
Senescence-like Phenotype After Chronic Exposure to Isoproterenol in Primary Quiescent Immune Cells

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of PBMC and T Cells
2.2. Isoproterenol Treatment
2.3. DNA Double-Strand Breaks
2.4. Telomere Length Determination
2.5. Telomerase Activity Measurements Using TRAP ELISA
2.6. Proliferations Analysis
2.7. Gene Expression
2.8. Senescence-Associated β-Galactosidase Activity
2.9. Cell Adhesion
2.10. Activity of Caspases 3 and 7
2.11. Statistics
3. Results
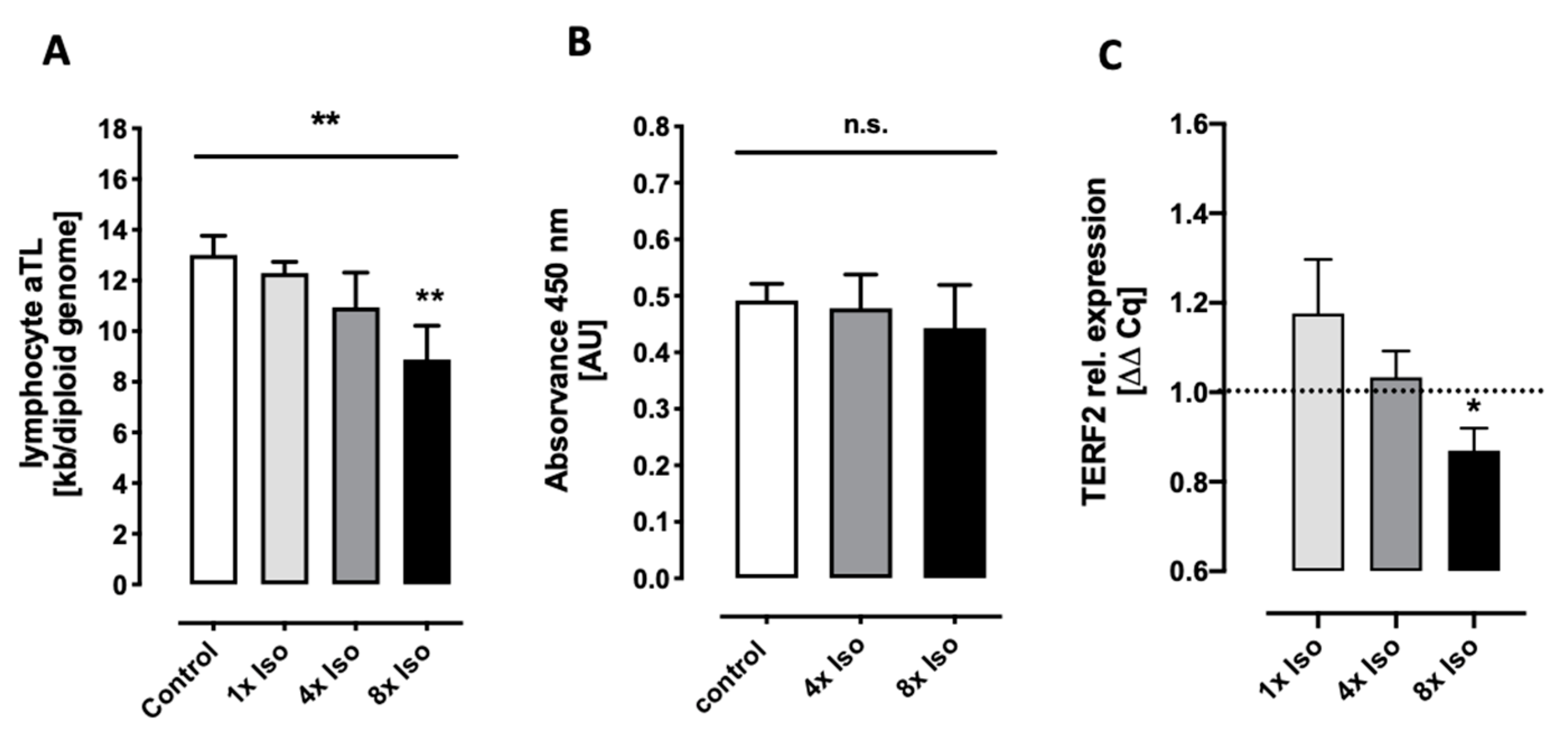
3.1. Isoproterenol Induces Telomere Shortening but Does Not Change Telomerase Activity
