
Metabolites and Metabolic Functional Changes—Potential Markers for Endothelial Cell Senescence


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. SA-β-Galactosidase Activity
2.3. Proliferation Assay
2.4. Immunocytochemistry
2.5. Tubulogenesis Assay
2.6. Wound Healing Assay
2.7. Protein Extraction and Quantification
2.8. Western Blotting
2.9. Telomere Length Measurement
2.10. Analysis of the Secretory Cytokines
2.11. Liquid Chromatography Mass Spectrometry (LC-MS) Metabolomics Analysis
2.12. Statistical Analysis
3. Results
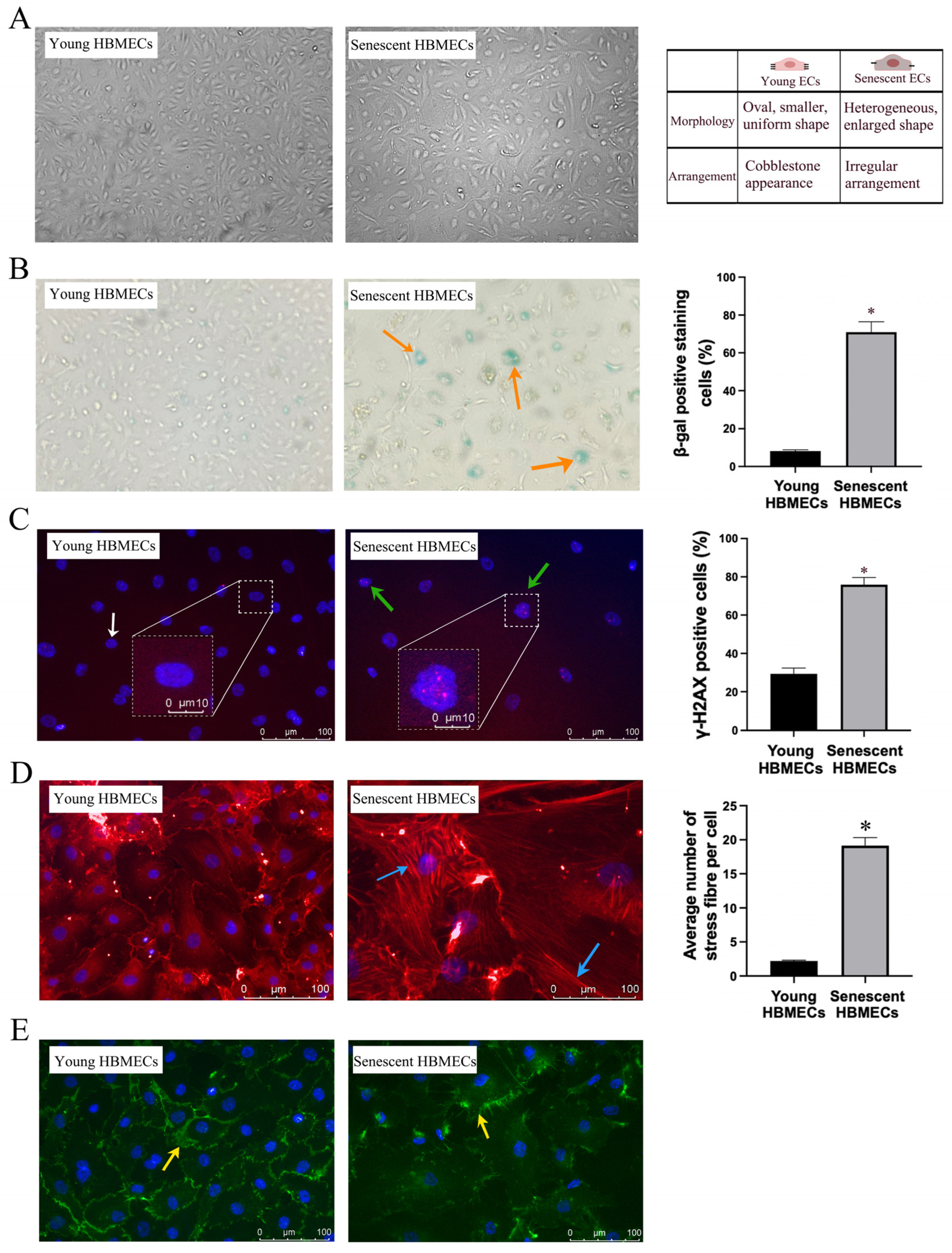
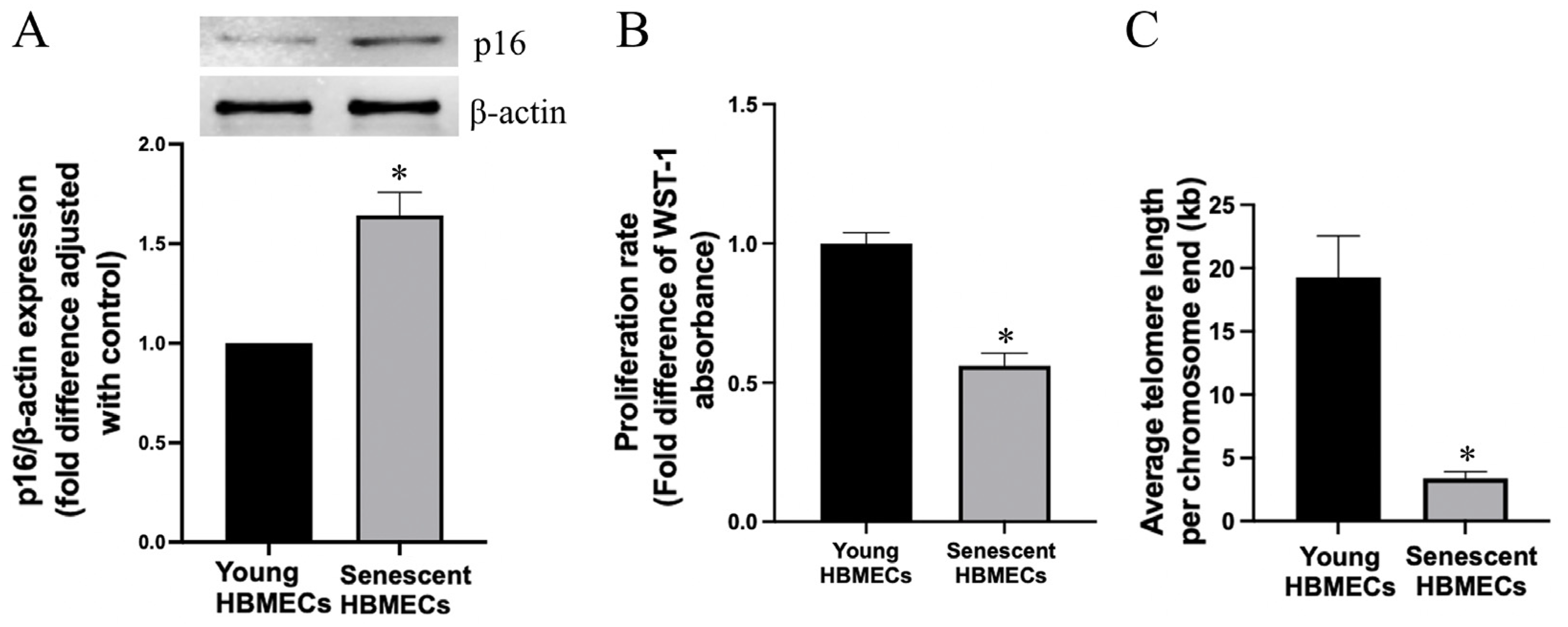
3.1. Late-Passage Endothelial Cells Exhibit Morphological Change and Express Markers of Cellular Senescence
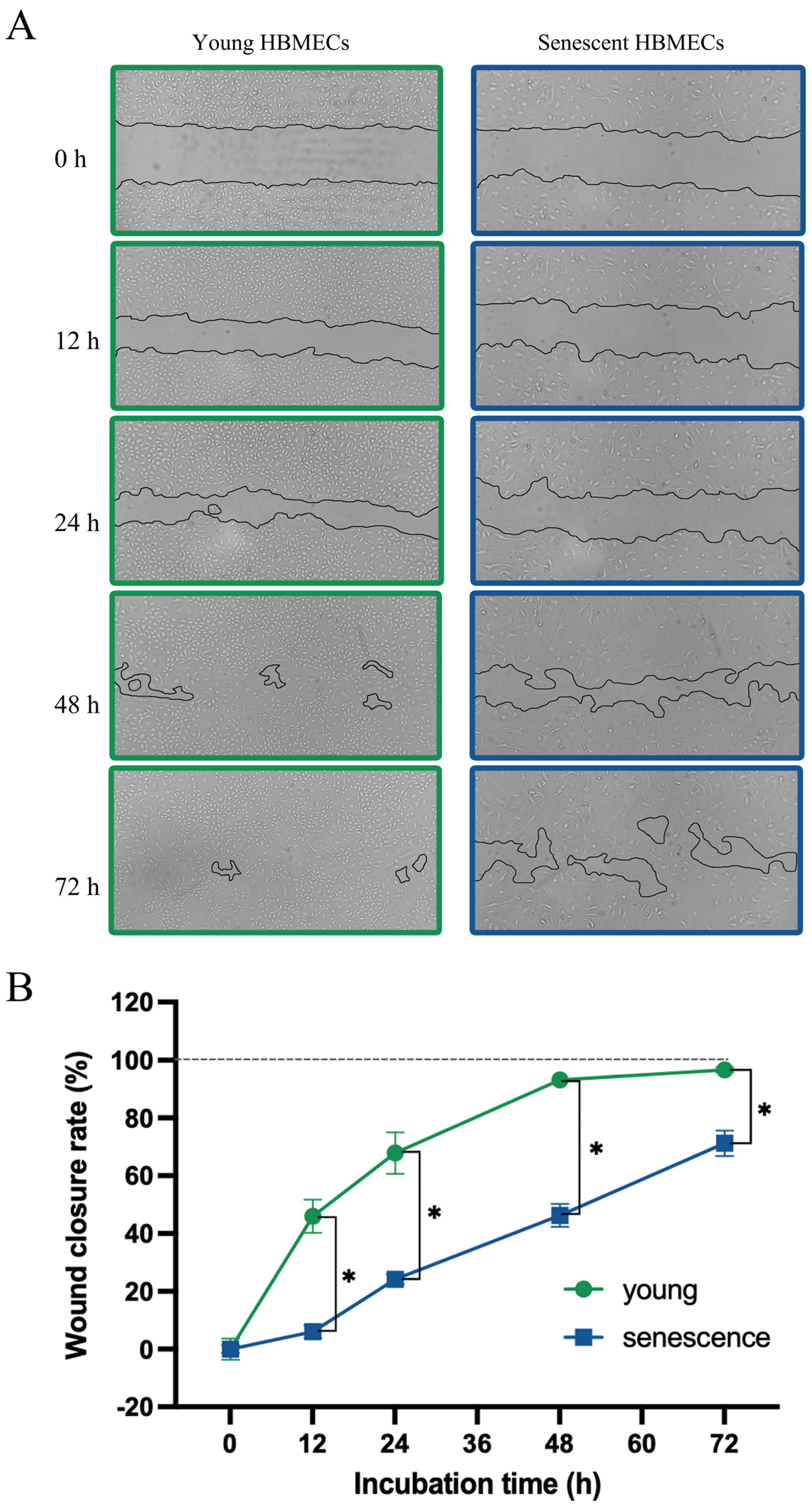
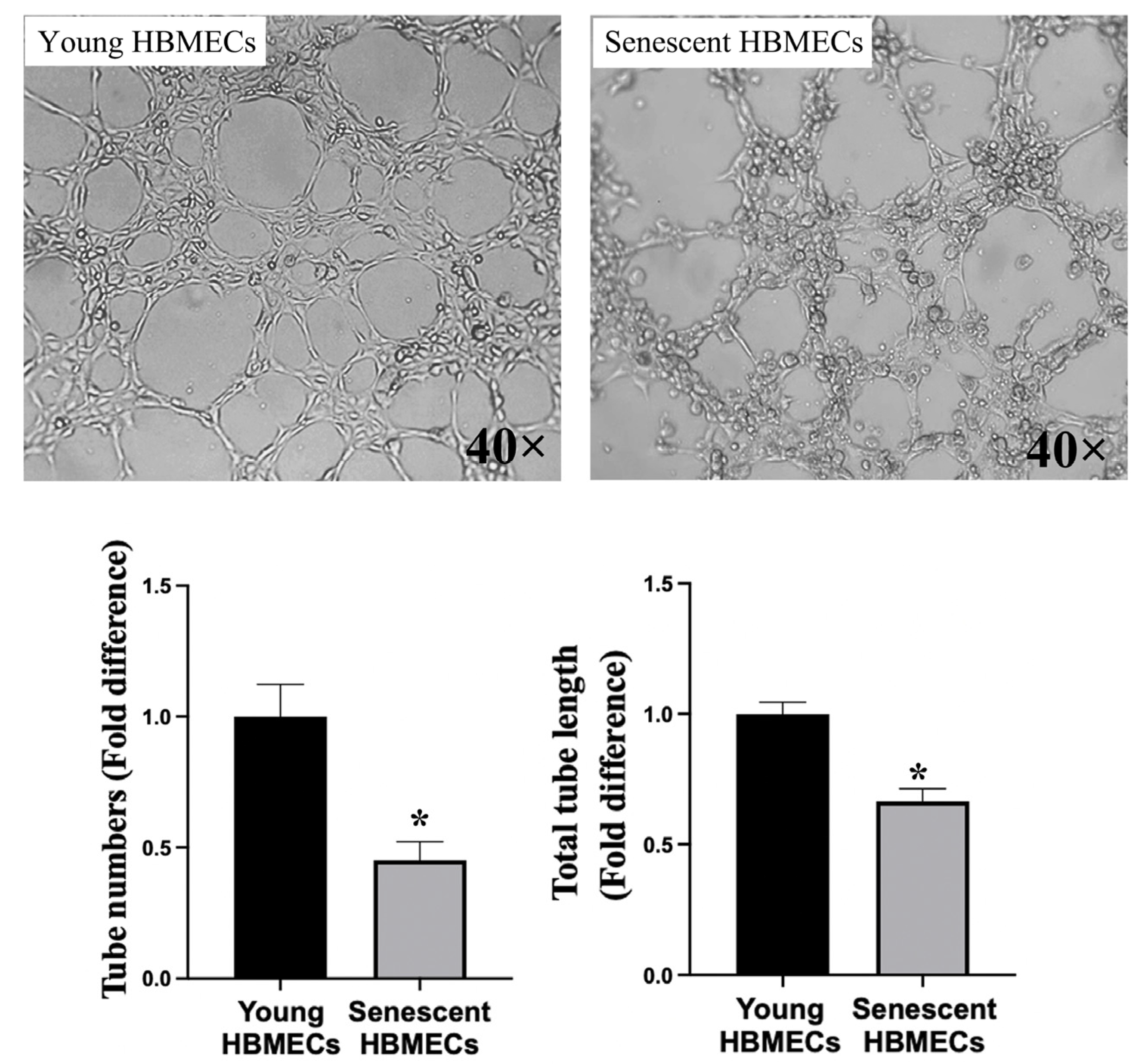
3.2. Senescent Endothelial Cells Lose Their Angiogenic Potential
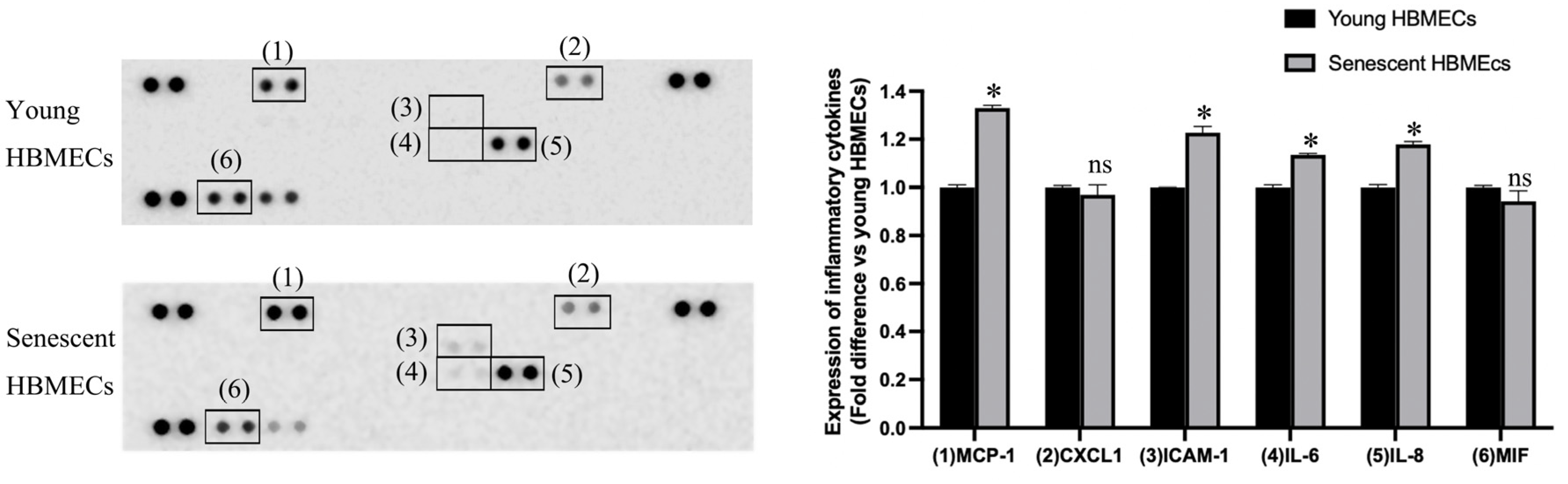
3.3. Production of Inflammatory Cytokines Is Elevated in Senescent HBMECs
3.4. Cellular Senescence Results in Changes in Metabolite Abundance in Endothelial Cells
3.5. Intracellular Metabolite Profile of HBMECs Changes with Senescence
3.6. The Profile of Metabolite Secretome of HBMECs Changes with Senescence
4. Discussion
Limitations and Future Work
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2021 Diseases and Injuries Collaborators. Global incidence, prevalence, years lived with disability (YLDs), disability-adjusted life-years (DALYs), and healthy life expectancy (HALE) for 371 diseases and injuries in 204 countries and territories and 811 subnational locations, 1990–2021: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2024, 403, 2133–2161. [Google Scholar] [CrossRef]
