
Adverse Effects of Aβ1-42 Oligomers: Impaired Contextual Memory and Altered Intrinsic Properties of CA1 Pyramidal Neurons

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Preparation of Aβ1-42 Oligomers
2.3. Stereotaxic Surgery and Microinjection of Oligomers
2.4. Behavioral Test Battery
2.4.1. Open-Field Test
2.4.2. Object Recognition Task
2.4.3. Y-Maze Task
2.4.4. Light–Dark Box Test
2.4.5. Social Recognition Task
2.4.6. Inhibitory Avoidance (IA) Task
2.4.7. Fear Conditioning Test
2.4.8. Flinch-Jump Test
2.5. Electrophysiology
2.6. Current Clamp Recordings
2.7. Congo Red Staining
2.8. Lecanemab for Immunostaining
2.9. Statistical Analysis
3. Results
3.1. Effects of Aβ1-42 Oligomers on IA Learning
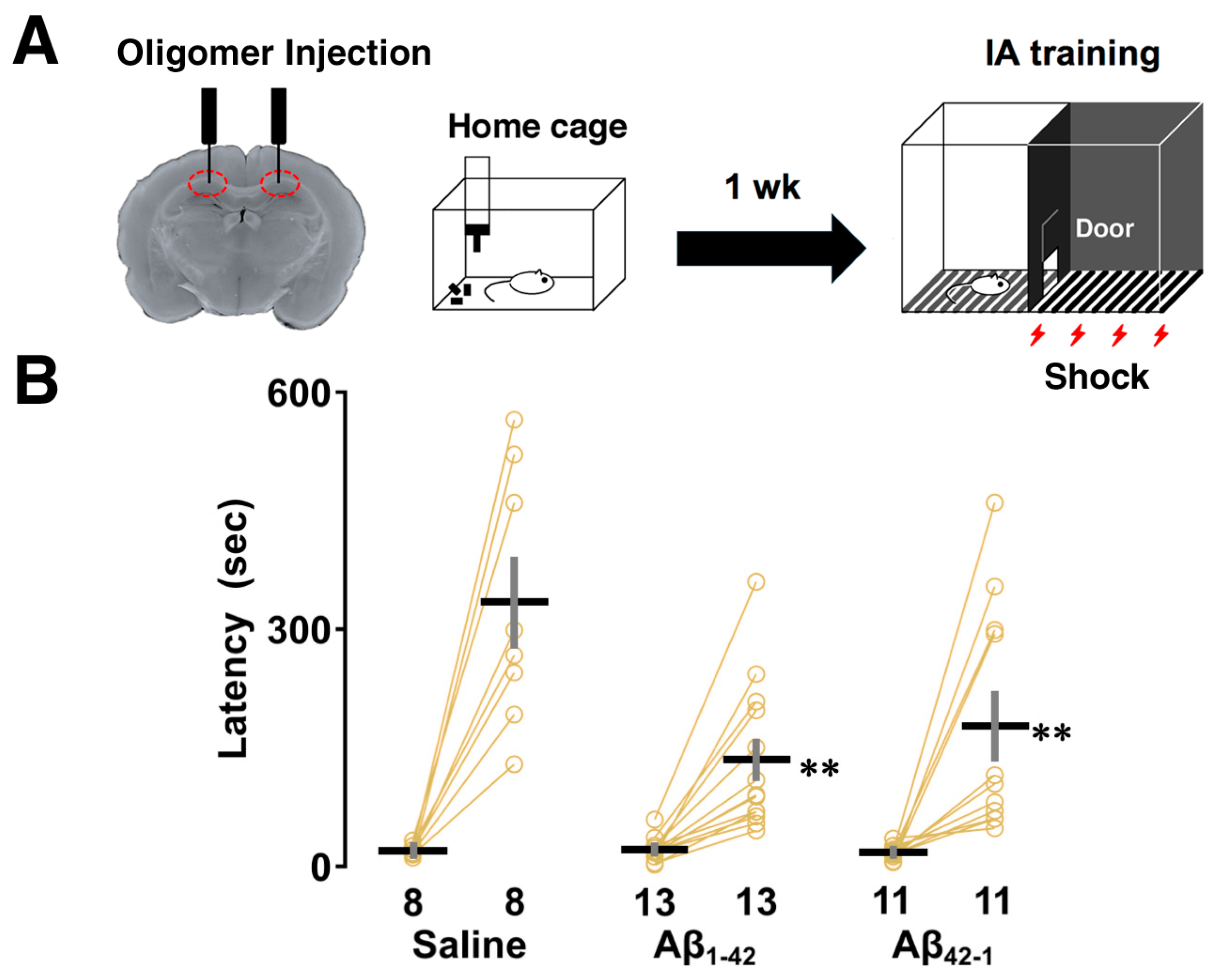
3.2. Effects of Aβ1-42 Oligomers on the Intrinsic Properties After IA Learning


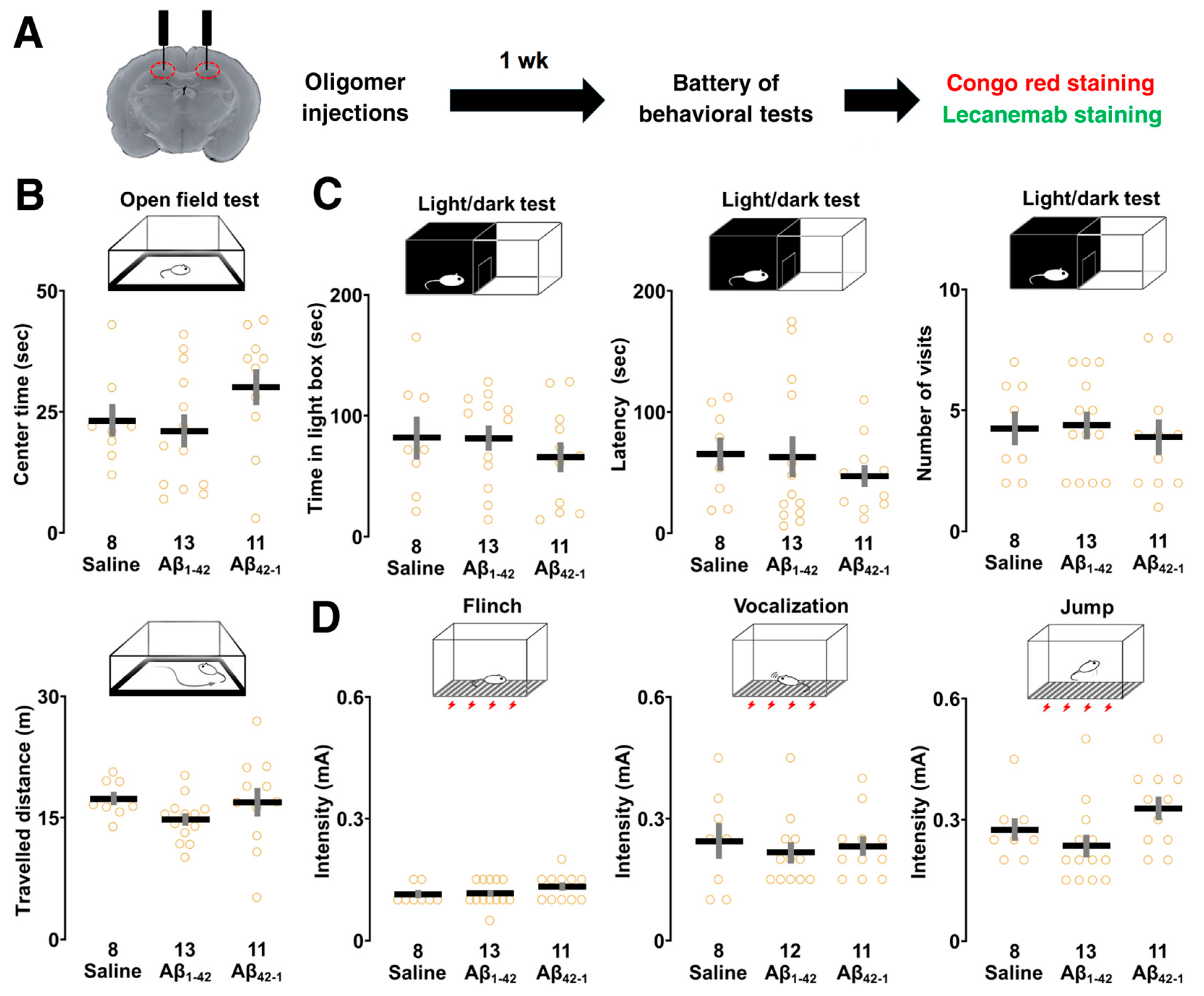
3.3. Effects of Aβ1-42 Oligomers on Sensory/Motor Functions, and Emotional State
3.4. Effects of Aβ1-42 Oligomers on Other Hippocampus-Dependent Tasks
3.5. Aβ Deposition in the CA1 Region
4. Discussion
4.1. Aβ1-42 Oligomers Impaired CA1-Dependent IA Learning
4.2. Aβ1-42 Oligomers Induced Neuronal Hyperexcitability After IA Learning
4.3. Aβ1-42 Oligomers Selectively Impaired Other Hippocampus-Dependent Tasks
4.4. Amyloid Deposition in the Target Area
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aCSF | Artificial cerebrospinal fluid |
| Aβ | Amyloid beta |
| AD | Alzheimer’s disease |
| AP | Anteroposterior |
| APP | Amyloid precursor protein |
| Cm | Membrane capacitance |
| CA1 | Cornu ammonis 1 |
| CSF | Cerebrospinal fluid |
| DG | Dentate gyrus |
| DMSO | Dimethy sulfoxide |
| DV | Dorsoventral |
| HFIP | 1,1,1,3,3,3-hexafluoro-2-propanol |
| IM | M-type potassium current |
| INaP | Persistent sodium current |
| IA task | Inhibitory avoidance task |
| ML | Mediolateral |
| Nav1.6 | Voltage-gated sodium channel 1.6 |
| PBS | Phosphate-buffered saline |
| Rm | Membrane resistance |
| RLZ | Riluzole |
| RMP | Resting membrane potential |
| Tau | Time constant |
| TASK current | TWIK-related acid-sensitive potassium current |
References
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Tran, L.; Lambert, M.P.; Glabe, C.G.; Klein, W.L.; LaFerla, F.M. Temporal profile of amyloid-β (Aβ) oligomerization in an in vivo model of Alzheimer disease. J. Biol. Chem. 2006, 281, 1599–1604. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Cheng, D.; Jouleh, B.; Torp, R.; LaFerla, F.M. Genetically augmenting tau levels does not modulate the onset or progression of Aβ pathology in transgenic mice. J. Neurochem. 