Abstract
Danon disease, an X-linked dominant vacuolar cardiomyopathy and skeletal myopathy, is caused by a primary deficiency of lysosome-associated membrane protein-2 (LAMP-2). This disease is one of the autophagy-related muscle diseases. Male patients present with the triad of cardiomyopathy, myopathy, and intellectual disability, while female patients present with cardiomyopathy. The disease’s leading cause of death is heart failure, and its prognostic factor is cardiomyopathy. Pathologically, the disease is characterized by the appearance of unique autophagic vacuoles with sarcolemmal features (AVSFs). Twenty-six families have been found to have this disease in Japan. It has been over 40 years since the first report of this disease by Danon et al. and over 20 years since the identification of the causative gene, LAMP2, by Nishino et al. Although the pathogenetic mechanism of Danon disease remains unestablished, the first clinical trials using AAV vectors have finally begun in recent years. The development of novel therapies is expected in the future.
1. Concept and History of Danon Disease
Danon disease (MIM# 300257) is an extremely rare myopathy, first described as a “lysosomal glycogen storage disease with normal acid maltase” by Danon al. in 1981 [1]. In 2000, Nishino et al. revealed a primary deficiency of LAMP-2 caused by a mutation in lysosome-associated membrane protein type 2 (LAMP2) [2]. Subsequently, in 2002, the authors reported for the first time that Danon disease showed X-linked dominant inheritance, with hypertrophic cardiomyopathy, myopathy, and intellectual disability as the triad of clinical symptoms in males and cardiomyopathy as the main symptom in females [3]. It is an extremely rare disease, with about 100 families found so far in the world [4] and 20 families found in a nationwide survey in Japan [5]. With the addition of six newly identified families reported after the survey, we have found 26 families in Japan. The cardiomyopathy of Danon disease is severe and is a prognostic factor for life. It has recently attracted attention as a differential diagnosis for hypertrophic cardiomyopathy of unknown cause [6,7,8], particularly if symptoms other than cardiomyopathy are mild.