- Cesare, M.D.; Bixby, H.; Gaziano, T.; Hadeed, L.; Kabudula, C.; McGhie, D.V.; Mwangi, J.; Pervan, B.; Perel, P.; Piñeiro, D.; et al. World Heart Report 2023: Confronting the World’s Number One Killer; World Heart Federation: Geneva, Switzerland, 2023. [Google Scholar]
- Townsend, N.; Kazakiewicz, D.; Lucy Wright, F.; Timmis, A.; Huculeci, R.; Torbica, A.; Gale, C.P.; Achenbach, S.; Weidinger, F.; Vardas, P. Epidemiology of cardiovascular disease in Europe. Nat. Rev. Cardiol. 2022, 19, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, W.S. The Economic Burden of Illness. JAMA Netw. Open 2023, 6, e232663. [Google Scholar] [CrossRef] [PubMed]
- Ya, J.; Bayraktutan, U. Vascular Ageing: Mechanisms, Risk Factors, and Treatment Strategies. Int. J. Mol. Sci. 2023, 24, 11538. [Google Scholar] [CrossRef] [PubMed]
- Bayraktutan, U. Reactive Oxygen Species, Nitric Oxide and Hypertensive Endothelial Dysfunction. Curr. Hypertens. Rev. 2005, 1, 201–215. [Google Scholar] [CrossRef]
- Gifre-Renom, L.; Daems, M.; Luttun, A.; Jones, E.A.V. Organ-Specific Endothelial Cell Differentiation and Impact of Microenvironmental Cues on Endothelial Heterogeneity. Int. J. Mol. Sci. 2022, 23, 1477. [Google Scholar] [CrossRef]
- Uwamori, H.; Ono, Y.; Yamashita, T.; Arai, K.; Sudo, R. Comparison of organ-specific endothelial cells in terms of microvascular formation and endothelial barrier functions. Microvasc. Res. 2019, 122, 60–70. [Google Scholar] [CrossRef]
- Segarra, M.; Aburto, M.R.; Acker-Palmer, A. Blood-Brain Barrier Dynamics to Maintain Brain Homeostasis. Trends Neurosci. 2021, 44, 393–405. [Google Scholar] [CrossRef]