2007, 102, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: A longitudinal study. JAMA Neurol. 2019, 76, 915. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Xu, X. γ-secretase catalyzes sequential cleavages of the Aβpp transmembrane domain. J. Alzheimer’s Dis. 2009, 16, 211–224. [Google Scholar] [CrossRef]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid β protein (Aβ) in Alzheimer’s disease brain. J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar] [CrossRef]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef]
- Yang, M.; Teplow, D.B. Amyloid β-protein monomer folding: Free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 2008, 384, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, D.; D’Hooge, R.; Staufenbiel, M.; Van Ginneken, C.; Van Meir, F.; De Deyn, P.P. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 2003, 17, 388–396. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of Aβ 1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Gandy, S.; Simon, A.J.; Steele, J.W.; Lublin, A.L.; Lah, J.J.; Walker, L.C.; Levey, A.I.; Krafft, G.A.; Levy, E.; Checler, F.; et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-β oligomers. Ann. Neurol. 2010, 68, 220–230. [Google Scholar] [CrossRef]
- Kuo, Y.-M.; Emmerling, M.R.; Vigo-Pelfrey, C.; Kasunic, T.C.; Kirkpatrick, J.B.; Murdoch, G.H.; Ball, M.J.; Roher, A.E. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 1996, 271, 4077–4081. [Google Scholar] [CrossRef]
- Roher, A.E.; Chaney, M.O.; Kuo, Y.-M.; Webster, S.D.; Stine, W.B.; Haverkamp, L.J.; Woods, A.S.; Cotter, R.J.; Tuohy, J.M.; Krafft, G.A.; et al. Morphology and toxicity of Aβ-(1-42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J. Biol. Chem. 1996, 271, 20631–20635. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef]
- Forny-Germano, L.; Lyra E Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.B.; et al. Alzheimer’s disease-like pathology induced by amyloid-β oligomers in nonhuman Primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Rolls, E.T. The storage and recall of memories in the hippocampo-cortical system. Cell Tissue Res. 2018, 373, 577–604. [Google Scholar] [CrossRef] [PubMed]
- Kesner, R.P.; Lee, I.; Gilbert, P. A behavioral assessment of hippocampal function based on a subregional analysis. Rev. Neurosci. 2004, 15, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kesner, R.P. Differential contribution of NMDA receptors in hippocampal subregions to spatial working memory. Nat. Neurosci. 2002, 5, 162–168. [Google Scholar] [CrossRef]