Although it has been over 40 years since the first report of Danon disease, the path to understanding its underlying mechanisms remains rocky. Indeed, the clinical outcome for patients who do not receive heart transplantations is extremely bleak. However, findings from LAMP-2-deficient animal models and patient-derived induced pluripotent stem cells have shed light on this pathway, and promising results from gene therapy point us in the direction of future treatments. We are confident that we will be able to improve its prognosis and overcome early death in the near future [9,10].
2. Clinical Symptoms
Danon disease is an X-linked dominant inheritance, and about half of the mothers of boys with the disease are symptomatic. The remainder are considered de novo mutations. Male patients present with the triad of cardiomyopathy, myopathy, and intellectual disability, while female patients present primarily with cardiomyopathy [3,4,5,6,11]. In males, onset occurs in teenage years and death occurs around age 30, while onset occurs later in females than in males, typically after age 30, and death occurs around age 40 [12]. Recently, early-onset female patients have been observed [6]. Both sexes inevitably present with myocardial damage and hypertrophic cardiomyopathy [8,13]. A high rate of cardiac conduction defects, such as Wolff–Parkinson–White syndrome, complicate the disease. Myopathy is characterized by muscle weakness and atrophy with a predominance of proximal muscles. Intellectual disability is present in about 60% of patients but is mild. In some patients, small vessel lesions in coronary arteries and cerebral arteries have been observed, with vascular stenosis caused by proliferation of vascular smooth muscle cells due to autophagy abnormalities [14].
3. Muscle Pathology
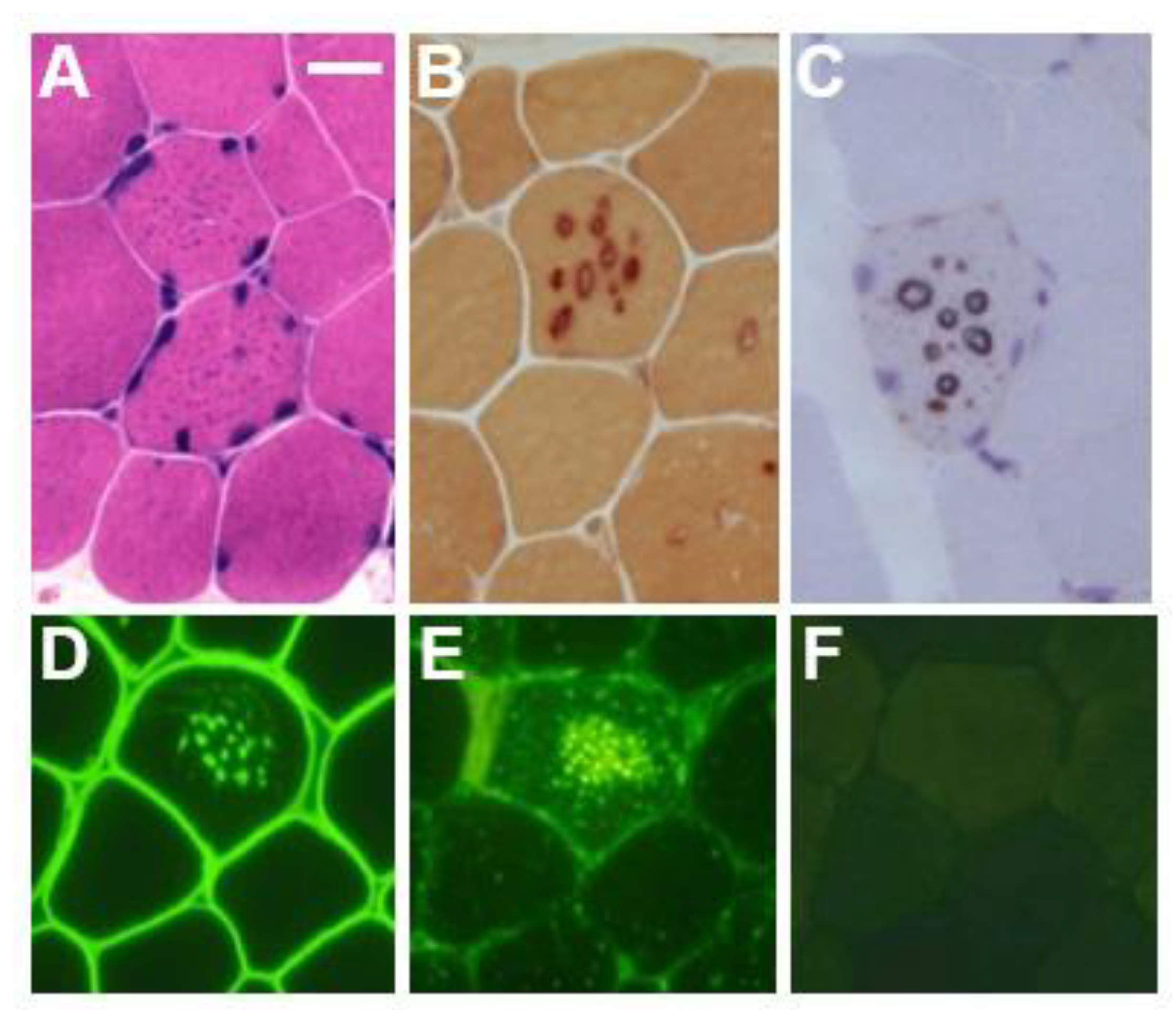
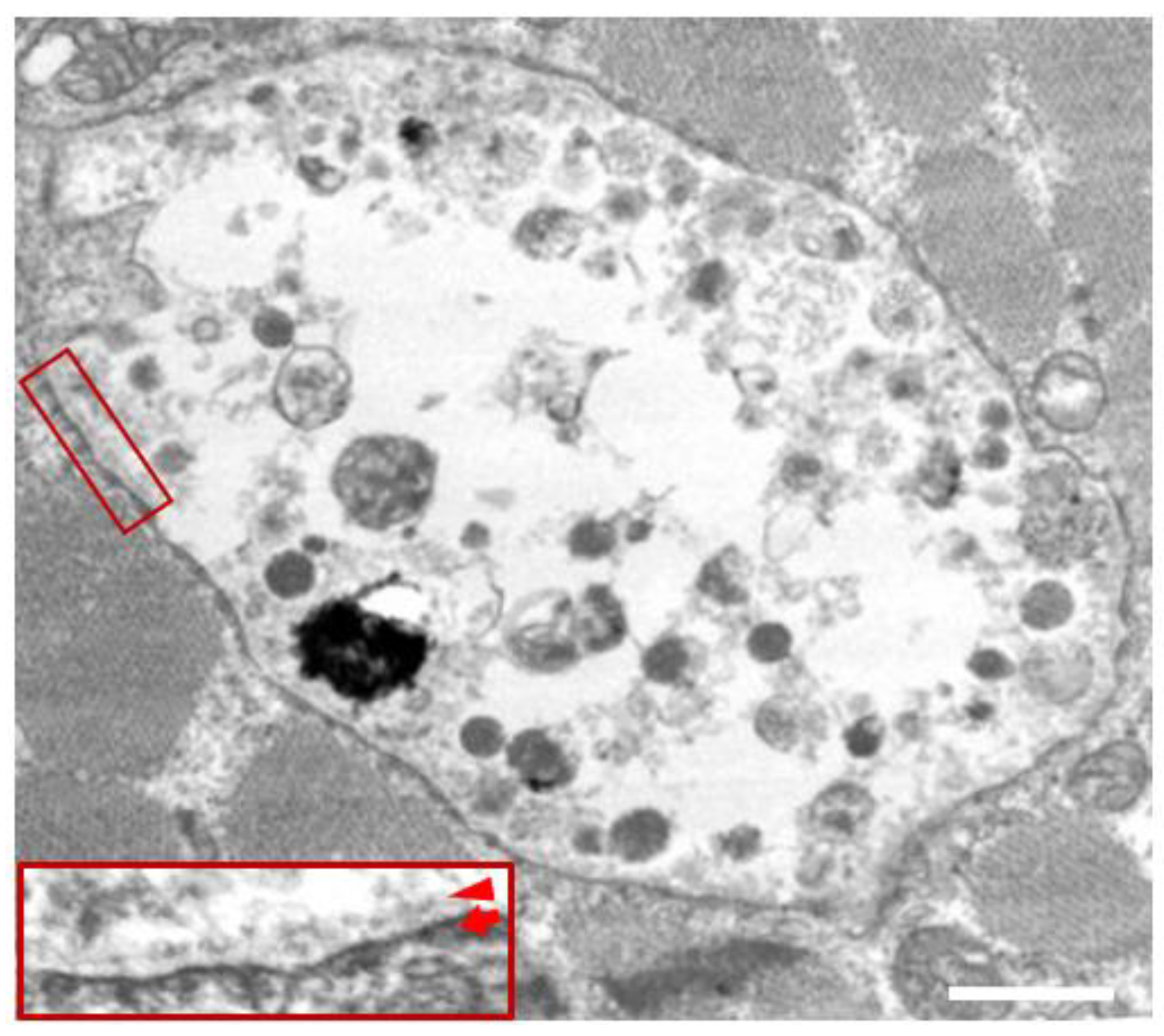
In the biopsied muscles of individuals with Danon disease, muscle fibers with small vacuoles are scattered throughout the muscle fascicles. The membranes of these small vacuoles show acetylcholine esterase (AChE) activity and the expression of sarcolemmal proteins such as dystrophin and sarcoglycan, which are normally found only at the neuromuscular junction (Figure 1). We call this autophagic vacuole with sarcolemmal proteins (AVSF) [15,16]. Our observations show the formation of AVSF to be a phenomenon in which cells create an extracellular environment inside themselves, but its mechanism and involvement in the disease’s pathogenesis remain unclear. Electron microscopic observations reveal a basement membrane along the inside of the vacuolar wall, characterized by autophagic vacuoles containing irregularly shaped abnormal structures and glycogen granules (Figure 2).