- Xiao, X.; Jiang, H.; Wei, H.; Zhou, Y.; Ji, X.; Zhou, C. Endothelial Senescence in Neurological Diseases. Aging Dis. 2023, 14, 2153–2166. [Google Scholar] [CrossRef]
- Ya, J.; Kadir, R.R.A.; Bayraktutan, U. Delay of endothelial cell senescence protects cerebral barrier against age-related dysfunction: Role of senolytics and senomorphics. Tissue Barriers 2023, 11, 2103353. [Google Scholar] [CrossRef]
- Malaquin, N.; Tu, V.; Rodier, F. Assessing Functional Roles of the Senescence-Associated Secretory Phenotype (SASP). Methods Mol. Biol. 2019, 1896, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Saada, Y.B.; Garcia, D.; Friguet, B. Effects of cellular senescence on metabolic pathways in non-immune and immune cells. Mech. Ageing Dev. 2021, 194, 111428. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Leung, S.W.S.; Shi, Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol. Sin. 2022, 43, 251–259. [Google Scholar] [CrossRef]
- Ya, J.; Bayraktutan, U. Senolytics and Senomorphics Targeting p38MAPK/NF-kappaB Pathway Protect Endothelial Cells from Oxidative Stress-Mediated Premature Senescence. Cells 2024, 13, 1292. [Google Scholar] [CrossRef]
- Kadir, R.R.A.; Alwjwaj, M.; McCarthy, Z.; Bayraktutan, U. Therapeutic hypothermia augments the restorative effects of PKC-beta and Nox2 inhibition on an in vitro model of human blood-brain barrier. Metab. Brain Dis. 2021, 36, 1817–1832. [Google Scholar] [CrossRef]
- Walsby-Tickle, J.; Gannon, J.; Hvinden, I.; Bardella, C.; Abboud, M.I.; Nazeer, A.; Hauton, D.; Pires, E.; Cadoux-Hudson, T.; Schofield, C.J.; et al. Anion-exchange chromatography mass spectrometry provides extensive coverage of primary metabolic pathways revealing altered metabolism in IDH1 mutant cells. Commun. Biol. 2020, 3, 247. [Google Scholar] [CrossRef]
