozlowski, M.; Hassa, P.O.; Hassler, M.; Ladurner, A.G.; Pearl, L.H.; Oliver, A.W. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012, 19, 685–692. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal 2010, 8, 31. [Google Scholar] [CrossRef]
- Shao, Z.; Lee, B.J.; Rouleau-Turcotte, É.; Langelier, M.-F.; Lin, X.; Estes, V.M.; Pascal, J.M.; Zha, S. Clinical PARP inhibitors do not abrogate PARP1 exchange at DNA damage sites in vivo. Nucleic Acids Res. 2020, 48, 9694–9709. [Google Scholar] [CrossRef]
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef]
- Kupczyk, P.; Simiczyjew, A.; Marczuk, J.; Dratkiewicz, E.; Beberok, A.; Rok, J.; Pieniazek, M.; Biecek, P.; Nevozhay, D.; Slowikowski, B.; et al. PARP1 as a Marker of an Aggressive Clinical Phenotype in Cutaneous Melanoma—A Clinical and an In Vitro Study. Cells 2021, 10, 286. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional Roles of PARP1 in Cancer. Mol. Cancer Res. 2014, 12, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Wang, P.Y.; Wang, Y.T.; Yang, G.F.; Zhang, A.; Miao, Z.H. An update on poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors: Opportunities and challenges in cancer therapy. J. Med. Chem. 2016, 59, 9575–9598. [Google Scholar] [CrossRef]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.D.; Xu, X.Q.; Peng, F.; Yu, J.Z.; Wu, H. The poly(ADP-ribose) polymerase-1 inhibitor 3-aminobenzamide suppresses cell growth and migration, enhancing suppressive effects of cisplatin in osteosarcoma cells. Oncol. Rep. 2011, 25, 1399–1405. [Google Scholar]
- Liang, B.Y.; Xiong, M.; Ji, G.B.; Zhang, E.L.; Zhang, Z.Y.; Dong, K.S.; Chen, X.P.; Huang, Z.Y. Synergistic Suppressive Effect of PARP-1 Inhibitor PJ34 and HDAC Inhibitor SAHA on Proliferation of Liver Cancer Cell. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 535–540. [Google Scholar] [CrossRef]
- El-Hamoly, T.; El-Denshary, E.S.; Saad, S.M.; El-Ghazaly, M.A. 3-aminobenzamide, a poly (ADP ribose) polymerase inhibitor, enhances wound healing in whole body gamma irradiated model. Wound Repair. Regen. 2015, 23, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Wasyluk, W.; Zwolak, A. PARP Inhibitors: An Innovative Approach to the Treatment of Inflammation and Metabolic Disorders in Sepsis. J. Inflamm. Res. 2021, 14, 1827–1844. [Google Scholar] [CrossRef]
- Canan, S.; Maegley, K.; Curtin, N.J. Strategies Employed for the Development of PARP Inhibitors. In Poly (ADP-Ribose) Polymerase: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2017; pp. 271–297. [Google Scholar]
- Chen, Y.; Zhang, L.; Hao, Q. Olaparib: A promising PARP inhibitor in ovarian cancer therapy. Arch. Gynecol. Obs. Obstet. 2013, 288, 367–374. [Google Scholar] [CrossRef]
- Jagtap, P.G.; Baloglu, E.; Southan, G.J.; Mabley, J.G.; Li, H.; Zhou, J.; Van Duzer, J.; Salzman, A.L.; Szabó, C. Discovery of potent poly(ADP-ribose) polymerase-1 inhibitors from the modification of indeno[1,2-c]isoquinolinone. J. Med. Chem. 2005, 48, 5100–5103. [Google Scholar] [CrossRef]
- Inbar-Rozensal, D.; Castiel, A.; Visochek, L.; Castel, D.; Dantzer, F.; Izraeli, S.; Cohen-Armon, M. A selective eradication of human nonhereditary breast cancer cells by phenanthridine-derived polyADP-ribose polymerase inhibitors. Breast Cancer Res. 2009, 11, R78. [Google Scholar] [CrossRef]
