

Molecular Thumbprints: Biological Signatures That Measure Loss of Identity

Abstract
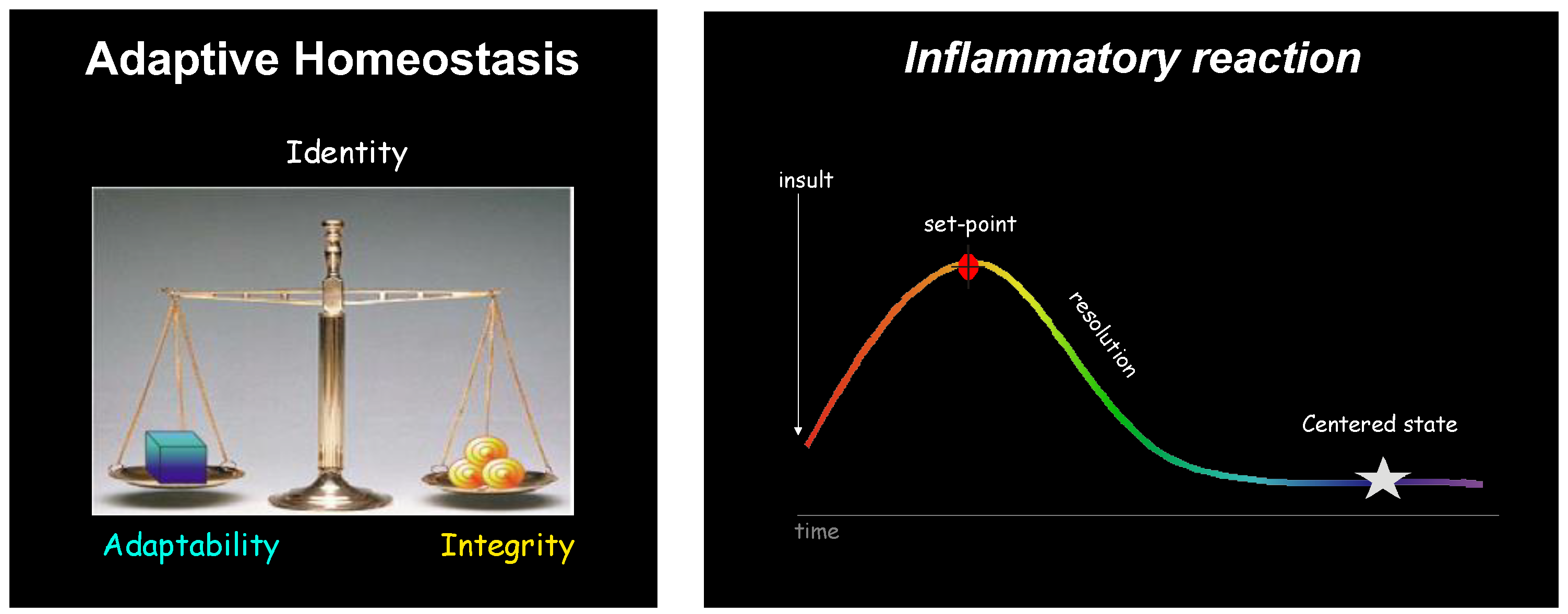
:1. Introduction
2. Materials and Methods
3. Results
3.1. Rat Model for a Thumbprint of Unresolved Colitis
3.2. Visualizing Unresolved Inflammation
3.3. Tangible Data
3.4. BLAST to Gene Identification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morris, G.P.; Beck, P.L.; Herridge, M.S.; Depew, W.T.; Szewczuk, M.R.; Wallace, J.L. Hapten-induced model of chronic inflam-mation and ulceration in the rat colon. Gastroenterology 1989, 96, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; MacNaughton, W.K.; Morris, G.P.; Beck, P.L. Inhibition of leukotriene synthesis markedly accelerates healing in a rat model of inflammatory bowel disease. Gastroenterology 1989, 96, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Zamuner, S.R.; Warrier, N.; Buret, A.G.; MacNaughton, W.K.; Wallace, J.L. Cyclooxygenase 2 mediates post-inflammatory colonic secretory and barrier dysfunction. Gut 2003, 52, 1714–1720. [Google Scholar] [CrossRef] [PubMed]
- Devchand, P.R.; Qiu, F.H.; Wada, K.; Serhan, C.N. Aspirin-triggered lipoxin A4 and lipoxin A4 up- regulate transcriptional corepressor NAB1 in human neutrophils. FASEB J. 2001, 15, 2736–2738. [Google Scholar]
- Devchand, P.R.; Hihi, A.K.; Perroud, M.; Schleuning, W.D.; Spiegelman, B.M.; Wahli, W. Chemical probes that differentially modulate peroxisome proliferator-activated receptor alpha and BLTR, nuclear and cell surface receptors for leukotriene B(4). J. Biol. Chem. 1999, 274, 23341–23348. [Google Scholar] [CrossRef]
- Liang, P.; Pardee, A.B. Analysing differential gene expression in cancer. Nat. Rev. Cancer 2003, 3, 869–876. [Google Scholar] [CrossRef]
- Wallace, J.L.; Devchand, P.R. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br. J. Pharmacol. 2005, 145, 275–282. [Google Scholar] [CrossRef]
- Potter, J.D. Colorectal cancer: Molecules and populations. J. Natl. Cancer Inst. 1999, 91, 916–932. [Google Scholar] [CrossRef]
- Glas, J.; Seiderer, J.; Czamara, D.; Pasciuto, G.; Diegelmann, J.; Wetzke, M.; Olszak, T.; Wolf, C.; Muller-Myhso, B.; Balschun, T.; et al. PTGER4 expression-modulating polymorphisms in the 5p13.1 region predispose to Chrohn’s disease and affect NFKB and XBP1 binding sites. PLoS ONE 2012, 7, e52873. [Google Scholar] [CrossRef]
- Prager, M.; Buttner, J.; Buning, C. PTGER4 modulating variants in Crohn’s disease. Int. J. Color. Dis. 2014, 29, 909–915. [Google Scholar] [CrossRef]
- Drew, D.A.; Kim, A.E.; Lin, Y.; Qu, C.; Morrison, J.; Lewinger, J.P.; Kawaguchi, E.; Wang, J.; Fu, Y.; Zemlianskaia, N.; et al. Two genome-wide interaction loci modify the association of non-steroidal anti-inflammatory drugs with colorectal cancer. Sci. Adv. 2024, 10, eadk3121. [Google Scholar] [CrossRef] [PubMed]