- Whitby, A.; Pabla, P.; Shastri, B.; Amugi, L.; Del Rio-Alvarez, A.; Kim, D.H.; Royo, L.; Armengol, C.; Dandapani, M. Characterisation of Aberrant Metabolic Pathways in Hepatoblastoma Using Liquid Chromatography and Tandem Mass Spectrometry (LC-MS/MS). Cancers 2023, 15, 5812. [Google Scholar] [CrossRef]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Ng, C.C.; Jones, O.; Fung, T.S.; Ryu, K.; Li, D.; Thompson, C.B. Lactate activates the mitochondrial electron transport chain independent of its metabolism. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.; Milligan, D.; Hughes, B.; Mizen, H.; Lopez-Dominguez, J.A.; Eduputa, U.; Tyler, E.J.; Serrano, M.; Bishop, C.L. Senescence-associated morphological profiles (SAMPs): An image-based phenotypic profiling method for evaluating the inter and intra model heterogeneity of senescence. Aging 2022, 14, 4220–4246. [Google Scholar] [CrossRef] [PubMed]
- Reskiawan, A.K.R.; Alwjwaj, M.; Ahmad Othman, O.; Rakkar, K.; Sprigg, N.; Bath, P.M.; Bayraktutan, U. Inhibition of oxidative stress delays senescence and augments functional capacity of endothelial progenitor cells. Brain Res. 2022, 1787, 147925. [Google Scholar] [CrossRef]
- Fan, L.M.; Geng, L.; Cahill-Smith, S.; Liu, F.; Douglas, G.; McKenzie, C.A.; Smith, C.; Brooks, G.; Channon, K.M.; Li, J.M. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J. Clin. Investig. 2019, 129, 3374–3386. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef]