- Malyuchenko, N.V.; Kotova, E.Y.; Kulaeva, O.I.; Kirpichnikov, M.P.; Studitskiy, V.M. PARP1 Inhibitors: Antitumor drug design. Acta Naturae 2015, 7, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Marsischky, G.T.; Wilson, B.A.; Collier, R.J. Role of Glutamic Acid 988 of Human Poly-ADP-ribose Polymerase in Polymer Formation. J. Biol. Chem. 1995, 270, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. The Development of Rucaparib/Rubraca®: A Story of the Synergy Between Science and Serendipity. Cancers 2020, 12, 564. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.; de Bono, J. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Kristeleit, R.; Lisyanskaya, A.; Fedenko, A.; Dvorkin, M.; de Melo, A.C.; Shparyk, Y.; Rakhmatullina, I.; Bondarenko, I.; Colombo, N.; Svintsitskiy, V.; et al. Rucaparib versus standard-of-care chemotherapy in patients with relapsed ovarian cancer and a deleterious BRCA1 or BRCA2 mutation (ARIEL4): An international, open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Maiorano, B.A.; Maiorano, M.F.P.; Maiello, E. Olaparib and advanced ovarian cancer: Summary of the past and looking into the future. Front. Pharmacol. 2023, 14, 1162665. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef]
- Wesolowski, R.; Stover, D.G.; Lustberg, M.B.; Shoben, A.; Zhao, M.; Mrozek, E.; Layman, R.M.; Macrae, E.; Duan, W.; Zhang, J.; et al. Phase I Study of Veliparib on an Intermittent and Continuous Schedule in Combination with Carboplatin in Metastatic Breast Cancer: A Safety and [18F]-Fluorothymidine Positron Emission Tomography Biomarker Study. Oncologist 2020, 25, e1158–e1169. [Google Scholar] [CrossRef]
- Sisay, M.; Edessa, D. PARP inhibitors as potential therapeutic agents for various cancers: Focus on niraparib and its first global approval for maintenance therapy of gynecologic cancers. Gynecol. Oncol. Res. Pr. 2017, 4, 18. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Eng. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Chi, Z.; Bai, Y.; Li, J.; Wang, K.; Xu, Y.; Luan, Y. Design, synthesis and antitumor activity study of PARP-1/HDAC dual targeting inhibitors. Bioorg. Med. Chem. Lett. 2022, 71, 128821. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Wang, J.; Mishail, D.; Wang, C.-Y. Recent advancements in PARP inhibitors-based targeted cancer therapy. Precis. Clin. Med. 2020, 3, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Fan, F.; Zhang, B.; Hu, Y.; Sun, C. Cardiovascular-specific mortality among multiple myeloma patients: A population-based study. Ther. Adv. Hematol. 2022, 13, 204062072210867. [Google Scholar] [CrossRef]
- Chan, C.Y.; Tan, K.V.; Cornelissen, B. PARP Inhibitors in Cancer Diagnosis and Therapy. Clin. Cancer Res. 2021, 27, 1585–1594. [Google Scholar] [CrossRef]