- Remondes, M.; Schuman, E.M. Role for a cortical input to hippocampal area CA1 in the consolidation of a long-term memory. Nature 2004, 431, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kesner, R.P. Differential contributions of dorsal hippocampal subregions to memory acquisition and retrieval in contextual fear-conditioning. Hippocampus 2004, 14, 301–310. [Google Scholar] [CrossRef]
- Mitsushima, D.; Ishihara, K.; Sano, A.; Kessels, H.W.; Takahashi, T. Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proc. Natl. Acad. Sci. USA 2011, 108, 12503–12508. [Google Scholar] [CrossRef]
- Ásgeirsdóttir, H.N.; Cohen, S.J.; Stackman, R.W., Jr. Object and place information processing by CA1 hippocampal neurons of C57BL/6J mice. J. Neurophysiol. 2020, 123, 1247–1264. [Google Scholar] [CrossRef]
- Heggland, I.; Storkaas, I.S.; Soligard, H.T.; Kobro-Flatmoen, A.; Witter, M.P. Stereological estimation of neuron number and plaque load in the hippocampal region of a transgenic rat model of A lzheimer’s disease. Eur. J. Neurosci. 2015, 41, 1245–1262. [Google Scholar] [CrossRef]
- Tamagnini, F.; Scullion, S.; Brown, J.T.; Randall, A.D. Intrinsic excitability changes induced by acute treatment of hippocampal CA1 pyramidal neurons with exogenous amyloid β peptide. Hippocampus 2015, 25, 786–797. [Google Scholar] [CrossRef]
- Fernandez-Perez, E.J.; Muñoz, B.; Bascuñan, D.A.; Peters, C.; Riffo-Lepe, N.O.; Espinoza, M.P.; Morgan, P.J.; Filippi, C.; Bourboulou, R.; Sengupta, U.; et al. Synaptic dysregulation and hyperexcitability induced by intracellular amyloid beta oligomers. Aging Cell 2021, 20, e13455. [Google Scholar] [CrossRef]
- Eslamizade, M.J.; Saffarzadeh, F.; Mousavi, S.M.M.; Meftahi, G.H.; Hosseinmardi, N.; Mehdizadeh, M.; Janahmadi, M. Alterations in CA1 pyramidal neuronal intrinsic excitability mediated by Ih channel currents in a rat model of amyloid beta pathology. Neuroscience 2015, 305, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Mo, H.; Kim, J.; Kim, J.W.; Nam, Y.; Rim, Y.A.; Ju, J.H. Mitigating effect of estrogen in Alzheimer’s disease-mimicking cerebral organoid. Front. Neurosci. 2022, 16, 816174. [Google Scholar] [CrossRef] [PubMed]
- Stine, W.B.; Dahlgren, K.N.; Krafft, G.A.; LaDu, M.J. In vitro characterization of conditions for amyloid-β Peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003, 278, 11612–11622. [Google Scholar] [CrossRef]