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.F. An evolving new paradigm: Endothelial cells—Conditional innate immune cells. J. Hematol. Oncol. 2013, 6, 61. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Bickel, M. The role of interleukin-8 in inflammation and mechanisms of regulation. J. Periodontol. 1993, 64, 456–460. [Google Scholar]
- Witkowska, A.M. Soluble ICAM-1: A marker of vascular inflammation and lifestyle. Cytokine 2005, 31, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dhalla, N.S. The Role of Pro-Inflammatory Cytokines in the Pathogenesis of Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 1082. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef]
- Calzada, E.; Onguka, O.; Claypool, S.M. Phosphatidylethanolamine Metabolism in Health and Disease. Int. Rev. Cell Mol. Biol. 2016, 321, 29–88. [Google Scholar] [CrossRef]
- Patel, S.K.; Bons, J.; Rose, J.P.; Chappel, J.R.; Beres, R.L.; Watson, M.A.; Webster, C.; Burton, J.B.; Bruderer, R.; Desprez, P.Y.; et al. Exosomes Released from Senescent Cells and Circulatory Exosomes Isolated from Human Plasma Reveal Aging-associated Proteomic and Lipid Signatures. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lee, S.J.; Yi, T.; Ahn, S.H.; Lim, D.K.; Hong, J.Y.; Cho, Y.K.; Lim, J.; Song, S.U.; Kwon, S.W. Senescing human bone-marrow-derived clonal mesenchymal stem cells have altered lysophospholipid composition and functionality. J. Proteome Res. 2014, 13, 1438–1449. [Google Scholar] [CrossRef]
- Patriarca, E.J.; Cermola, F.; D’Aniello, C.; Fico, A.; Guardiola, O.; De Cesare, D.; Minchiotti, G. The Multifaceted Roles of Proline in Cell Behavior. Front. Cell Dev. Biol. 2021, 9, 728576. [Google Scholar] [CrossRef]