- Friedlander, M.; Lee, Y.C.; Tew, W.P. Managing Adverse Effects Associated with Poly (ADP-ribose) Polymerase Inhibitors in Ovarian Cancer: A Synthesis of Clinical Trial and Real-World Data. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390876. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; de Lorenzo, S.B.; Flatten, K.S.; Poirier, G.G.; Kaufmann, S.H. Failure of iniparib to inhibit poly(ADP-ribose) polymerase in vitro. Clin. Cancer Res. 2012, 18, 1655–1662. [Google Scholar] [CrossRef]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Yelamos, J.; Farres, J.; Llacuna, L.; Ampurdanes, C.; Martin-Caballero, J. PARP-1 and PARP-2: New players in tumour development. Am. J. Cancer Res. 2011, 1, 328–346. [Google Scholar]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Dilmac, S.; Ozpolat, B. Mechanisms of PARP-Inhibitor-Resistance in BRCA-Mutated Breast Cancer and New Therapeutic Approaches. Cancers 2023, 15, 3642. [Google Scholar] [CrossRef]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Krais, J.J.; Johnson, N. BRCA1 Mutations in Cancer: Coordinating Deficiencies in Homologous Recombination with Tumorigenesis. Cancer Res. 2020, 80, 4601–4609. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.-H.; Seow, K.-M.; Chen, K.-H. The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 8125. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Lin, X.; Jiang, W.; Rudolph, J.; Lee, B.J.; Luger, K.; Zha, S. PARP inhibitors trap PARP2 and alter the mode of recruitment of PARP2 at DNA damage sites. Nucleic Acids Res. 2022, 50, 3958–3973. [Google Scholar] [CrossRef]
- Jurkovicova, D.; Neophytou, C.M.; Gašparović, A.Č.; Gonçalves, A.C. DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. Int. J. Mol. Sci. 2022, 23, 14672. [Google Scholar] [CrossRef]
- Bhamidipati, D.; Haro-Silerio, J.I.; Yap, T.A.; Ngoi, N. PARP inhibitors: Enhancing efficacy through rational combinations. Br. J. Cancer 2023, 129, 904–916. [Google Scholar] [CrossRef]
- Rivero Belenchón, I.; Congregado Ruiz, C.B.; Saez, C.; Osman García, I.; Medina López, R.A. Parp Inhibitors and Radiotherapy: A New Combination for Prostate Cancer (Systematic Review). Int. J. Mol. Sci. 2023, 24, 12978. [Google Scholar] [CrossRef]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.; Hyodo, M.; Fujimori, H.; Shimizu, A.; Yoshioka, K. Sensitization of Cancer Cells to Radiation and Topoisomerase I Inhibitor Camptothecin Using Inhibitors of PARP and Other Signaling Molecules. Cancers 2018, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Chu, A.; Song, R.; Liu, S.; Chai, T.; Wang, X.; Liu, Z. PARP inhibitors combined with radiotherapy: Are we ready? Front. Pharmacol. 2023, 14, 1234973. [Google Scholar] [CrossRef] [PubMed]
- Vormoor, B.; Curtin, N.J. Poly(ADP-ribose) polymerase inhibitors in Ewing sarcoma. Curr. Opin. Oncol. 2014, 26, 428–433. [Google Scholar] [CrossRef]
- Angel, M.; Zarba, M.; Sade, J.P. PARP inhibitors as a radiosensitizer: A future promising approach in prostate cancer? Ecancermedicalscience 2021, 15, ed118. [Google Scholar] [CrossRef]
- Loap, P.; Loirat, D.; Berger, F.; Rodrigues, M.; Bazire, L.; Pierga, J.-Y.; Vincent-Salomon, A.; Laki, F.; Boudali, L.; Raizonville, L.; et al. Concurrent Olaparib and Radiotherapy in Patients with Triple-Negative Breast Cancer. JAMA Oncol. 2022, 8, 1802. [Google Scholar] [CrossRef]