- Na, R.Y.; Jung, D.; Stakenborg, M.; Jang, H.; Gu, G.J.; Jeong, M.R.; Suh, S.Y.; Kim, H.J.; Park, K.J.; Im, J.P.; et al. Prostaglandin E2 receptor PTGER4-expressing macrophages promote intestinal epithelial barrier regeneration upon inflammation. Gut 2021, 70, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Argmann, C.; Hou, R.; Ungaro, R.C.; Irizar, H.; Al-Taie, Z.; Huang, R.; Kosoy, R.; Venkat, S.; Song, W.M.; Di, A.F.; et al. Biopsy and blood-based molecular biomarker of inflammation in IBD. Gut 2023, 72, 1271–1287. [Google Scholar] [CrossRef] [PubMed]
- Breyer, R.M.; Bagdassarian, C.K.; Myers, S.A.; Breyer, M.D. Prostanoid receptors: Subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 661–690. [Google Scholar] [CrossRef]
- Ricciotti, E.; Haines, P.G.; Chai, W.; FitzGerald, G.A. Prostanoids in cardiac and vascular remodelling. Ateriosclerosis Thromb. Vasc. Biol. 2024, 44, 558–583. [Google Scholar] [CrossRef]
- Zamuner, S.R.; Bak, A.W.; Devchand, P.R.; Wallace, J.L. Predisposition to colorectal cancer in rats with resolved colitis: Role of cyclooxygenase-2-derived prostaglandin D2. Am. J. Pathol. 2005, 167, 1293–1300. [Google Scholar] [CrossRef]
- Forman, J.E.; Sorg, J.M.; McGuinnis, K.S.; Rigas, B.; Williams, J.L.; Clapper, M.L.; Gonzalez, F.J.; Peters, J.M. Regulation of pe-roxisome proliferator-activated receptor b/d by the APC/beta-CATENIN pathway and NSAIDs. Mol. Carcinog. 2009, 48, 942–952. [Google Scholar] [CrossRef]
- Sammuelsson, B.; Dahlen, S.E.; Lindgren, J.A.; Rouzer, C.A.; Serhan, C.N. Leukotrienes and lipoxins: Structures, biosynthesis and biological effects. Science 1987, 237, 1171–1176. [Google Scholar] [CrossRef]
- Devchand, P.R. Lipoxin agonists: Turn right! to path of resolving neutrophil. Mem. Inst. Oswaldo Cruz. 2005, 100 (Suppl. S1), 55–57. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Flower, R.J. The development of COX2 inhibitors. Nat. Rev. Drug Discov. 2003, 2, 179–191. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, G.A. COX-2 and beyond: Approaches to prostaglandin inhibition in human disease. Nat. Rev. Drug Discov. 2003, 2, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Miller, M.J. Nitric oxide in mucosal defense: A little goes a long way. Gastroenterology 2000, 119, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Ajuebor, M.N.; Singh, A.; Wallace, J.L. Cyclooxygenase-2-derived prostaglandin D2 is an early anti- inflammatory signal in experimental colitis. Am. J. Physiol. 2000, 279, G238–G244. [Google Scholar] [CrossRef]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty acids, eicosanoids, and hypoli-pidemic agents identified as ligands of peroxisome proliferator- activated receptors by coactivator-dependent receptorligand assay. Mol. Endocrinol. 1998, 11, 779–791. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactionswith peroxisome proliferator-acti-vated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxi-some proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vasquez, M.; Gonzalez, F.J.; Wahli, W. The PPARa–leukotriene B4 pathway to inflam-mation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Forman, J.E.; Chang, W.-C.L.; Palkar, P.S.; Zhu, B.; Borland, M.G.; Williams, J.L.; Kramer, L.R.; Clapper, M.L.; Gonzalez, F.J.; Peter, J.M. Functional characterization of peroxisome proliferator-activated receptor b/d expression in colon cancer. Mol. Carcinog. 2011, 50, 884–900. [Google Scholar] [CrossRef]