- Choudhury, D.; Rong, N.; Senthil Kumar, H.V.; Swedick, S.; Samuel, R.Z.; Mehrotra, P.; Toftegaard, J.; Rajabian, N.; Thiyagarajan, R.; Podder, A.K.; et al. Proline restores mitochondrial function and reverses aging hallmarks in senescent cells. Cell Rep. 2024, 43, 113738. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3-dependent Ca2+ release in mouse brain endothelial cells. J. Cell Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Reactive Oxygen Species Detection in Senescent Cells. Methods Mol. Biol. 2019, 1896, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rebollo, E.; Franzen, J.; Goetzke, R.; Hollmann, J.; Ostrowska, A.; Oliverio, M.; Sieben, T.; Rath, B.; Kornfeld, J.W.; Wagner, W. Senescence-Associated Metabolomic Phenotype in Primary and iPSC-Derived Mesenchymal Stromal Cells. Stem. Cell Rep. 2020, 14, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Hamsanathan, S.; Anthonymuthu, T.; Prosser, D.; Lokshin, A.; Greenspan, S.L.; Resnick, N.M.; Perera, S.; Okawa, S.; Narasimhan, G.; Gurkar, A.U. A molecular index for biological age identified from the metabolome and senescence-associated secretome in humans. Aging Cell 2024, 23, e14104. [Google Scholar] [CrossRef]
 and young
and young  HBMEC lysate and culture media samples. OPLS-DA of cell lysate (R2Y = 0.980; Q2 = 0.969) and media (R2Y = 0.995; Q2 = 0.985) shows a good fit of data and the predictive ability of the model without overfitting.
and young HBMEC lysate and culture media samples. OPLS-DA of cell lysate (R2Y = 0.980; Q2 = 0.969) and media (R2Y = 0.995; Q2 = 0.985) shows a good fit of data and the predictive ability of the model without overfitting.