- Powell, C.; Mikropoulos, C.; Kaye, S.B.; Nutting, C.M.; Bhide, S.A.; Newbold, K.; Harrington, K.J. Pre-clinical and clinical evaluation of PARP inhibitors as tumour-specific radiosensitisers. Cancer Treat. Rev. 2010, 36, 566–575. [Google Scholar] [CrossRef]
- Lesueur, P.; Chevalier, F.; El-Habr, E.A.; Junier, M.-P.; Chneiweiss, H.; Castera, L.; Müller, E.; Stefan, D.; Saintigny, Y. Radiosensitization Effect of Talazoparib, a Parp Inhibitor, on Glioblastoma Stem Cells Exposed to Low and High Linear Energy Transfer Radiation. Sci. Rep. 2018, 8, 3664. [Google Scholar] [CrossRef]
- Kleinberg, L.; Ye, X.; Supko, J.; Stevens, G.H.J.; Shu, H.-K.; Mikkelsen, T.; Lieberman, F.; Lesser, G.J.; Lee, E.; Grossman, S.A. A multi-site phase I trial of Veliparib with standard radiation and temozolomide in patients with newly diagnosed glioblastoma multiforme (GBM). J. Neurooncol. 2023, 165, 499–507. [Google Scholar] [CrossRef]
- Stupp, R.; Gander, M.; Leyvraz, S.; Newlands, E. Current and future developments in the use of temozolomide for the treatment of brain tumours. Lancet Oncol. 2001, 2, 552–560. [Google Scholar] [CrossRef]
- Boulton, S.; Pemberton, L.; Porteous, J.; Curtin, N.; Griffin, R.; Golding, B.; Durkacz, B. Potentiation of temozolomide-induced cytotoxicity: A comparative study of the biological effects of poly(ADP-ribose) polymerase inhibitors. Br. J. Cancer 1995, 72, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Schaff, L.R.; Kushnirsky, M.; Lin, A.L.; Nandakumar, S.; Grommes, C.; Miller, A.M.; Gavrilovic, I.T.; Nolan, C.; Pentsova, E.; Mellinghoff, I.K.; et al. Combination Olaparib and Temozolomide for the Treatment of Glioma. Neurology 2022, 99, 750–755. [Google Scholar] [CrossRef]
- Meador, C.B.; Digumarthy, S.; Yeap, B.Y.; Farago, A.F.; Heist, R.S.; Marcoux, J.P.; Rangachari, D.; Barbie, D.A.; Piotrowska, Z. Phase I/II investigator-initiated study of olaparib and temozolomide in SCLC: Updated analysis and CNS outcomes. J. Clin. Oncol. 2022, 40, 8565. [Google Scholar] [CrossRef]
- Mweempwa, A.; Wilson, M.K. Mechanisms of resistance to PARP inhibitors—An evolving challenge in oncology. Cancer Drug Resist. 2019, 2, 608–617. [Google Scholar] [CrossRef]
- Lupo, B.; Trusolino, L. Inhibition of poly(ADP-ribosyl)ation in cancer: Old and new paradigms revisited. Biochim. Biophys. Acta -Rev. Cancer 2014, 1846, 201–215. [Google Scholar] [CrossRef]
- Xu, J.; Keenan, T.E.; Overmoyer, B.; Tung, N.M.; Gelman, R.S.; Habin, K.; Garber, J.E.; Ellisen, L.W.; Winer, E.P.; Goss, P.E.; et al. Phase II trial of veliparib and temozolomide in metastatic breast cancer patients with and without BRCA1/2 mutations. Breast Cancer Res. Treat. 2021, 189, 641–651. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Ao, D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. Med. Comm. 2021, 2, 654–691. [Google Scholar] [CrossRef]
- O’Leary, J.; Muggia, F.M. Camptothecins: A review of their development and schedules of administration. Eur. J. Cancer 1998, 34, 1500–1508. [Google Scholar] [CrossRef]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for Poly(ADP-ribose) Polymerase (PARP) Inhibitors in Combination Therapy with Camptothecins or Temozolomide Based on PARP Trapping versus Catalytic Inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef]