- Sakimoto, Y.; Shintani, A.; Yoshiura, D.; Goshima, M.; Kida, H.; Mitsushima, D. A critical period for learning and plastic changes at hippocampal CA1 synapses. Sci. Rep. 2022, 12, 7199. [Google Scholar] [CrossRef]
- Beninger, R.J.; Jhamandas, K.; Boegman, R.J.; El-Defrawy, S.R. Effects of scopolamine and unilateral lesions of the basal forebrain on T-maze spatial discrimination and alternation in rats. Pharmacol. Biochem. Behav. 1986, 24, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Hiramatsu, M.; Noda, Y.; Mamiya, T.; Murai, M.; Kameyama, T.; Komori, Y.; Nikai, T.; Sugihara, H.; Nabeshima, T. Role of nitric oxide and cyclic GMP in the dizocilpine-induced impairment of spontaneous alternation behavior in mice. Neuroscience 1996, 74, 365–374. [Google Scholar] [CrossRef]
- Arrant, A.E.; Schramm-Sapyta, N.L.; Kuhn, C.M. Use of the light/dark test for anxiety in adult and adolescent male rats. Behav. Brain Res. 2013, 256, 119–127. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, H.; Han, P.-L. Striatal inhibition of MeCP2 or TSC1 produces sociability deficits and repetitive behaviors. Exp. Neurobiol. 2018, 27, 539–549. [Google Scholar] [CrossRef]
- Takase, K.; Sakimoto, Y.; Kimura, F.; Mitsushima, D. Developmental trajectory of contextual learning and 24-h acetylcholine release in the hippocampus. Sci. Rep. 2014, 4, 3738. [Google Scholar] [CrossRef]
- Koppensteiner, P.; Trinchese, F.; Fà, M.; Puzzo, D.; Gulisano, W.; Yan, S.; Poussin, A.; Liu, S.; Orozco, I.; Dale, E.; et al. Time-dependent reversal of synaptic plasticity induced by physiological concentrations of oligomeric Aβ42: An early index of Alzheimer’s disease. Sci. Rep. 2016, 6, 32553. [Google Scholar] [CrossRef]
- Lehner, M.; Wisłowska-Stanek, A.; Maciejak, P.; Szyndler, J.; Sobolewska, A.; Krzaścik, P.; Płaźnik, A. The relationship between pain sensitivity and conditioned fear response in rats. Acta Neurobiol. Exp. 2010, 70, 56–66. [Google Scholar] [CrossRef]
- Kida, H.; Sakimoto, Y.; Mitsushima, D. Slice patch clamp technique for analyzing learning-induced plasticity. J. Vis. Exp. 2017, 129, 55876. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Bain in Stereotaxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2006. [Google Scholar]
- Cerbai, F.; Lana, D.; Nosi, D.; Petkova-Kirova, P.; Zecchi, S.; Brothers, H.M.; Wenk, G.L.; Giovannini, M.G. The neuron-astrocyte-microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS ONE 2012, 7, e45250. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Ugolini, F.; Nosi, D.; Wenk, G.L.; Giovannini, M.G. Alterations in the interplay between neurons, astrocytes and microglia in the rat dentate gyrus in experimental models of neurodegeneration. Front. Aging Neurosci. 2017, 9, 296. [Google Scholar] [CrossRef] [PubMed]