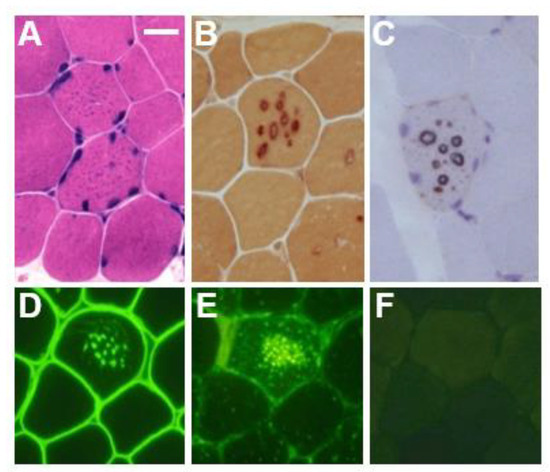
Figure 1.
Muscle pathology in a male patient with Danon disease. Basophilic tiny autophagic vacuoles (arrows) are scattered in muscle fibers stained with hematoxylin and eosin (A). The small vacuoles have acid phosphatase activity (B) and acetylcholine esterase activity (C). Immunohistochemical staining shows the expression of sarcolemmal proteins such as dystrophin (D) in the autophagic vacuolar membrane (AVSF). LIMP-1, a marker of lysosomes, is strongly expressed (E) and LAMP-2 is absent (F). A–F: Bar = 30 μm; D–F: serial sections. With permission from our previous report [5].
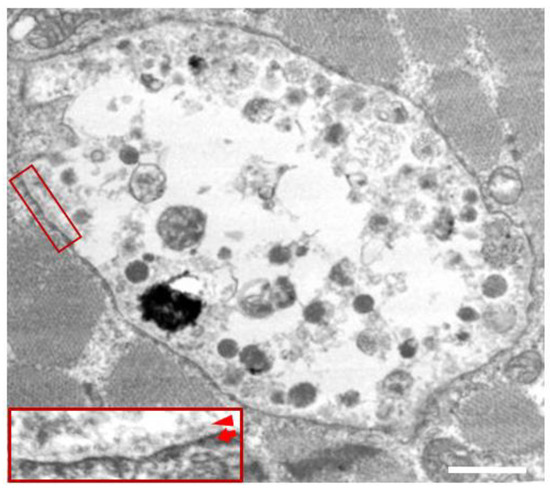
Figure 2.
Electron microscopy in the skeletal muscle of a male patient with Danon disease. The vacuoles have autophagic nature as indicated by the presence of electron-dense granular materials, myeloid bodies, and variable cytoplasmic debris. In addition, basal lamina (red arrowhead) is likely to be observed along the inner surface of an autophagic vacuole (red arrow), suggesting further evidence that the vacuolar membrane has sarcolemmal features. Bar = 1 nm. Published with permission from our previous report [5].
Immunohistochemical and western blot analyses reveal the complete absence of LAMP-2 protein in skeletal muscle and cardiac muscle regardless of a specific LAMP2 gene mutation in male patients [3,15] (Figure 1). Other lysosomal membrane proteins, such as lysosomal integral membrane protein-I (LIMP-1), are associated with the autophagic vacuoles in Danon disease [3,15]. Western blot analysis of LAMP-2 staining for females differs from that of male patients. Normal LAMP-2 expression has been observed in some female patients [16,17], and two female patients showed decreased LAMP-2 staining (40%), most likely due to ‘LAMP-2 haploinsufficiency’ [5,6,11], suggesting that this test may be inconclusive in females. On the other hand, the deletion of LAMP-2 protein was observed in the muscle of a few girls with early onset myopathy and cardiomyopathy [18], which may reflect an extremely skewed X-chromosome inactivation pattern (XCI) that favors the mutant allele. In fact, successful XCI pattern–disease severity correlations have been observed in females [19,20,21]. More recently, the irregular distribution of LAMP-2 in cardiac muscle fibers has been raised as a major determinant of the development of cardiomyopathy in females [22,23].
4. Biochemical and Genetic Characteristics of LAMP-2 and Autophagy
LAMP-2 is a component of the lysosomal membrane and, together with its complement LAMP-1, may protect the lysosomal membrane and cytoplasm from proteolytic enzymes in the lysosomal lumen. In contrast to the constant expression of LAMP-1, the expression of LAMP-2 increases under various conditions. LAMP2 is composed of nine exons, with exons 1–8 and part of exon 9 constituting the lumenal domain and the remainder of exon 9 constituting both the transmembrane and cytoplasmic domains. Selective splicing of exon 9 results in 9A/9B/9C, with three isoforms, LAMP-2A/2B/2C, respectively. LAMP-2A is universally expressed in tissues, while LAMP-2B is known to be expressed mainly in the cardiac muscle, skeletal muscle, and brain and may play an important role in the pathogenesis of this disease. The role of LAMP-2C is still unclear. In Danon disease, autophagy, which occurs constantly in these regions, is thought to cease at the final stage of fusion between autophagosomes and lysosomes [24].