- Das, S.K.; Rehman, I.; Ghosh, A.; Sengupta, S.; Majumdar, P.; Jana, B.; Das, B.B. Poly(ADP-ribose) polymers regulate DNA topoisomerase I (Top1) nuclear dynamics and camptothecin sensitivity in living cells. Nucleic Acids Res. 2016, 44, 8363–8375. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Yap, T.A.; de Bono, J.S. Poly(ADP-ribose) polymerase inhibitors in cancer treatment: A clinical perspective. Eur. J. Cancer 2010, 46, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef]
- Buck, J.; Dyer, P.J.C.; Hii, H.; Carline, B.; Kuchibhotla, M.; Byrne, J.; Howlett, M.; Whitehouse, J.; Ebert, M.A.; McDonald, K.L.; et al. Veliparib Is an Effective Radiosensitizing Agent in a Preclinical Model of Medulloblastoma. Front. Mol. Biosci. 2021, 8, 633344. [Google Scholar] [CrossRef]
- Bernges, F.; Zeller, W.J. Combination effects of poly(ADP-ribose) polymerase inhibitors and DNA-damaging agents in ovarian tumor cell lines—With special reference to cisplatin. J. Cancer Res. Clin. Oncol. 1996, 122, 665–670. [Google Scholar] [CrossRef]
- Cohen-Armon, M. The modified phenanthridine PJ34 unveils an exclusive cell-death mechanism in human cancer cells. Cancers 2020, 12, 1628. [Google Scholar] [CrossRef] [PubMed]
- Yusoh, N.A.; Ahmad, H.; Gill, M.R. Combining PARP Inhibition with Platinum, Ruthenium or Gold Complexes for Cancer Therapy. ChemMedChem 2020, 15, 2121–2135. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Shirbhate, E.; Singh, V.; Jahoriya, V.; Mishra, A.; Veerasamy, R.; Tiwari, A.K.; Rajak, H. Dual inhibitors of HDAC and other epigenetic regulators: A novel strategy for cancer treatment. Eur. J. Med. Chem. 2024, 263, 115938. [Google Scholar] [CrossRef]
- Ramos, L.; Truong, S.; Zhai, B.; Joshi, J.; Ghaidi, F.; Lizardo, M.M.; Shyp, T.; Kung, S.H.Y.; Rezakhanlou, A.M.; Oo, H.Z.; et al. A Bifunctional PARP-HDAC Inhibitor with Activity in Ewing Sarcoma. Clin. Cancer Res. 2023, 29, 3541–3553. [Google Scholar] [CrossRef]
- Lechner, S.; Malgapo, M.I.P.; Grätz, C.; Steimbach, R.R.; Baron, A.; Rüther, P.; Nadal, S.; Stumpf, C.; Loos, C.; Ku, X.; et al. Target deconvolution of HDAC pharmacopoeia reveals MBLAC2 as common off-target. Nat. Chem. Biol. 2022, 18, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Chen, S.; Sun, Q.; Wang, N.; Li, D.; Miao, S.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Olaparib hydroxamic acid derivatives as dual PARP and HDAC inhibitors for cancer therapy. Bioorg. Med. Chem. 2017, 25, 4100–4109. [Google Scholar] [CrossRef]
- Bondar, D.; Bragina, O.; Lee, J.Y.; Semenyuta, I.; Järving, I.; Brovarets, V.; Wipf, P.; Bahar, I.; Karpichev, Y. Hydroxamic Acids as PARP-1 Inhibitors: Molecular Design and Anticancer Activity of Novel Phenanthridinones. Helv. Chim. Acta 2023, 106, e202300133. [Google Scholar] [CrossRef]
- Colson, K.; Doss, D.S.; Swift, R.; Tariman, J.; Thomas, T.E. Bortezomib, a Newly Approved Proteasome Inhibitor for the Treatment of Multiple Myeloma: Nursing Implications. Clin. J. Oncol. Nurs. 2004, 8, 473–480. [Google Scholar] [CrossRef]