- Horsley, J.R.; Jovcevski, B.; Wegener, K.L.; Yu, J.; Pukala, T.L.; Abell, A.D. Rationally designed peptide-based inhibitor of Aβ42 fibril formation and toxicity: A potential therapeutic strategy for Alzheimer’s disease. Biochem. J. 2020, 477, 2039–2054. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Gordon, M.N.; Morgan, D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat. Protoc. 2006, 1, 1591–1595. [Google Scholar] [CrossRef]
- Vadukul, D.M.; Gbajumo, O.; Marshall, K.E.; Serpell, L.C. Amyloidogenicity and toxicity of the reverse and scrambled variants of amyloid-β 1-42. FEBS Lett. 2017, 591, 822–830. [Google Scholar] [CrossRef]
- Marshall, K.E.; Vadukul, D.M.; Dahal, L.; Theisen, A.; Fowler, M.W.; Al-Hilaly, Y.; Ford, L.; Kemenes, G.; Day, I.J.; Staras, K.; et al. A critical role for the self-assembly of amyloid-β1-42 in neurodegeneration. Sci. Rep. 2016, 6, 30182. [Google Scholar] [CrossRef]
- Kravenska, Y.; Nieznanska, H.; Nieznanski, K.; Lukyanetz, E.; Szewczyk, A.; Koprowski, P. The monomers, oligomers, and fibrils of amyloid-β inhibit the activity of mitoBKCa channels by a membrane-mediated mechanism. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183337. [Google Scholar] [CrossRef]
- Hirakura, Y.; Lin, M.C.; Kagan, B.L. Alzheimer amyloid abeta1-42 channels: Effects of solvent, pH, and Congo Red. J. Neurosci. Res. 1999, 57, 458–466. [Google Scholar] [CrossRef]
- Koh, M.T.; Haberman, R.P.; Foti, S.; McCown, T.J.; Gallagher, M. Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive Impairment. Neuropsychopharmacology 2010, 35, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Yassa, M.A.; Stark, S.M.; Bakker, A.; Albert, M.S.; Gallagher, M.; Stark, C.E.L. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. NeuroImage 2010, 51, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.-Q.; Yao, W.; Yan, J.-Z.; Jin, C.; Yin, J.-J.; Yuan, J.; Yu, S.; Cheng, Z. Amyloid β causes excitation/inhibition imbalance through dopamine receptor 1-dependent disruption of fast-spiking GABAergic input in anterior cingulate cortex. Sci. Rep. 2018, 8, 302. [Google Scholar] [CrossRef]