More recently, in addition to autophagy dysfunction, the involvement of mitophagy defects, oxidative stress, and energy deficiency have been reported to play important pathophysiological roles in the development of Danon disease [25,26,27,28,29]. Dysfunction of mitophagy may be an early pathological hallmark of Danon disease with important upstream effects such as impaired mitochondrial metabolism. These results suggest a link between lysosomal and autophagic dysfunction due to LAMP-2 deficiency and mitochondrial turnover defects [27]. Indeed, the downregulation of main mitophagy target genes at the mRNA level was evident. Moreover, mitochondria were partially co-localized with p62+ vacuoles, suggesting a hallmark of Danon disease—the impaired lysosomal clearance of dysfunctional mitochondria [29,30]. As a common final pathway for non-selective autophagy as well as mitophagy, dysfunctional lysosomes may inhibit mitochondrial degradation after fusion with autophagosomes [31].
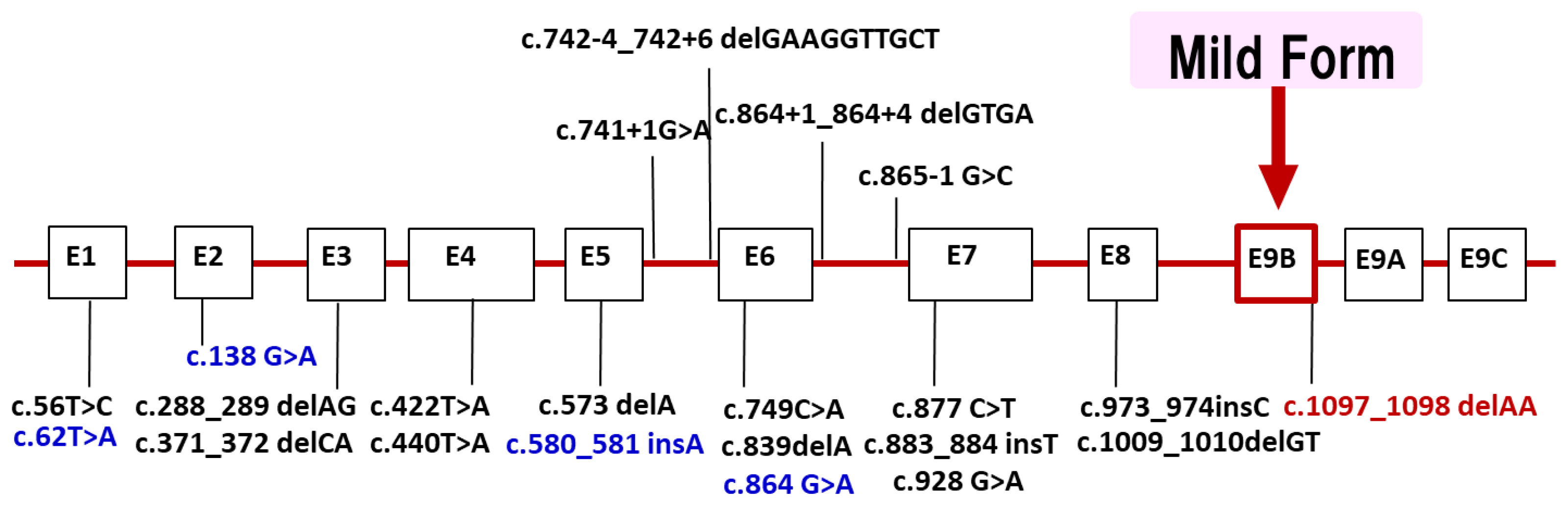
LAMP2 is located in Xq24. Most of the reported mutations are stop codons or out-of-frame, which are thought to cleave the protein and cause loss of the transmembrane and cytoplasmic domains. About 100 families [4] have been found so far in the world, and in Japan’s nationwide survey conducted by the authors in 2018, 20 families were found [5]. We have added patients newly found and reported since the 2018 national survey in Japan. Currently, we have identified 22 different LAMP2 mutations in 26 families (Figure 3). Half of the probands showed de novo mutations. The distribution of mutations widely ranged from exon 1 to 9. We found four families with mutations in exon 9B (c.1097_1098 delAA) that encodes LAMP-2B, which is considered genetically important. Recently, we reported that male patients over 50 years of age with mutations in exon 9B showed very mild clinical symptoms, including cardiomyopathy [32]. Our findings suggest the presence of factors in exon 9B that may prevent severe clinical symptoms, but they have not yet been established.
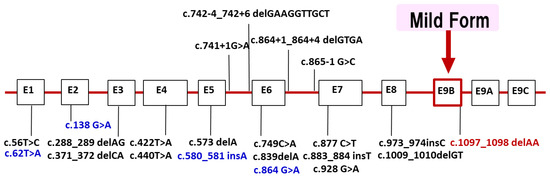
Figure 3.
LAMP2 mutations in Japanese patients with Danon disease. We identified 22 different LAMP2 mutations in 26 families with Danon disease. Four mutations (blue) were identified after the nationwide survey in 2018. The distribution of mutations widely ranged from exon 1 to 9. Four families with mutations in exon 9B (red) which encodes LAMP-2B showed markedly mild or no cardiomyopathy. Exons are to scale and introns are not to scale.