- Useini, A.; Schwerin, I.K.; Kunze, G.; Strater, N. Structural studies on the binding mode of bisphenols to PPARgamma. Biomolecules 2024, 14, 640. [Google Scholar]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Fenton, M.J.; Clark, B.D.; Collins, K.L.; Webb, A.C.; Rich, A.; Auron, P.E. Transcriptional regulation of the human prointerleukin 1 beta gene. J. Immunol. 1988, 138, 3972–3979. [Google Scholar] [CrossRef]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed]
- Devchand, P.R.; Ziouzenkova, O.; Plutzky, J. Oxidative stress and peroxisome proliferator-activated receptors: Reversing the curse? Circ. Res. 2004, 95, 1137–1139. [Google Scholar] [CrossRef] [PubMed]
- Archambault, A.N.; Lin, Y.; Jeon, J.; Harrison, T.A.; Bishop, D.T.; Brenner, H.; Casey, G.; Chan, A.T.; Chang-Claude, J.; Figueiredo, J.C.; et al. Non-genetic determinants of risk for early onset colorectal cancer. JNCI Cancer Sectr. 2021, 5, pkab029. [Google Scholar] [CrossRef]
- Cannon, W.B. Organisation for physiological homeostasis. Physiol. Rev. 1929, 3, 399–431. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Low Signal | High Signal |
2A-1 Sv2b Synaptic vesicle glycoprotein 2b Transcript 1 5271-5872 | 5A-1 Pallidin Syntaxin-13 interacting protein 1871-2041 1770-1846 |
14B-3 Sim to region on Chr4 on mouse 76075-75953 | 5A’-1 Aligns to mChr8 BAC clone Possibly a MHC gene |
23A-1 Fra-2 Fos-related antigen DNA 105-234 | 5C-2 Rat homologue of mKIAA1930 4091-4386 |
| 4B-1 Sephs 1 Selenophosphate synthetase | 6A-1 DNA Primase Subunit 49 1171-1374 1407-1591 |
15B-2 Homology to region in mChr1 | 11A-1 Ptger EP4 receptor 2619-2811 |
| 15A-4 Sim to region on Chr11 on mouse 63633-751. 63766-882 | |
| 21B-2 Rbbp5 Retinoblastoma binding protein 5 Similar to mChr1 23480-23132 in rat 766-897;898-970 |
Low Signal | High Signal |
2B-w Sv2b Synaptic vesicle glycoprotein 2b Transcript 2 5271-5942 | 5D-1 Rat homologue of m.musc cDNA BC051083 1095-1329 |
| 21A-2 Similar to region on Chr 15 on mouse 168293-7953 | 8A-1 Rat homologue of MICAL3 Microtubule-associated monooxygenase calponin Lim-domain containing 3 1546-1694 |
| 13B-1 Receptor for hyaluron-mediated motility Coded on mitochrondrial genome 308-144 | |
| 14A-4 Image clone 7123360 2688-2770 2519-2631 78bp homologous to interferon-induced P44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devchand, P.R.; Dicay, M.; Wallace, J.L. Molecular Thumbprints: Biological Signatures That Measure Loss of Identity. Biomolecules 2024, 14, 1271. https://doi.org/10.3390/biom14101271
Devchand PR, Dicay M, Wallace JL. Molecular Thumbprints: Biological Signatures That Measure Loss of Identity. Biomolecules. 2024; 14(10):1271. https://doi.org/10.3390/biom14101271
Chicago/Turabian StyleDevchand, Pallavi R., Michael Dicay, and John L. Wallace. 2024. "Molecular Thumbprints: Biological Signatures That Measure Loss of Identity" Biomolecules 14, no. 10: 1271. https://doi.org/10.3390/biom14101271