- George, A.A.; Vieira, J.M.; Xavier-Jackson, C.; Gee, M.T.; Cirrito, J.R.; Bimonte-Nelson, H.A.; Picciotto, M.R.; Lukas, R.J.; Whiteaker, P. Implications of oligomeric amyloid-beta (oAβ42) signaling through α7β2-nicotinic acetylcholine receptors (nAChRs) on basal forebrain cholinergic neuronal intrinsic excitability and cognitive decline. J. Neurosci. 2021, 41, 555–575. [Google Scholar] [CrossRef]
- Taverna, S.; Tkatch, T.; Metz, A.E.; Martina, M. Differential expression of TASK channels between horizontal interneurons and pyramidal cells of rat hippocampus. J. Neurosci. 2005, 25, 9162–9170. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Vervaeke, K.; Storm, J.F. Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J. Physiol. 2002, 545, 783–805. [Google Scholar] [CrossRef]
- Matsumura, R.; Yamamoto, H.; Hayakawa, T.; Katsurabayashi, S.; Niwano, M.; Hirano-Iwata, A. Dependence and homeostasis of membrane impedance on cell morphology in cultured hippocampal neurons. Sci. Rep. 2018, 8, 9905. [Google Scholar] [CrossRef]
- Ungureanu, A.-A.; Benilova, I.; Krylychkina, O.; Braeken, D.; De Strooper, B.; Van Haesendonck, C.; Dotti, C.G.; Bartic, C. Amyloid beta oligomers induce neuronal elasticity changes in age-dependent manner: A force spectroscopy study on living hippocampal neurons. Sci. Rep. 2016, 6, 25841. [Google Scholar] [CrossRef]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef]
- Nimmrich, V.; Grimm, C.; Draguhn, A.; Barghorn, S.; Lehmann, A.; Schoemaker, H.; Hillen, H.; Gross, G.; Ebert, U.; Bruehl, C. Amyloid β oligomers (Aβ1–42 globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-Type calcium currents. J. Neurosci. 2008, 28, 788–797. [Google Scholar] [CrossRef]
- Ferrara, N.C.; Jarome, T.J.; Cullen, P.K.; Orsi, S.A.; Kwapis, J.L.; Trask, S.; Pullins, S.E.; Helmstetter, F.J. GluR2 endocytosis-dependent protein degradation in the amygdala mediates memory updating. Sci. Rep. 2019, 9, 5180. [Google Scholar] [CrossRef] [PubMed]
- Piromalli Girado, D.; Miranda, M.; Giachero, M.; Weisstaub, N.; Bekinschtein, P. Endocytosis is required for consolidation of pattern-separated memories in the perirhinal cortex. Front. Syst. Neurosci. 2023, 17, 1043664. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ueta, Y.; Wang, L.; Yamamoto, R.; Inoue, N.; Inokuchi, K.; Aiba, A.; Yonekura, H.; Kato, N. Suppression of a neocortical potassium channel activity by intracellular amyloid-β and its rescue with Homer1a. J. Neurosci. 2011, 31, 11100–11109. [Google Scholar] [CrossRef]