5. Definitive Diagnosis
There are no internationally established diagnostic criteria for Danon disease. However, diagnostic criteria for Danon disease were developed by the “Autophagocytic Vacuolar Myopathy Study Group” and the “Study Group on Rare and Intractable Muscle Diseases” of the Japanese Ministry of Health, Labour and Welfare, taking into consideration the necessary and sufficient conditions for diagnosis (Table 1). The diagnostic criteria were approved by the Japan Society of Neurology. The diagnosis of Danon disease is confirmed by clinical features and muscle pathology, as well as LAMP-2 deficiency in biopsied muscle and/or western blot analysis, and LAMP2 gene analysis. In female patients, LAMP-2 protein varies from deficient to normal, but LAMP-2 haploinsufficiency is also considered as a contributing factor [6,11,15]. As Danon disease is extremely rare, it is practically difficult to suspect Danon disease and perform genetic analysis based on clinical symptoms alone. For diagnosis, it is important to perform a muscle biopsy and find AVSF in the muscle fibers. In fact, the three main criteria for diagnosis are (A) clinical features; (B) muscle pathology; and (C) evaluation of LAMP-2. A “definite diagnosis” is defined as a patient who meets at least one of criteria A or B and also meets criterion C. Clinically, differential diagnosis includes other myopathies and muscular dystrophies and cardiomyopathies of other definite causes. Pathologically, it is important to differentiate the diseases from other muscle diseases with autophagic vacuoles.

Table 1.
Diagnostic criteria for Danon disease. (Japanese Ministry of Health, Labour and Welfare, “Autophagic Vacuolar Myopathy Study Group” (2012)).
As radical treatment has yet to be established for Danon disease, early diagnosis is essential for the planning of appropriate treatments to prevent sudden cardiac death prior to heart transplantation [12,33,34,35,36]. In fact, the frequency of Danon disease is 4–6% in children with hypertrophic cardiomyopathy [33], suggesting that it is one of the major causes of hypertrophic cardiomyopathy. Given that most patients die of heart failure or arrhythmia, palpitations or chest pain are prominent clinical features and the most important prognostic factors. Heart transplantation significantly improves survival, but only 17.6% of patients with Danon disease undergo this procedure [12]. Therefore, disease awareness and early diagnosis of Danon disease are critical.
6. Treatment and Prognosis
In Danon disease, cardiomyopathy is the determinant of life prognosis, and the cause of death is sudden death due to heart failure or cardiac conduction defects [7,8,15]. Currently, the only radical therapy is heart transplantation, and catheter ablation or implantable cardioverter defibrillators should be considered at the early stages of the disease [37]. Since transplantation is desired within two years of the onset of heart failure, early diagnosis is required. Furthermore, since there is a possibility that Danon disease patients are hidden among patients with hypertrophic cardiomyopathy of unknown cause, genetic analysis should be considered early on if this disease is suspected. In addition, since female patients with Danon disease have only cardiomyopathy and no or minimal skeletal muscle symptoms, an undiagnosed family history of the disease may be considered. We suggest further investigations of relatives who are at risk of developing the disease and believe that careful follow-up observation is particularly important for asymptomatic adult women. As there is a risk of sudden death in female patients, it is necessary to consider the application of catheter ablation or defibrillators, as well as early drug therapy. The degree of intellectual disability is mild and not life-threatening, but it is necessary to consider providing support for patients’ learning and mental health, as well as education. This can include referring patients to local intellectual disability support centers and connecting them with relevant mental health professionals.
Since this disease is extremely rare, the accuracy of the current diagnostic criteria will need to be verified through an accumulation of patients and collaboration with other medical departments. Furthermore, future issues in the policy of intractable diseases include nationwide epidemiological surveys, clarification of natural history based on the results of these surveys, identification of new patients, and the establishment of a registry.
7. Research for Novel Therapeutic Strategies of Danon Disease
Vigorous efforts are now being made on various fronts to develop new treatment methods. An initial step was to use animal models. LAMP2 knockout (KO) mice generated by a German group demonstrated that LAMP-2 deficiency causes Danon disease [24]. Approximately 50% of LAMP2 KO mice died between 20 and 40 days of age, regardless of sex or genetic background. LAMP2 KO mice showed autophagic vacuoles in various organs, including the heart, skeletal muscle, liver, pancreas, spleen, and kidney; the surviving mice were small and showed cardiac hypertrophy. KO mice that died early often showed small intestinal stenosis and pancreatic lesions, which may be the result of the KO mice showing multiorgan involvement. This suggests that Danon disease patients may develop multiorgan involvement in addition to the triad of signs. On the other hand, LAMP1 deficient mice show normal lysosomal morphology and function and do not develop any symptoms [38]. This suggests that LAMP-1 and LAMP-2, which are homologous to each other, may play different roles, as LAMP-2 serves a compensatory hyperfunction in LAMP1 KO mice whereas LAMP-1 is not hyperfunctional in LAMP2 KO mice [24].
Subsequently, rat models of Danon disease were developed in 2017 and zebrafish models were developed in 2019 [39,40]. These models showed the phenotypes of Danon disease, including cardiomyopathy and myopathy and the presence of autophagic vacuoles. In addition, mTOR inhibition was shown to normalize features of the LAMP2 KO, including ejection fraction, β-adrenergic response, and actomyosin activation kinetics, demonstrating the potential of new intervention strategies related to mTOR [40]. Breakthroughs in patient-derived pluripotent stem cell technology have opened new avenues for the elucidation of genetic diseases, including Danon disease, and it has been used as a powerful tool [10]. Using a small amount of blood collected from patients, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM) were cultured to provide important insights into development. This technique has been widely utilized by research groups around the world and has contributed significantly to research advances [41]. In vitro and in vivo studies since 2018 have revealed the role of mitochondrial dysfunction and fragmentation in pathological mechanisms and potential targets for therapeutic intervention [14,28]. Notably, in patient-derived hiPSC-CMs, correction of the LAMP2 mutation with single-stranded DNA using CRISPR-Cas9 resulted in increases in ATP production, oxygen consumption rate, and maximal respiratory rate comparable to those in normal hiPSC-CMs. These breakthrough results suggest that this correction may improve the partial phenotype of Danon disease in vitro [28].