HBMEC lysate and culture media samples. OPLS-DA of cell lysate (R2Y = 0.980; Q2 = 0.969) and media (R2Y = 0.995; Q2 = 0.985) shows a good fit of data and the predictive ability of the model without overfitting.
and young HBMEC lysate and culture media samples. OPLS-DA of cell lysate (R2Y = 0.980; Q2 = 0.969) and media (R2Y = 0.995; Q2 = 0.985) shows a good fit of data and the predictive ability of the model without overfitting.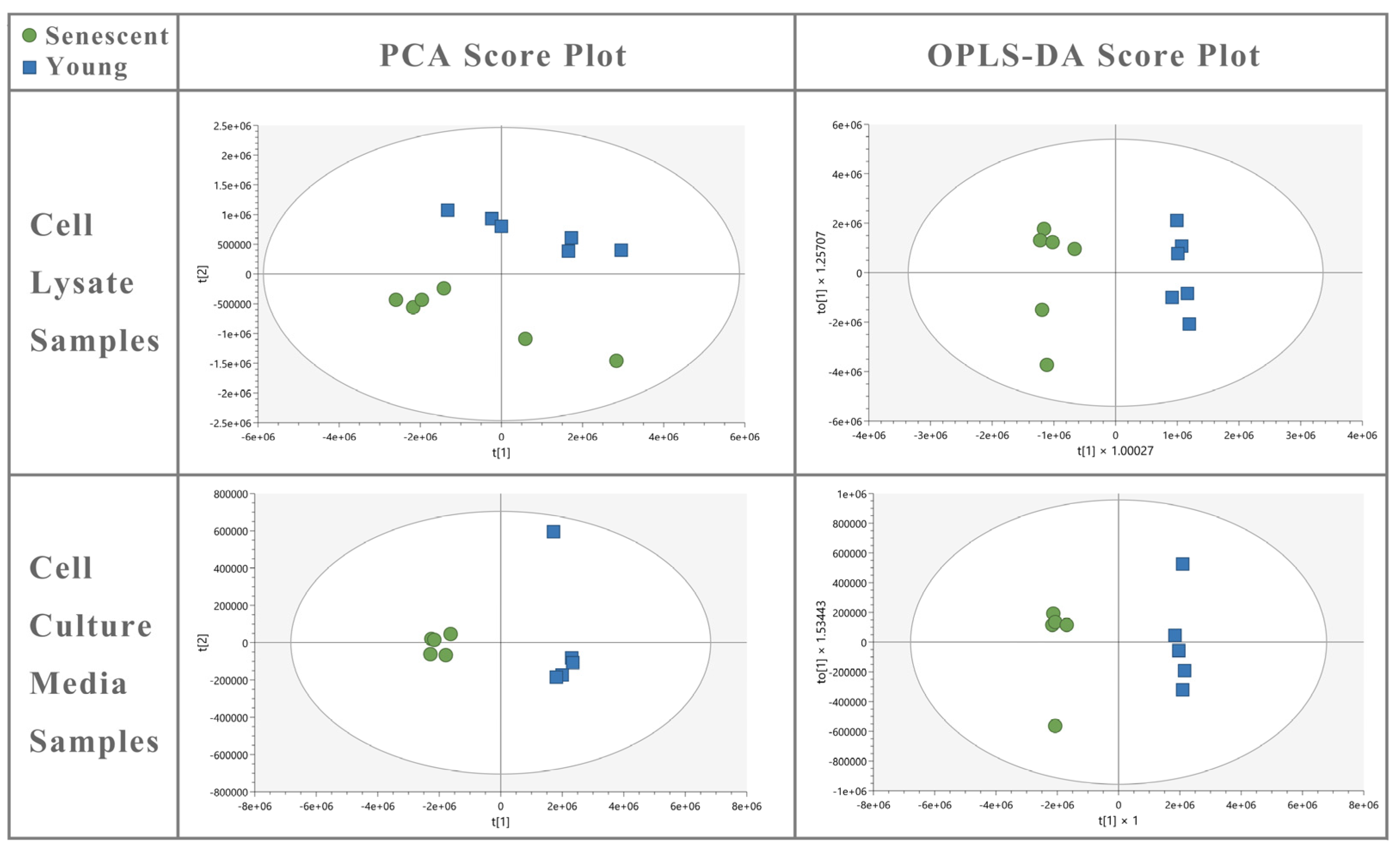

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotation | Formula | ID Confidence Level * | Fold Change (Senescent/Young) | FDR Corrected p-Value | VIP Value |
|---|---|---|---|---|---|
| Hypotaurine | C2H7NO2S | 3 | 0.49 | 8.45 × 10−9 | 0.38 |
| N-Acetyl-L-glutamate | C7H11NO5 | 2 | 0.15 | 6.51 × 10−7 | 0.70 |
| Taurine | C2H7NO3S | 2 | 0.60 | 1.67 × 10−6 | 0.81 |
| 2-Aminoprop-2-enoate | C3H5NO2 | 4 | 0.52 | 6.53 × 10−6 | 0.18 |
| L-Pyroglutamic acid | C5H7NO3 | 2 | 0.63 | 6.53 × 10−6 | 0.47 |
| (Z)-2-Aminobutenoate | C4H7NO3 | 4 | 0.66 | 1.77 × 10−5 | 0.40 |
| Ethanolamine phosphate | C2H8NO4P | 2 | 7.29 | 2.63 × 10−5 | 1.43 |
| UDP-N-acetyl-D-galactosamine | C17H27N3O17P2 | 4 | 0.57 | 3.99 × 10−5 | 0.36 |
| N-Acetyl-L-methionine | C7H13NO3S | 4 | 0.40 | 4.21 × 10−5 | 0.41 |
| N-Methyl-L-glutamate | C6H11NO4 | 4 | 0.71 | 8.62 × 10−5 | 0.21 |
| L-Aspartate | C4H7NO4 | 2 | 0.53 | 8.62 × 10−5 | 1.23 |
| L-Glutamate | C5H9NO4 | 2 | 0.64 | 8.62 × 10−5 | 2.56 |
| O-Acetyl-L-serine | C5H9NO4 | 2 | 0.62 | 9.36 × 10−5 | 0.43 |
| NAD+ | C21H29N7O14P2 | 2 | 0.65 | 9.78 × 10−5 | 0.25 |
| O-Acetyl-L-homoserine | C6H11NO4 | 4 | 0.48 | 1.87 × 10−5 | 0.31 |
| N-Acetyl-L-aspartate | C6H9NO5 | 4 | 1.82 | 3.56 × 10−4 | 0.57 |
| sn-Glycero-3-phosphoethanolamine | C5H14NO6P | 4 | 0.79 | 3.70 × 10−4 | 0.51 |
| Malate | C4H6O5 | 2 | 0.59 | 3.70 × 10−4 | 0.99 |
| β-Alanine | C3H7NO2 | 4 | 0.50 | 7.16 × 10−4 | 0.68 |
| 2-Oxoglutarate | C5H6O5 | 4 | 0.65 | 4.41 × 10−3 | 0.34 |
| L-Proline | C5H9NO2 | 2 | 0.55 | 5.77 × 10−3 | 4.59 |