- Ciccone, R.; Franco, C.; Piccialli, I.; Boscia, F.; Casamassa, A.; De Rosa, V.; Cepparulo, P.; Cataldi, M.; Annunziato, L.; Pannaccione, A. Amyloid β-induced upregulation of Nav1.6 underlies neuronal hyperactivity in Tg2576 Alzheimer’s disease mouse model. Sci. Rep. 2019, 9, 13592. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Kedia, N.; Illes-Toth, E.; Haralampiev, I.; Prisner, S.; Herrmann, A.; Wanker, E.E.; Bieschke, J. Amyloid-β (1-42) aggregation initiates its cellular uptake and cytotoxicity. J. Biol. Chem. 2016, 291, 19590–19606. [Google Scholar] [CrossRef]
- Chatelier, A.; Zhao, J.; Bois, P.; Chahine, M. Biophysical characterisation of the persistent sodium current of the Nav1.6 neuronal sodium channel: A single-channel analysis. Pflügers Arch.-Eur. J. Physiol. 2010, 460, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Vervaeke, K.; Hu, H.; Graham, L.J.; Storm, J.F. Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron 2006, 49, 257–270. [Google Scholar] [CrossRef]
- Ren, S.; Chen, P.; Jiang, H.; Mi, Z.; Xu, F.; Hu, B.; Zhang, J.; Zhu, Z. Persistent sodium currents contribute to Aβ1-42-induced hyperexcitation of hippocampal CA1 pyramidal neurons. Neurosci. Lett. 2014, 580, 62–67. [Google Scholar] [CrossRef]
- Min-Kaung-Wint-Mon; Kimura, R.; Mitsuhsima, D. Riluzole improves hippocampus-dependent learning deficits caused by Aβ1–42 oligomers. Abstract for the 102nd Annual Meeting of the Physiological Society of Japan. J. Physiol. Sci. 2025, 75 (Suppl. S1). in press. [Google Scholar]
- Sipos, E.; Kurunczi, A.; Kasza, Á.; Horváth, J.; Felszeghy, K.; Laroche, S.; Toldi, J.; Párducz, Á.; Penke, B.; Penke, Z. β-Amyloid pathology in the entorhinal cortex of rats induces memory deficits: Implications for Alzheimer’s disease. Neuroscience 2007, 147, 28–36. [Google Scholar] [CrossRef]
- Chang, K.-W.; Zong, H.-F.; Ma, K.-G.; Zhai, W.-Y.; Yang, W.-N.; Hu, X.-D.; Xu, J.-H.; Chen, X.-L.; Ji, S.-F.; Qian, Y.-H. Activation of α7 nicotinic acetylcholine receptor alleviates Aβ1-42-induced neurotoxicity via downregulation of p38 and JNK MAPK signaling pathways. Neurochem. Int. 2008, 120, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.; Marcussen, A.B.; Wörtwein, G.; Knudsen, G.M.; Aznar, S. Aβ (1–42) injection causes memory impairment, lowered cortical and serum BDNF levels, and decreased hippocampal 5-HT2A levels. Exp. Neurol. 2008, 210, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, J.; Caillierez, R.; Zommer, N.; Alves-Pires, C.; Benilova, I.; Blum, D.; De Strooper, B.; Buee, L. Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid- 1-42 oligomers are revealed in vivo by using a novel animal model. J. Neurosci. 