On the other hand, recent years have also seen advances in the pharmacological aspects of Danon disease treatment strategies. Some studies show the efficacy of conventional drug therapies used to control heart failure and ventricular remodeling, mainly inhibitors of the renin–angiotensin–aldosterone system (RAAS), in particular Ramipril [42]. In fact, there have been several reports on the effects of RAAS on autophagy and its role in cardiac hypertrophy across various models [43]. However, the causal relationship between oxidative stress and cardiac phenotype and whether antioxidants play any role remain unclear.
In an in vivo study using LAMP2 KO mice in 2020, Manso et al. reported an innovative method to correct LAMP-2B deficiency using recombinant adeno-associated virus (AAV9) as a vector [44]. They observed that human LAMP-2B was constructed on AAV9 and that LAMP-2B protein was persistently expressed; intravenous injection of AAV9-LAMP-2B into LAMP2 KO mice resulted in dose-dependent recovery of LAMP-2 protein in the heart, liver and skeletal muscle. Survival of the gene-treated aged mice clearly improved [44]. Based on this study, the first clinical trials of promising gene therapy on human patients with Danon disease started in the United States in 2019 (NCT03882437) [45,46]. The trials were part of a nonrandomized, open-label phase 1 study to evaluate the safety and toxicity of gene therapy with AAV9-LAMP-2B (RP-A501). Secondary outcomes at the end of three years included assessments of cardiac and skeletal muscle gene transfer induced by therapy, histological evaluation of cardiomyocytes, and the clinical assessment of cardiomyopathy. As of 2024, phase 2 trials are underway (NCT06092034) [46].
In addition, recent studies have shown that the efficacy of the RAAS system with ramipril is equivalent to that of gene therapy, and the protective effect of drug combination therapy is expected to be even greater than that of ramipril alone, suggesting a need to extend the drug therapy that is currently applied to Danon disease patients with heart failure [47]. Further elucidation of the functions and molecular mechanisms of autophagy and lysosomes in Danon disease is expected to elucidate the disease’s pathomechanisms and lead to the development of therapeutic methods.
8. Conclusions
Over 40 years since the first report of Danon disease and over 20 years since the identification of its causative gene have passed, but heart transplantation remains the only curative treatment, and the path to elucidating the underlying pathophysiology is steep. However, dramatic advances have been made in clinical, genetic, and basic research over the past 25 years, and the pathophysiology caused by autophagy abnormalities is becoming clearer [48,49]. The postulated pathomechanism underlining Danon disease may be impaired mitochondrial metabolism due to dysfunction of mitophagy, as well as lysosomal and autophagy dysfunction due to LAMP-2 deficiency. After fusion with autophagosomes, dysfunctional lysosomes may inhibit mitochondrial degradation through non-selective autophagy and mitophagy. The development of model animals such as mice, rats, and zebrafish, as well as findings from patient-derived iPS cells, provide a ray of hope on Danon disease research. As these studies demonstrate, correction of the LAMP2 mutation resulted in increases in ATP production, oxygen consumption rate, and maximum respiratory rate, all of which are comparable to normal in vitro, indicating that this correction may improve the partial phenotype of Danon disease and reveal potential targets for therapeutic intervention. Furthermore, the first clinical trials of promising gene therapy in humans finally began in 2019, with phase 2 trials currently underway as of 2024. We hope that the prognosis and survival rate will improve in the near future.
Author Contributions
Conceptualization: K.S. and I.N.; supervision: I.N.; writing—original draft: K.S.; writing—review and editing: K.S. and I.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a Grant-in-Aid for Research (23FC1014) on Intractable Diseases from the Ministry of Health, Labor, and Welfare of Japan.
Acknowledgments
We thank Ikuya Nonaka from the National Center of Neurology and Psychiatry for his enthusiastic guidance for more than 25 years.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| AVSF | autophagic vacuoles with sarcolemmal features |
| CK | creatine kinase |
| LAMP-2 | lysosome-associated membrane protein-2 |
| LIMP-1 | lysosomal integral membrane protein-1 |
References
- Danon, M.J.; Oh, S.J.; DiMauro, S.; Manaligod, J.R.; Eastwood, A.; Naidu, S.; Schliselfeld, L.H. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981, 31, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Yamamoto, A.; Murayama, K.; Oh, S.J.; Takahashi, M.; Mora, M.; Riggs, J.E.; Colomer, J.; Iturriaga, C.; Meloni, A.; et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002, 58, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Cenacchi, G.; Papa, V.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Danon disease: Review of natural history and recent advances. Neuropathol. Appl. Neurobiol. 2020, 46, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Komaki, H.; Eura, N.; Shiota, T.; Onoue, K.; Tsukaguchi, H.; Minami, N.; Ogawa, M.; Kiriyama, T.; Kataoka, H.; et al. A Nationwide Survey on Danon Disease in Japan. Int. J. Mol. Sci. 2018, 19, 3507. [Google Scholar] [CrossRef]