| sn-Glycero-3-Phosphocholine | C8H20NO6P | 2 | 0.82 | 8.36 × 10−3 | 2.93 |
| Pantothenate | C9H17NO5 | 4 | 0.74 | 1.10 × 10−2 | 0.31 |
| Palmitoyl ethanolamide | C18H37NO2 | 4 | 0.69 | 1.47 × 10−2 | 0.25 |
| AMP | C10H14N5O7P | 2 | 0.75 | 2.03 × 10−2 | 0.11 |
| O-Acetylcarnitine | C9H17NO4 | 2 | 0.59 | 3.17 × 10−2 | 0.39 |
| sn-Glycerol 3-phosphate | C3H9O6P | 4 | 0.85 | 4.21 × 10−2 | 0.18 |
| Choline phosphate | C5H14NO4P | 2 | 1.24 | 4.69 × 10−2 | 1.46 |
| (R)-2-Hydroxyglutarate | C5H8O5 | 2 | 0.79 | 4.80 × 10−2 | 0.19 |
| Annotation | Formula | ID Confidence Level * | Fold Change (Senescent/Young) | FDR Correct d p-Value | VIP Value |
|---|---|---|---|---|---|
| Guanine | C5H5N5O | 4 | 0.52 | 3.94 × 10−6 | 0.20 |
| Lactate | C3H6O3 | 2 | 0.56 | 2.64 × 10−5 | 5.72 |
| Hypoxanthine | C5H4N4O | 2 | 0.93 | 1.02 × 10−4 | 0.55 |
| L-Thyronine | C15H15NO4 | 4 | 1.41 | 1.57 × 10−4 | 0.03 |
| 2′-Deoxycytidine | C9H13N3O4 | 3 | 0.35 | 2.73 × 10−4 | 0.15 |
| 3/4-Methyl-2-oxopentanoate | C6H10O3 | 4 | 1.44 | 1.90 × 10−3 | 0.69 |
| 3-Methyl-2-oxobutanoate | C5H8O3 | 4 | 1.31 | 3.43 × 10−3 | 0.31 |
| O-Succinyl-homoserine | C8H13NO6 | 4 | 0.76 | 1.06 × 10−2 | 0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ya, J.; Whitby, A.; Bayraktutan, U. Metabolites and Metabolic Functional Changes—Potential Markers for Endothelial Cell Senescence. Biomolecules 2024, 14, 1476. https://doi.org/10.3390/biom14111476
Ya J, Whitby A, Bayraktutan U. Metabolites and Metabolic Functional Changes—Potential Markers for Endothelial Cell Senescence. Biomolecules. 2024; 14(11):1476. https://doi.org/10.3390/biom14111476
Chicago/Turabian StyleYa, Jingyuan, Alison Whitby, and Ulvi Bayraktutan. 2024. "Metabolites and Metabolic Functional Changes—Potential Markers for Endothelial Cell Senescence" Biomolecules 14, no. 11: 1476. https://doi.org/10.3390/biom14111476
APA StyleYa, J., Whitby, A., & Bayraktutan, U. (2024). Metabolites and Metabolic Functional Changes—Potential Markers for Endothelial Cell Senescence. Biomolecules, 14(11), 1476. https://doi.org/10.3390/biom14111476