- Gupta, N.; Hanley, M.J.; Diderichsen, P.M.; Yang, H.; Ke, A.; Teng, Z.; Labotka, R.; Berg, D.; Patel, C.; Liu, G.; et al. Model-Informed Drug Development for Ixazomib, an Oral Proteasome Inhibitor. Clin. Pharmacol. Ther. 2019, 105, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.J.; Clasen, S.; Hwang, W.-T.; Garfall, A.; Vogl, D.T.; Carver, J.; O’Quinn, R.; Cohen, A.D.; Stadtmauer, E.A.; Ky, B.; et al. Carfilzomib-Associated Cardiovascular Adverse Events. JAMA Oncol. 2018, 4, e174519. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Lee, Y.-Y.; Park, J.-Y.; Shim, S.-H.; Kim, S.I.; Kong, T.-W.; Lim, C.K.; Cho, H.W.; Suh, D.H. Major clinical research advances in gynecologic cancer in 2022: Highlight on late-line PARP inhibitor withdrawal in ovarian cancer, the impact of ARIEL-4, and SOLO-3. J. Gynecol. Oncol. 2023, 34, e51. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Krivak, T.C.; Kabil, N.; Munley, J.; Moore, K.N. PARP Inhibitors in Ovarian Cancer: A Review. Target. Oncol. 2023, 18, 471–503. [Google Scholar] [CrossRef]
- Grygorenko, O.O.; Volochnyuk, D.M.; Ryabukhin, S.V.; Judd, D.B. The Symbiotic Relationship Between Drug Discovery and Organic Chemistry. Chem. Eur. J. 2020, 26, 1196–1237. [Google Scholar] [CrossRef]
- Puhlmann, N.; Vidaurre, R.; Kümmerer, K. Designing greener active pharmaceutical ingredients: Insights from pharmaceutical industry into drug discovery and development. Eur. J. Pharm. Sci. 2024, 192, 106614. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Li, J.J. (Eds.) The Art of Drug Synthesis; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Beutler, J.A. Natural Products as a Foundation for Drug Discovery. Curr. Protoc. Pharmacol. 2019, 86, e67. [Google Scholar] [CrossRef] [PubMed]
- Constable, D.J.C.; Jimenez-Gonzalez, C.; Henderson, R.K. Perspective on Solvent Use in the Pharmaceutical Industry. Org. Process Res. Dev. 2007, 11, 133–137. [Google Scholar] [CrossRef]
- Procopio, D.; Siciliano, C.; Trombino, S.; Dumitrescu, D.E.; Suciu, F.; Di Gioia, M.L. Green solvents for the formation of amide linkages. Org. Biomol. Chem. 2022, 20, 1137–1149. [Google Scholar] [CrossRef]
- Castiello, C.; Junghanns, P.; Mergel, A.; Jacob, C.; Ducho, C.; Valente, S.; Rotili, D.; Fioravanti, R.; Zwergel, C.; Mai, A. GreenMedChem: The challenge in the next decade toward eco-friendly compounds and processes in drug design. Green Chem. 2023, 25, 2109–2169. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Zimmerman, J.B.; Anastas, P.T.; Erythropel, H.C.; Leitner, W. Designing for a green chemistry future. Science 2020, 367, 397–400. [Google Scholar] [CrossRef]
- Erythropel, H.C.; Zimmerman, J.B.; de Winter, T.M.; Petitjean, L.; Melnikov, F.; Lam, C.H.; Lounsbury, A.W.; Mellor, K.E.; Janković, N.Z.; Tu, Q.; et al. The Green ChemisTREE: 20 years after taking root with the 12 principles. Green Chem. 2018, 20, 1929–1961. [Google Scholar] [CrossRef]
- Saxena, K.; Balani, S.; Srivastava, P. The role of pharmaceutical industry in building resilient health system. Front. Public. Health 2022, 10, 964899. [Google Scholar] [CrossRef]
- de Oliveira Souza, H.; dos Santos Costa, R.; Quadra, G.R.; dos Santos Fernandez, M.A. Pharmaceutical pollution and sustainable development goals: Going the right way? Sustain. Chem. Pharm. 2021, 21, 100428. [Google Scholar] [CrossRef]