- Sugie, K.; Yoshizawa, H.; Onoue, K.; Nakanishi, Y.; Eura, N.; Ogawa, M.; Nakano, T.; Sakaguchi, Y.; Hayashi, Y.K.; Kishimoto, T.; et al. Early onset of cardiomyopathy and intellectual disability in a girl with Danon disease associated with a de novo novel mutation of the LAMP-2 gene. Neuropathology 2016, 36, 561–565. [Google Scholar] [CrossRef]
- Maron, B.J.; Roberts, W.C.; Arad, M.; Haas, T.S.; Spirito, P.; Wright, G.B.; Almquist, A.K.; Baffa, J.M.; Saul, J.P.; Ho, C.Y. Clinical outcome and phenotypic expression in LAMP-2 cardiomyopathy. JAMA 2009, 301, 1253–1259. [Google Scholar] [CrossRef]
- Miani, D.; Taylor, M.; Mestroni, L.; D’Aurizio, F.; Finato, N.; Fanin, M.; Brigido, S.; Proclemer, A. Sudden death associated with danon disease in women. Am. J. Cardiol. 2012, 109, 406–411. [Google Scholar] [CrossRef]
- Sugie, K. Editorial commentary: Highlighting the ray of hope in Danon disease research after 40 years. Trends Cardiovasc. Med. 2023, 33, 90–91. [Google Scholar] [CrossRef]
- Zhai, Y.; Miao, J.; Peng, Y.; Wang, Y.; Dong, J.; Zhao, X. Clinical features of Danon disease and insights gained from LAMP-2 deficiency models. Trends Cardiovasc. Med. 2023, 33, 81–89. [Google Scholar] [CrossRef]
- Sugie, K.; Koori, T.; Yamamoto, A.; Ogawa, M.; Hirano, M.; Inoue, K.; Nonaka, I.; Nishino, I. Characterization of Danon disease in a male patient and his affected mother. Neuromuscul. Disord. 2003, 13, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Boucek, D.; Jirikowic, J.; Taylor, M. Natural history of Danon disease. Genet. Med. 2011, 13, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H., Jr.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Noguchi, S.; Sugie, K.; Matsuo, Y.; Nguyen, C.T.H.; Koito, H.; Shiojima, I.; Nishino, I.; Tsukaguchi, H. Small-Vessel Vasculopathy Due to Aberrant Autophagy in LAMP-2 Deficiency. Sci. Rep. 2018, 8, 3326. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Noguchi, S.; Kozuka, Y.; Arikawa-Hirasawa, E.; Tanaka, M.; Yan, C.; Saftig, P.; von Figura, K.; Hirano, M.; Ueno, S.; et al. Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J. Neuropathol. Exp. Neurol. 2005, 64, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Sugie, K.; Nishino, I. Lysosomal Membrane Disorders: LAMP-2 Deficiency. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 567–574. [Google Scholar]
- Fanin, M.; Nascimbeni, A.C.; Fulizio, L.; Spinazzi, M.; Melacini, P.; Angelini, C. Generalized LAMP-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in Danon disease. Am. J. Pathol. 2006, 168, 1309–1320. [Google Scholar] [CrossRef]
- Kim, H.; Cho, A.; Lim, B.C.; Kim, M.J.; Kim, K.J.; Nishino, I.; Hwang, Y.S.; Chae, J.H. A 13-year-old girl with proximal weakness and hypertrophic cardiomyopathy with Danon disease. Muscle Nerve 2010, 41, 879–882. [Google Scholar] [CrossRef]
- Majer, F.; Vlaskova, H.; Krol, L.; Kalina, T.; Kubanek, M.; Stolnaya, L.; Dvorakova, L.; Elleder, M.; Sikora, J. Danon disease: A focus on processing of the novel LAMP2 mutation and comments on the beneficial use of peripheral white blood cells in the diagnosis of LAMP2 deficiency. Gene 2012, 498, 183–195. [Google Scholar] [CrossRef]
- Xu, J.; Wang, L.; Liu, X.; Dai, Q. A novel LAMP2 p.G93R mutation associated with mild Danon disease presenting with familial hypertrophic cardiomyopathy. Mol. Genet. Genom. Med. 2019, 7, e00941. [Google Scholar] [CrossRef]
- Majer, F.; Kousal, B.; Dusek, P.; Piherova, L.; Reboun, M.; Mihalova, R.; Gurka, J.; Krebsova, A.; Vlaskova, H.; Dvorakova, L.; et al. Alu-mediated Xq24 deletion encompassing CUL4B, LAMP2, TMEM255A, and ZBTB33 genes causes Danon disease in a female patient. Am. J. Med. Genet. A 2020, 182, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Casamayor Polo, L.; Bravo García-Morato, M.; Enguita Valls, A.B.; Ruiz-Bravo, E.; Muñoz-Cabello, P.; Ibáñez, K.; Rodríguez-Laguna, L.; Martín-Arenas, R.; Ortega, M.; et al. Molecular and histologic insights on early onset cardiomyopathy in Danon disease females. Clin. Genet. 2021, 99, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Hedberg Oldfors, C.; Máthé, G.; Thomson, K.; Tulinius, M.; Karason, K.; Östman-Smith, I.; Oldfors, A. Early onset cardiomyopathy in females with Danon disease. Neuromuscul. Disord. 2015, 25, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lüllmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Alcalai, R.; Arad, M.; Wakimoto, H.; Yadin, D.; Gorham, J.; Wang, L.; Burns, E.; Maron, B.J.; Roberts, W.C.; Konno, T.; et al. LAMP2 Cardiomyopathy: Consequences of Impaired Autophagy in the Heart. J. Am. Heart Assoc. 2021, 10, e018829. [Google Scholar] [CrossRef]
- Hashem, S.I.; Perry, C.N.; Bauer, M.; Han, S.; Clegg, S.D.; Ouyang, K.; Deacon, D.C.; Spinharney, M.; Panopoulos, A.D.; Izpisua Belmonte, J.C.; et al. Brief report: Oxidative stress mediates cardiomyocyte apoptosis in a human model of danon disease and heart failure. Stem Cells 2015, 33, 2343–2350. [Google Scholar] [CrossRef]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef]