2012, 32, 7852–7861. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ji, W.; Zhu, Z.; Wu, Y.; Zhang, Z.; Qu, S. Rhynchophylline suppresses soluble Aβ1-42-induced impairment of spatial cognition function via inhibiting excessive activation of extrasynaptic NR2B-containing NMDA receptors. Neuropharmacology 2018, 135, 100–112. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, J.; Ruan, Z.; Tian, S.; Ma, Y.; Zhu, J.; Li, G. Intrahippocampal injection of Aβ 1-42 inhibits neurogenesis and down-regulates IFN-γ and NF-κB expression in hippocampus of adult mouse brain. Amyloid 2013, 20, 13–20. [Google Scholar] [CrossRef]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab-binding profiles to different forms of amyloid-Beta might explain efficacy and side effects in clinical trials for Alzheimer’s disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef]
- O’Hare, E.; Weldon, D.T.; Mantyh, P.W.; Ghilardi, J.R.; Finke, M.P.; Kuskowski, M.A.; Maggio, J.E.; Shephard, R.A.; Cleary, J. Delayed behavioral effects following intrahippocampal injection of aggregated Aβ (1–42). Brain Res. 1999, 815, 1–10. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min-Kaung-Wint-Mon; Kida, H.; Kanehisa, I.; Kurose, M.; Ishikawa, J.; Sakimoto, Y.; Paw-Min-Thein-Oo; Kimura, R.; Mitsushima, D. Adverse Effects of Aβ1-42 Oligomers: Impaired Contextual Memory and Altered Intrinsic Properties of CA1 Pyramidal Neurons. Biomolecules 2024, 14, 1425. https://doi.org/10.3390/biom14111425
Min-Kaung-Wint-Mon, Kida H, Kanehisa I, Kurose M, Ishikawa J, Sakimoto Y, Paw-Min-Thein-Oo, Kimura R, Mitsushima D. Adverse Effects of Aβ1-42 Oligomers: Impaired Contextual Memory and Altered Intrinsic Properties of CA1 Pyramidal Neurons. Biomolecules. 2024; 14(11):1425. https://doi.org/10.3390/biom14111425
Chicago/Turabian StyleMin-Kaung-Wint-Mon, Hiroyuki Kida, Itsuki Kanehisa, Masahiko Kurose, Junko Ishikawa, Yuya Sakimoto, Paw-Min-Thein-Oo, Ryoichi Kimura, and Dai Mitsushima. 2024. "Adverse Effects of Aβ1-42 Oligomers: Impaired Contextual Memory and Altered Intrinsic Properties of CA1 Pyramidal Neurons" Biomolecules 14, no. 11: 1425. https://doi.org/10.3390/biom14111425
APA StyleMin-Kaung-Wint-Mon, Kida, H., Kanehisa, I., Kurose, M., Ishikawa, J., Sakimoto, Y., Paw-Min-Thein-Oo, Kimura, R., & Mitsushima, D. (2024). Adverse Effects of Aβ1-42 Oligomers: Impaired Contextual Memory and Altered Intrinsic Properties of CA1 Pyramidal Neurons. Biomolecules, 14(11), 1425. https://doi.org/10.3390/biom14111425