- Wollensack, L.; Budzinski, K.; Backmann, J. Defossilization of pharmaceutical manufacturing. Curr. Opin. Green Sustain. Chem. 2022, 33, 100586. [Google Scholar] [CrossRef]
- Veleva, V.R.; Cue, B.W.; Todorova, S. Benchmarking Green Chemistry Adoption by the Global Pharmaceutical Supply Chain. ACS Sustain. Chem. Eng. 2018, 6, 2–14. [Google Scholar] [CrossRef]
- Borovika, A.; Albrecht, J.; Li, J.; Wells, A.S.; Briddell, C.; Dillon, B.R.; Diorazio, L.J.; Gage, J.R.; Gallou, F.; Koenig, S.G.; et al. The PMI Predictor app to enable green-by-design chemical synthesis. Nat. Sustain. 2019, 2, 1034–1040. [Google Scholar] [CrossRef]
- Sherer, E.C.; Bagchi, A.; Kosjek, B.; Maloney, K.M.; Peng, Z.; Robaire, S.A.; Sheridan, R.P.; Metwally, E.; Campeau, L.-C. Driving aspirational Process Mass Intensity using simple structure-based prediction. Org. Process Res. Dev. 2022, 26, 1405–1410. [Google Scholar] [CrossRef]
- Jimenez-Gonzalez, C.; Ponder, C.S.; Broxterman, Q.B.; Manley, J.B. Using the Right Green Yardstick: Why Process Mass Intensity is used in the pharmaceutical industry to drive more sustainable processes. Org. Process Res. Dev. 2011, 15, 912–917. [Google Scholar] [CrossRef]
- Benison, C.H.; Payne, P.R. Manufacturing mass intensity: 15 Years of Process Mass Intensity and development of the metric into plant cleaning and beyond. Curr. Res. Green. Sustain. Chem. 2022, 5, 100229. [Google Scholar] [CrossRef]
- Jimenez-Gonzalez, C.; Lund, C. Green metrics in pharmaceutical development. Curr. Opin. Green. Sustain. Chem. 2022, 33, 100564. [Google Scholar] [CrossRef]
- Luu, D.-N.; Gachet, H.; Maier, C.-J.; Maranzana, N.; Aoussat, A. Eco-design and medicine: Opportunities to implement eco-design in the pharmaceutical R&D process. J. Clean. Prod. 2022, 365, 132785. [Google Scholar]
- Gomollón-Bel, F. Ten Chemical Innovations That Will Change Our World: IUPAC identifies emerging technologies in Chemistry with potential to make our planet more sustainable. Chem. Int. 2019, 41, 12–17. [Google Scholar] [CrossRef]
- Milanović, I.; Biliškov, N.; Užarević, K.; Lukin, S.; Etter, M.; Halasz, I. Mechanochemical Synthesis and Thermal Dehydrogenation of Novel Calcium-Containing Bimetallic Amidoboranes. ACS Sustain. Chem. Eng. 2021, 9, 2089–2099. [Google Scholar] [CrossRef]
- Friščić, T.; Mottillo, C.; Titi, H.M. Mechanochemistry for Synthesis. Angew. Chem. Int. Ed. 2020, 59, 1018–1029. [Google Scholar] [CrossRef]
- Ardila-Fierro, K.J.; Hernández, J.G. Sustainability Assessment of Mechanochemistry by Using the Twelve Principles of Green Chemistry. ChemSusChem 2021, 14, 2145–2162. [Google Scholar] [CrossRef] [PubMed]
- Michalchuk, A.A.L.; Boldyreva, E.V.; Belenguer, A.M.; Emmerling, F.; Boldyrev, V.V. Tribochemistry, Mechanical Alloying, Mechanochemistry: What is in a Name? Front. Chem. 2021, 9, 685789. [Google Scholar] [CrossRef] [PubMed]
- Nikonovich, T.; Jarg, T.; Martõnova, J.; Kudrjašov, A.; Merzhyievskyi, D.; Kudrjašova, M.; Gallou, F.; Aav, R.; Kananovich, D. Protecting-group-free mechanosynthesis of amides from hydroxycarboxylic acids: Application to the synthesis of imatinib. RSC Mechanochemistry 2024, 1, 189–195. [Google Scholar] [CrossRef]