- Chi, C.; Leonard, A.; Knight, W.E.; Beussman, K.M.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.A.; Small, E.M.; et al. LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565. [Google Scholar] [CrossRef]
- Kleefeld, F.; Hentschel, A.; von Moers, A.; Hahn, K.; Horvath, R.; Goebel, H.H.; Preusse, C.; Schallner, J.; Schuelke, M.; Roos, A.; et al. Beyond vacuolar pathology: Multiomic profiling of Danon disease reveals dysfunctional mitochondrial homeostasis. Neuropathol. Appl. Neurobiol. 2023, 49, e12920. [Google Scholar] [CrossRef]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Yasui, T.; Nagaoka, U.; Oya, Y.; Uruha, A.; Karashima, J.; Funai, A.; Miyamoto, K.; Matsubara, S.; Sugaya, K.; Takahashi, K.; et al. Mild form of Danon disease: Two case reports. Neuromuscul. Disord. 2021, 31, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Tanimoto, K.; Hirayama-Yamada, K.; Tsuda, E.; Ayusawa, M.; Nunoda, S.; Hosaki, A.; Kimura, A. Genetic background of Japanese patients with pediatric hypertrophic restrictive cardiomyopathy. J. Hum. Genet. 2018, 63, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Shimozono, T.; Ueno, K.; Shiokawa, N.; Ohno, S.; Kawano, Y. Early diagnosis of infantile Danon disease complicated by tetralogy of Fallot. Pediatr. Int. 2021, 63, 988–990. [Google Scholar] [CrossRef] [PubMed]
- Gandaeva, L.; Sonicheva-Paterson, N.; McKenna, W.J.; Savostyanov, K.; Myasnikov, R.; Pushkov, A.; Zhanin, I.; Barskiy, V.; Zharova, O.; Silnova, I.; et al. Clinical features of pediatric Danon disease and the importance of early diagnosis. Int. J. Cardiol. 2023, 389, 131189. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Luo, S.; Cai, S.; Hong, W.; Guo, Y.; Wu, J.; Liu, T.; Zhao, C.; Li, F.; Huang, H.; et al. Identification of LAMP2 Mutations in Early-Onset Danon Disease with Hypertrophic Cardiomyopathy by Targeted Next-Generation Sequencing. Am. J. Cardiol. 2016, 118, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.N.; Battikha, C.; John, S.; Lin, A.; Bui, Q.M.; Brambatti, M.; Storm, G.; Boynton, K.; Medina-Hernandez, D.; Garcia-Alvarez, A.; et al. Cardiac Transplantation in Danon Disease. J. Card. Fail. 2022, 28, 664–669. [Google Scholar] [CrossRef]
- Andrejewski, N.; Punnonen, E.L.; Guhde, G.; Tanaka, Y.; Lüllmann-Rauch, R.; Hartmann, D.; von Figura, K.; Saftig, P. Normal lysosomal morphology and function in LAMP-1-deficient mice. J. Biol. Chem. 1999, 274, 12692–12701. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Cai, W.; Shi, Y.; Zhou, X.; Guo, G.; Guo, C.; Huang, X.; Han, Z.; Zhang, S.; et al. A Critical Evaluation of Liver Pathology in Humans with Danon Disease and Experimental Correlates in a Rat Model of LAMP-2 Deficiency. Clin. Rev. Allergy Immunol. 2017, 53, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Dvornikov, A.V.; Wang, M.; Yang, J.; Zhu, P.; Le, T.; Lin, X.; Cao, H.; Xu, X. Phenotyping an adult zebrafish lamp2 cardiomyopathy model identifies mTOR inhibition as a candidate therapy. J. Mol. Cell Cardiol. 2019, 133, 199–208. [Google Scholar] [CrossRef]
- Li, X.; Fu, W.; Guo, G.; Liu, M.; Du, W.; Zhao, J.; Liu, Y.; Wang, L.; Dong, J.; Zhao, X. A heterozygous MYH7 (c. 2156G > A) mutant human induced pluripotent stem cell line (ZZUNEUi020-A) generated from a patient with hypertrophic cardiomyopathy. Stem Cell Res. 2021, 51, 102158. [Google Scholar] [CrossRef]
- Yadin, D.; Guetta, T.; Petrover, Z.; Alcalai, R.; Seidman, J.; Seidman, C.E.; Ofek, E.; Kornowski, R.; Hochhauser, E.; Arad, M. Effect of pharmacological heart failure drugs and gene therapy on Danon’s cardiomyopathy. Biochem. Pharmacol. 2023, 215, 115735. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ma, B.; Han, X. The role of autophagy in angiotensin II-induced pathological cardiac hyper-trophy. J. Mol. Endocrinol. 2016, 57, R143–R152. [Google Scholar] [CrossRef] [PubMed]
- Manso, A.M.; Hashem, S.I.; Nelson, B.C.; Gault, E.; Soto-Hermida, A.; Villarruel, E.; Brambatti, M.; Bogomolovas, J.; Bushway, P.J.; Chen, C.; et al. Systemic AAV9.LAMP2B injection reverses metabolic and physiologic multiorgan dysfunction in a murine model of Danon disease. Sci. Transl. Med. 2020, 12, eaax1744. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.; Eshraghian, E.; Battiprolu, P.; Ricks, D.; Yarabe, P.; Schwartz, J.; Patel, K.; Shah, G.; Trevejo, J. Results from first-in-human clinical trial of RP-A501 (AAV9: LAMP2B) gene therapy treatment for Danon disease. Circulation 2021, 144 (Suppl. S1), A10727. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov (accessed on 30 August 2024).
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, 1757–1780. [Google Scholar] [CrossRef]
- Sugie, K. Autophagic vacuolar myopathy: Danon disease and related myopathies. Neurol. Clin. Neurosci. 2021, 10, 273–278. [Google Scholar] [CrossRef]
- Sugie, K. Autophagy Dysfunction in Alzheimer’s Disease and Dementia; Hamano, T., Mutoh, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 207–224. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).