- Kayumov, M.; Jia, L.; Pardaev, A.; Song, S.-S.; Mirzaakhmedov, S.; Ding, C.; Cheng, Y.-J.; Zhang, R.I.; Bao, X.; Miao, Z.-H.; et al. Design, synthesis and pharmacological evaluation of new PARP1 inhibitors by merging pharmacophores of olaparib and the natural product alantolactone. Eur. J. Med. Chem. 2022, 240, 114574. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tan, H.-B.; Zhang, Y.-J.; Tang, D.-Y.; Zhan, F.; Li, H.; Chen, Z.-Z.; Xu, Z.-G. Catalyst-Free One-Pot Synthesis of Densely Substituted Pyrazole-Pyrazines as Anti-Colorectal Cancer Agents. Sci. Rep. 2020, 10, 9281. [Google Scholar] [CrossRef]
- Hughes, D.L. Applications of Flow Chemistry in the Pharmaceutical Industry—Highlights of the Recent Patent Literature. Org. Process Res. Dev. 2020, 24, 1850–1860. [Google Scholar] [CrossRef]
- Burange, A.S.; Osman, S.M.; Luque, R. Understanding flow chemistry for the production of active pharmaceutical ingredients. iScience 2022, 25, 103892. [Google Scholar] [CrossRef]
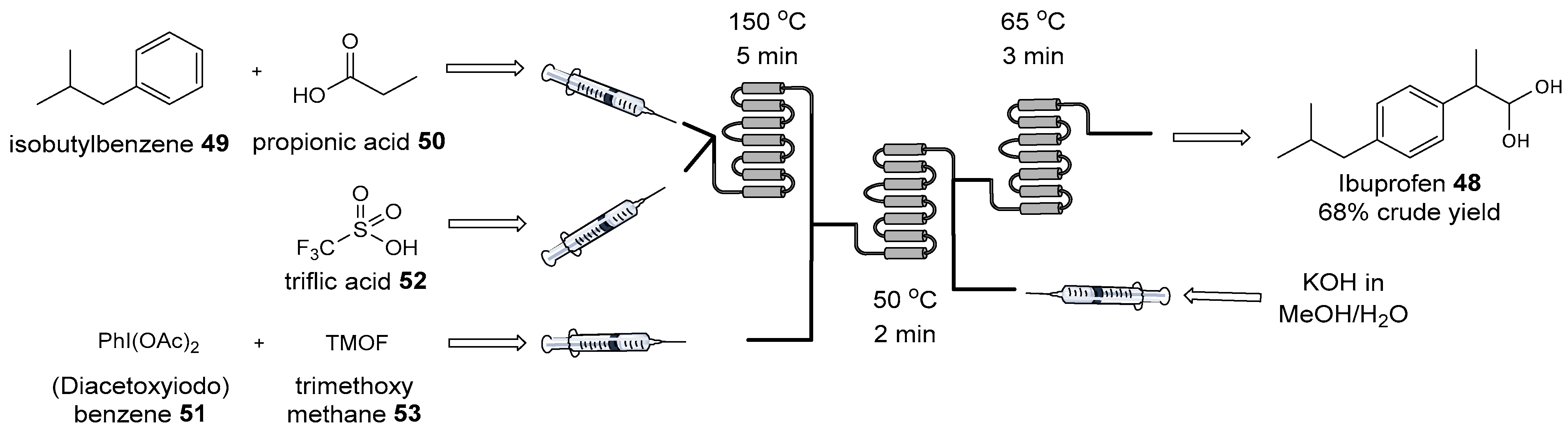
- Bogdan, A.R.; Poe, S.L.; Kubis, D.C.; Broadwater, S.J.; McQuade, D.T. The Continuous-Flow Synthesis of Ibuprofen. Angew. Chem. Int. Ed. 2009, 48, 8547–8550. [Google Scholar] [CrossRef]
- Baumann, M.; Moody, T.S.; Smyth, M.; Wharry, S. A Perspective on Continuous Flow Chemistry in the Pharmaceutical Industry. Org. Process Res. Dev. 2020, 24, 1802–1813. [Google Scholar] [CrossRef]
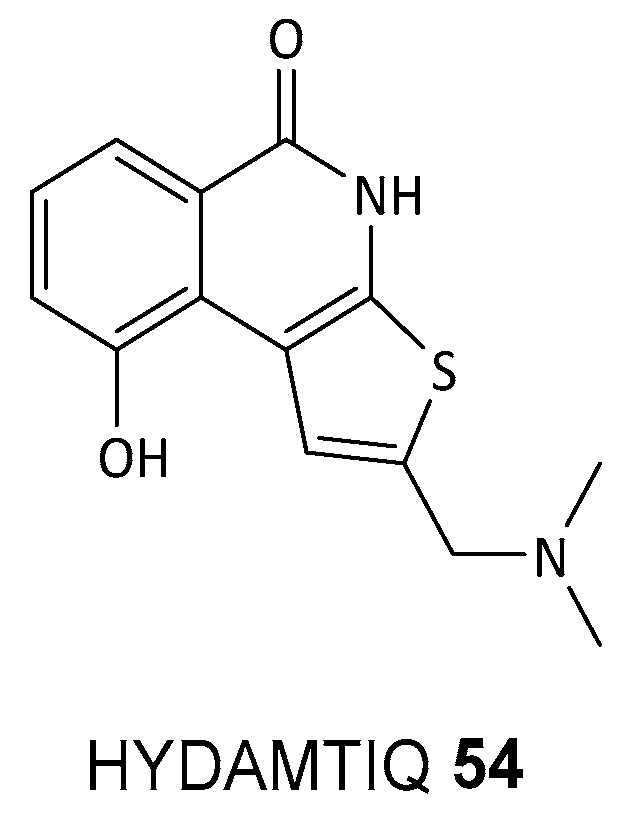
- Filipponi, P.; Cerra, B.; Piccinno, A.; Camaioni, E.; Gioiello, A. Continuous Flow Synthesis of the PARP-1/2 Inhibitor HYDAMTIQ: Synthetic Strategy, Optimization, and Green Metrics Evaluation. Org. Process Res. Dev. 2024, 28, 1648–1656. [Google Scholar] [CrossRef]
- Mini, E.; Landini, I.; Lucarini, L.; Lapucci, A.; Napoli, C.; Perrone, G.; Tassi, R.; Masini, E.; Moroni, F.; Nobili, S. The inhibitory effects of HYDAMTIQ, a novel PARP inhibitor, on growth in human tumor cell lines with defective DNA damage response pathways. Oncol. Res. 2017, 25, 1441–1451. [Google Scholar] [CrossRef] [PubMed]

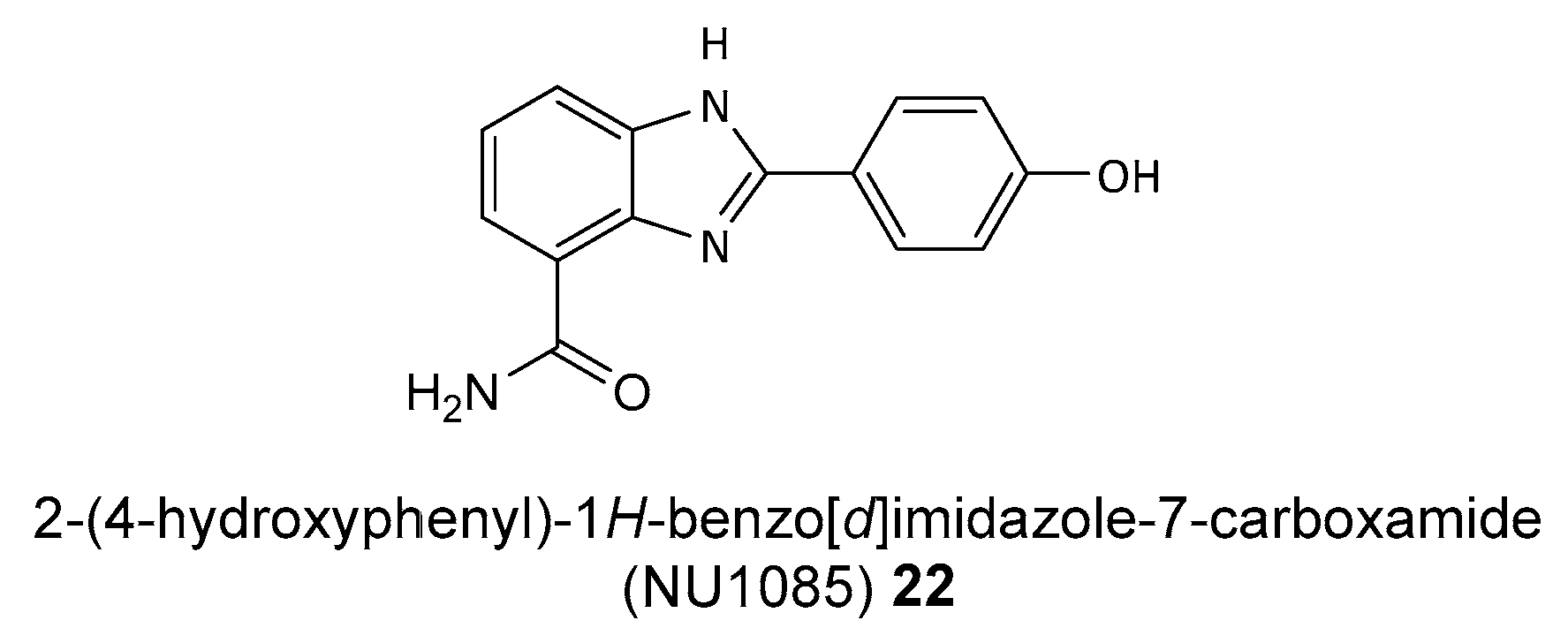
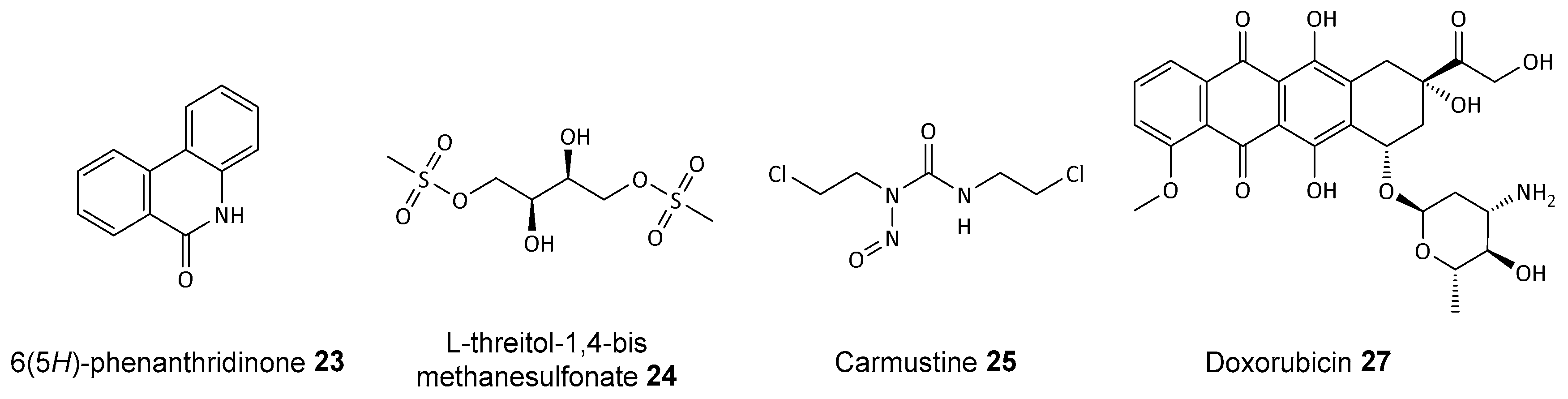
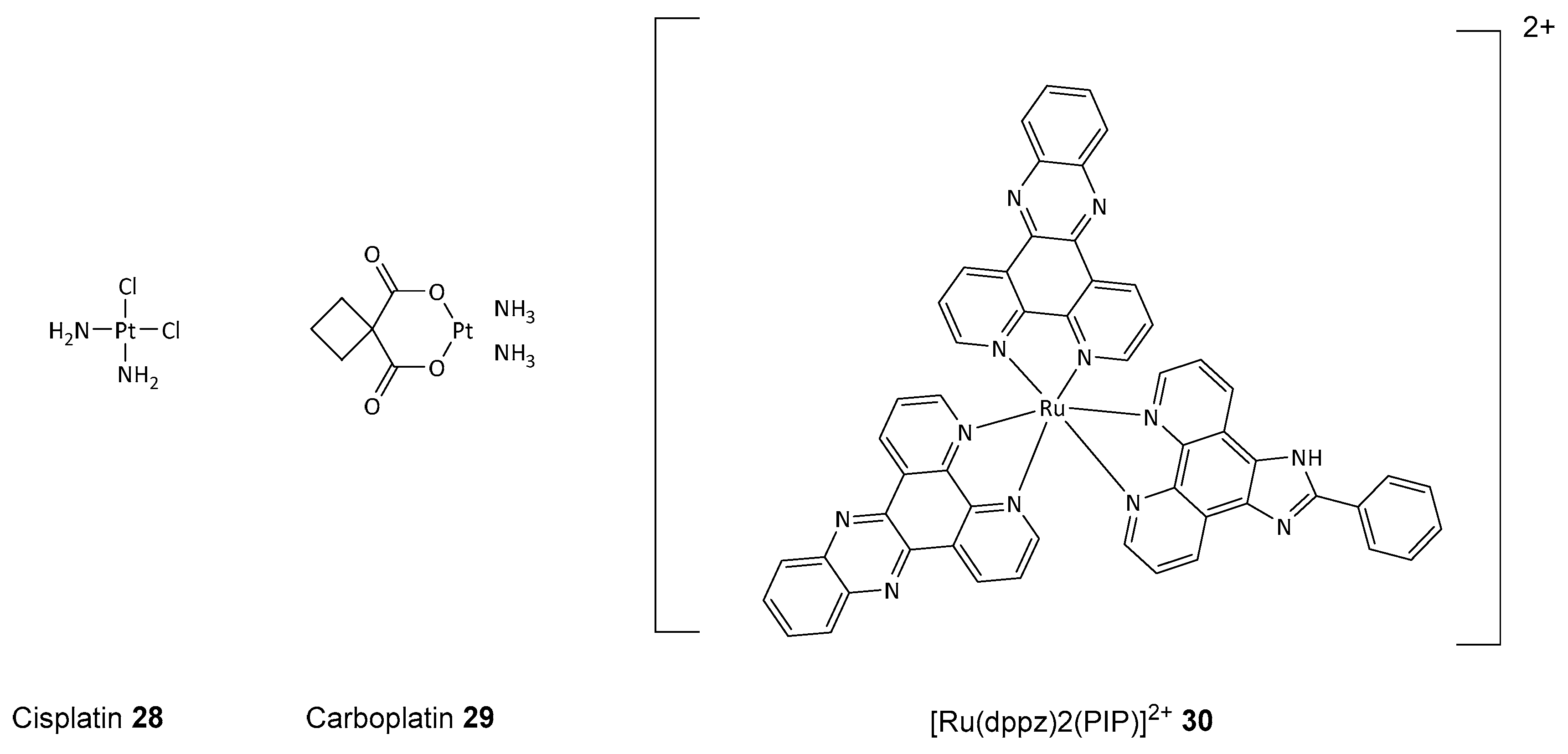
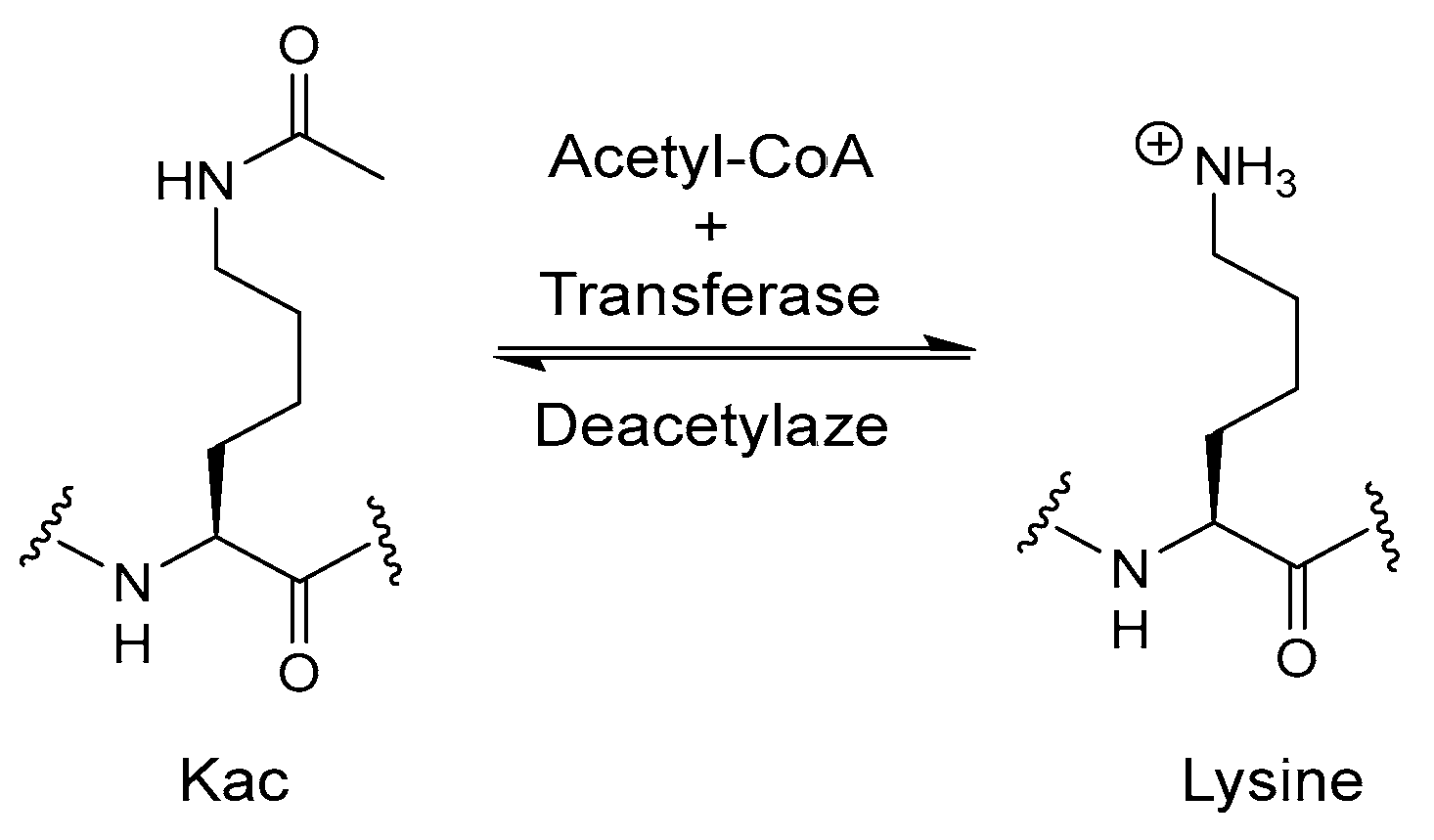

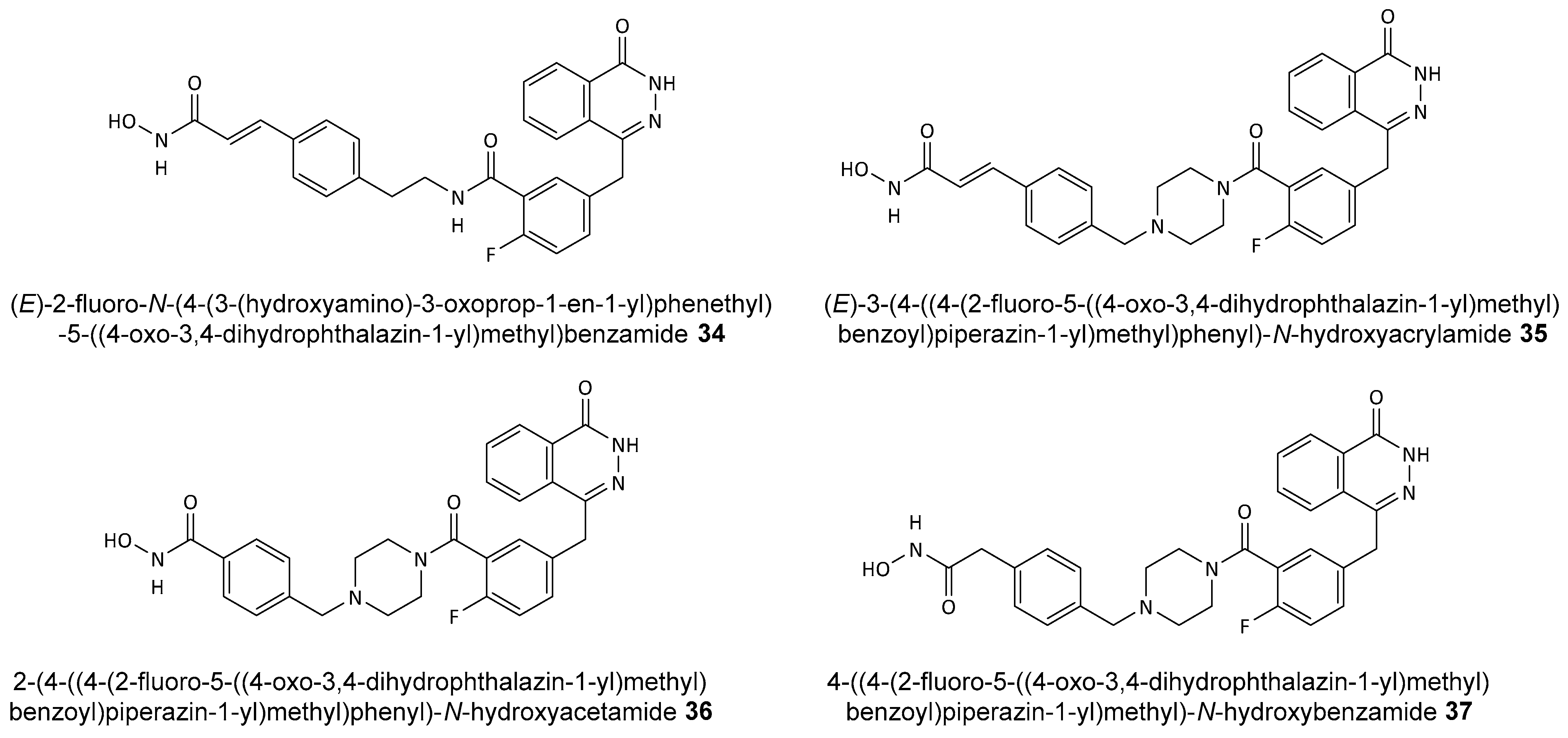
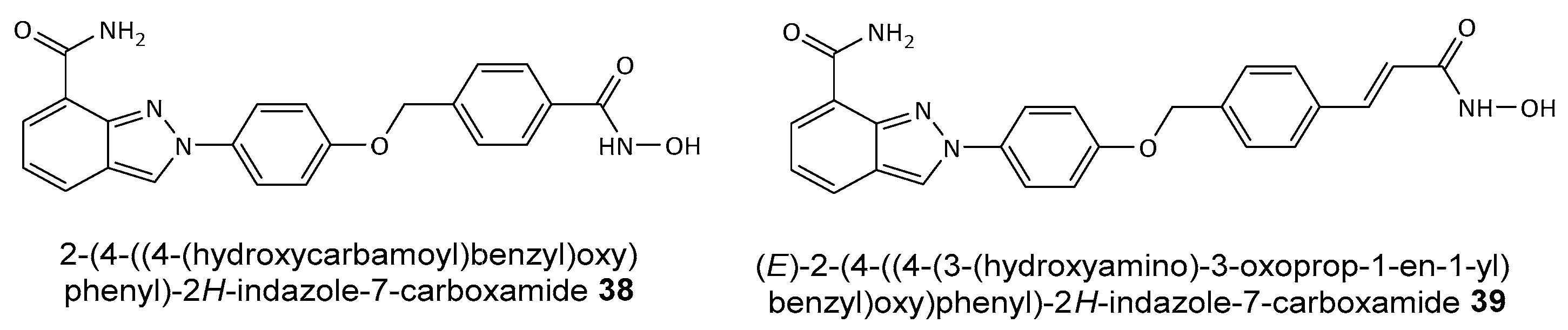
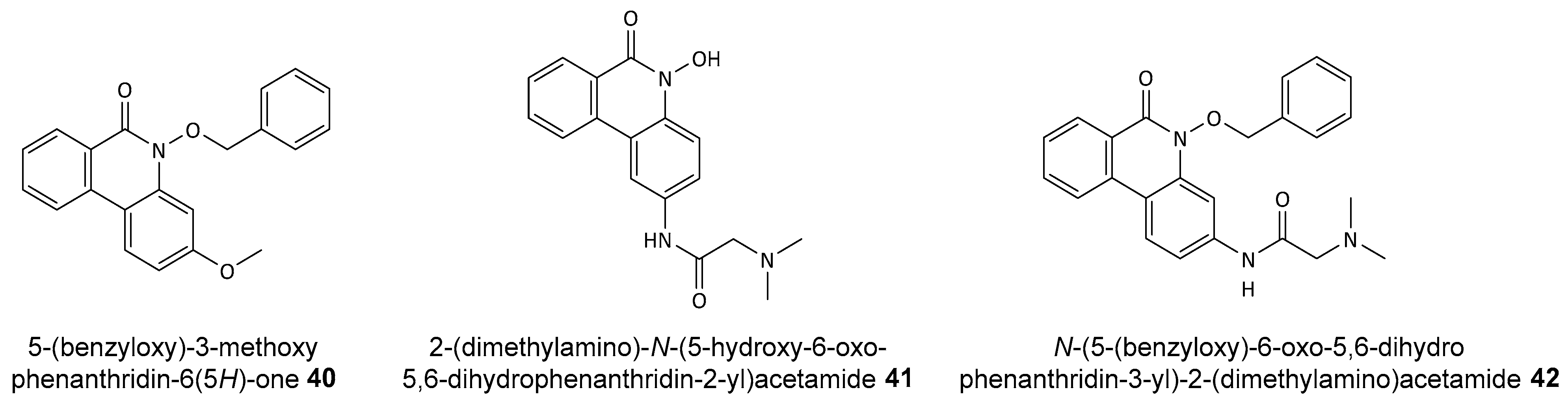
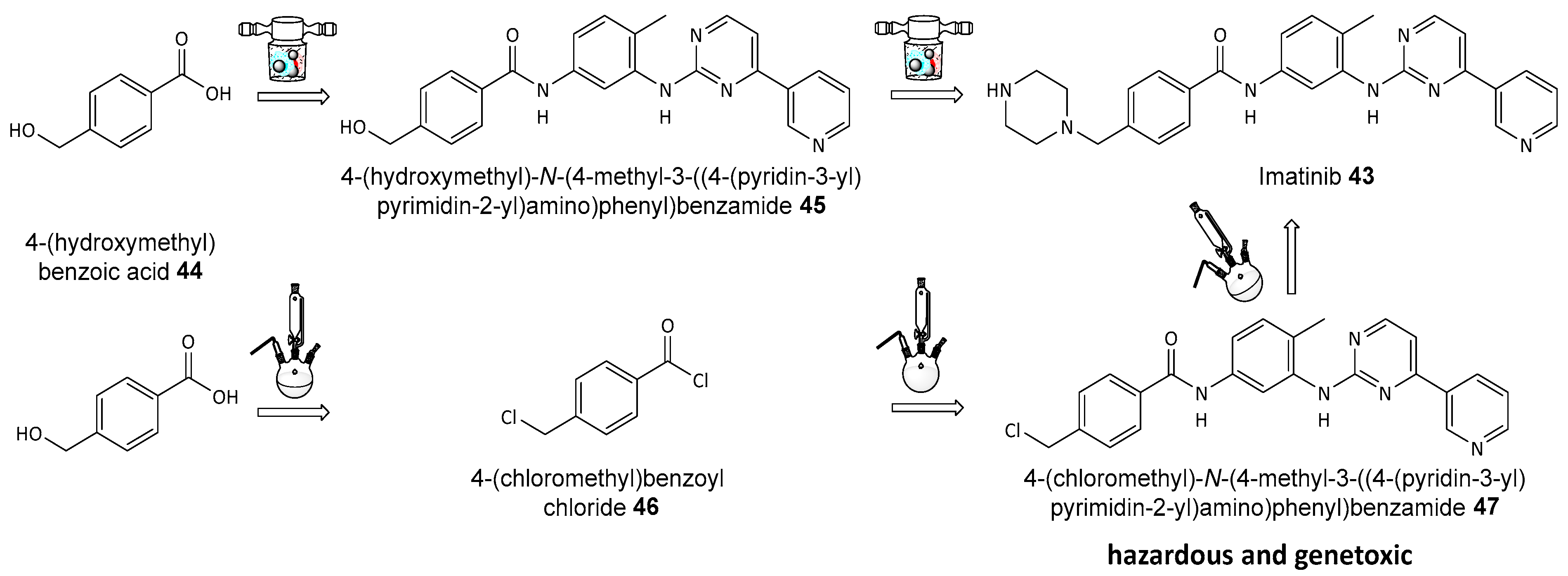
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondar, D.; Karpichev, Y. Poly(ADP-Ribose) Polymerase (PARP) Inhibitors for Cancer Therapy: Advances, Challenges, and Future Directions. Biomolecules 2024, 14, 1269. https://doi.org/10.3390/biom14101269
Bondar D, Karpichev Y. Poly(ADP-Ribose) Polymerase (PARP) Inhibitors for Cancer Therapy: Advances, Challenges, and Future Directions. Biomolecules. 2024; 14(10):1269. https://doi.org/10.3390/biom14101269
Chicago/Turabian StyleBondar, Denys, and Yevgen Karpichev. 2024. "Poly(ADP-Ribose) Polymerase (PARP) Inhibitors for Cancer Therapy: Advances, Challenges, and Future Directions" Biomolecules 14, no. 10: 1269. https://doi.org/10.3390/biom14101269