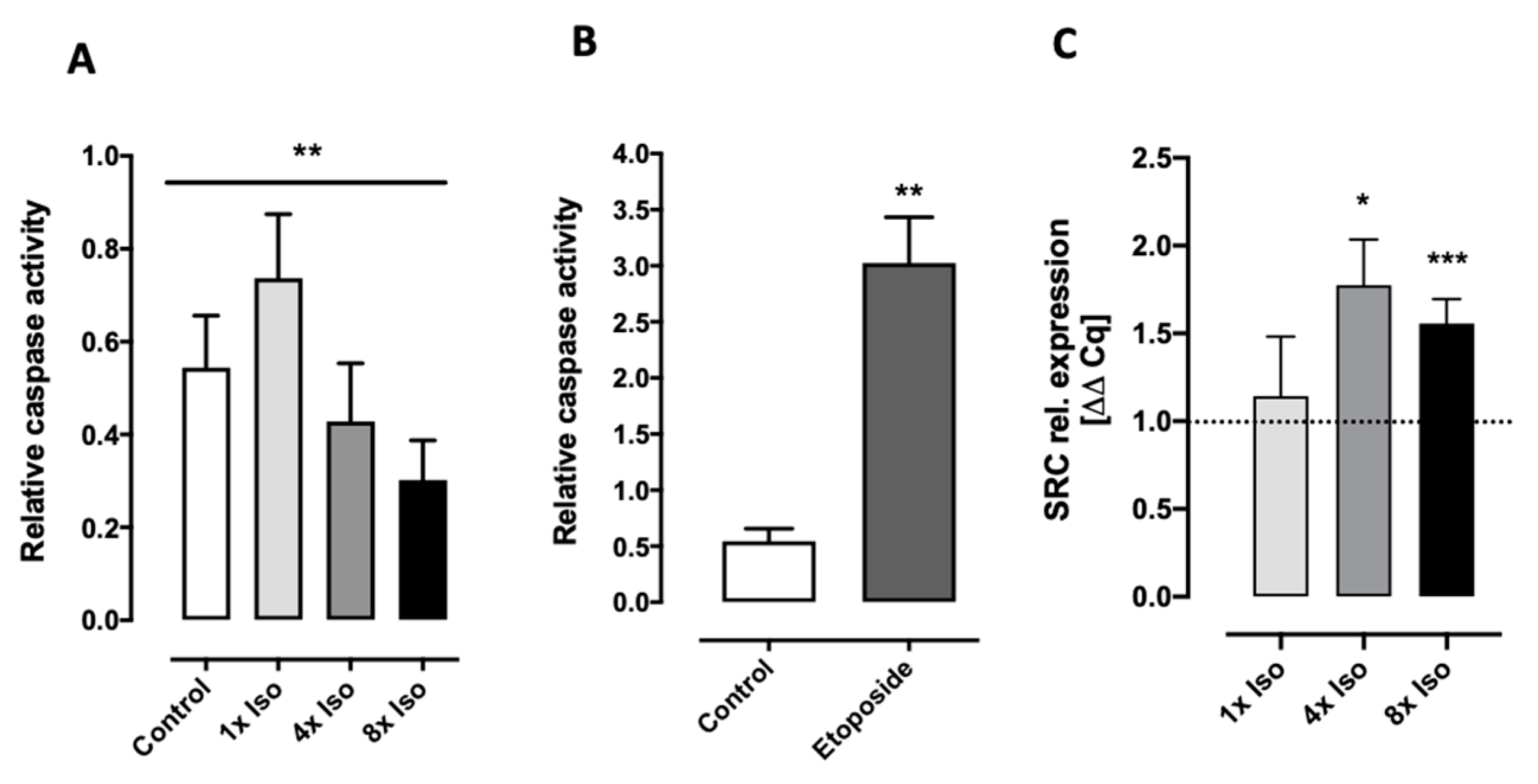
3.2. Isoproterenol Induces DNA Double-Strand Breaks and Reduced Caspase 3 and 7 Activity
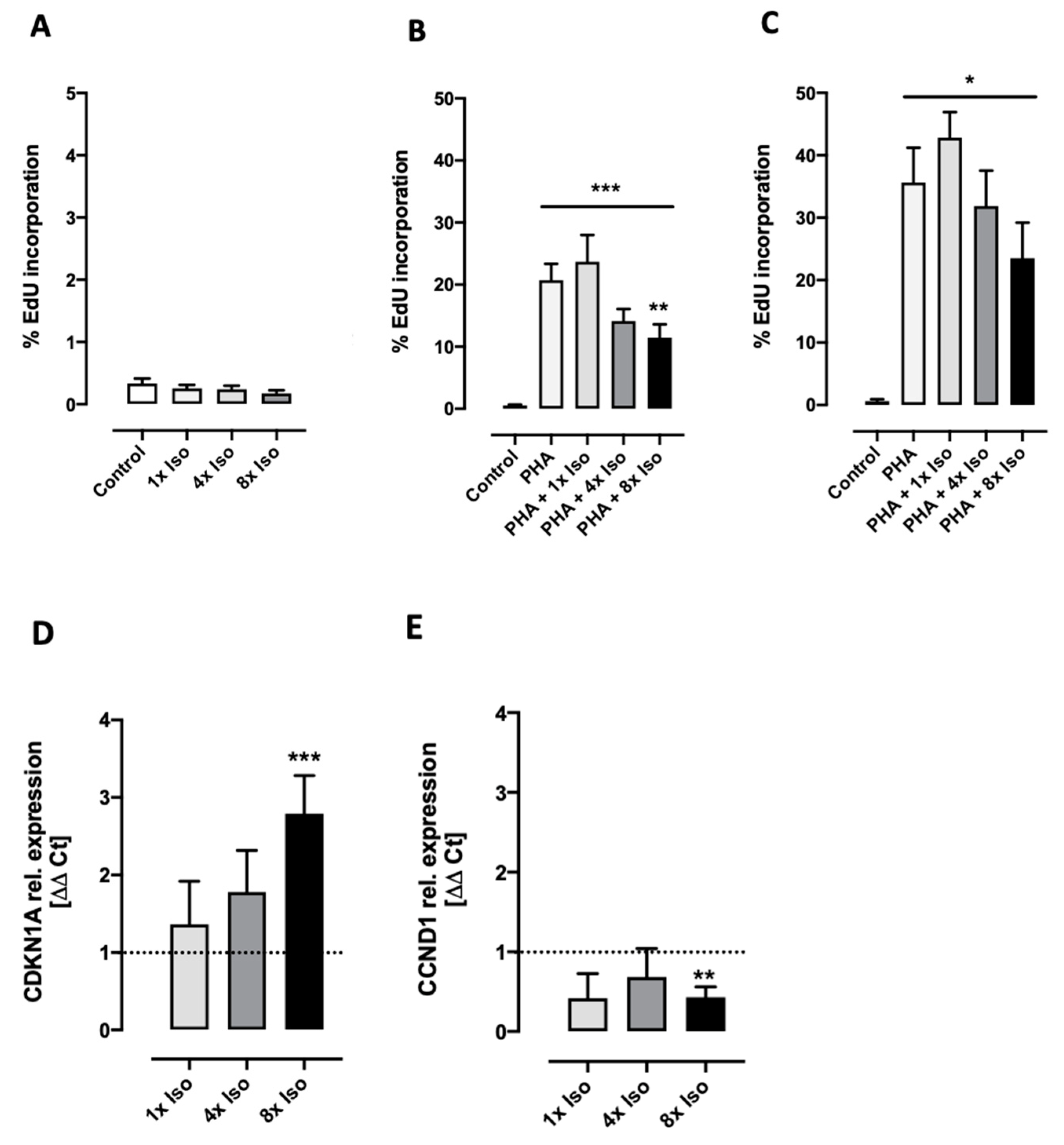
3.3. Isoproterenol Inhibits PHA-Induced Cell Proliferation
3.4. Isoproterenol Induces Cell Adhesion
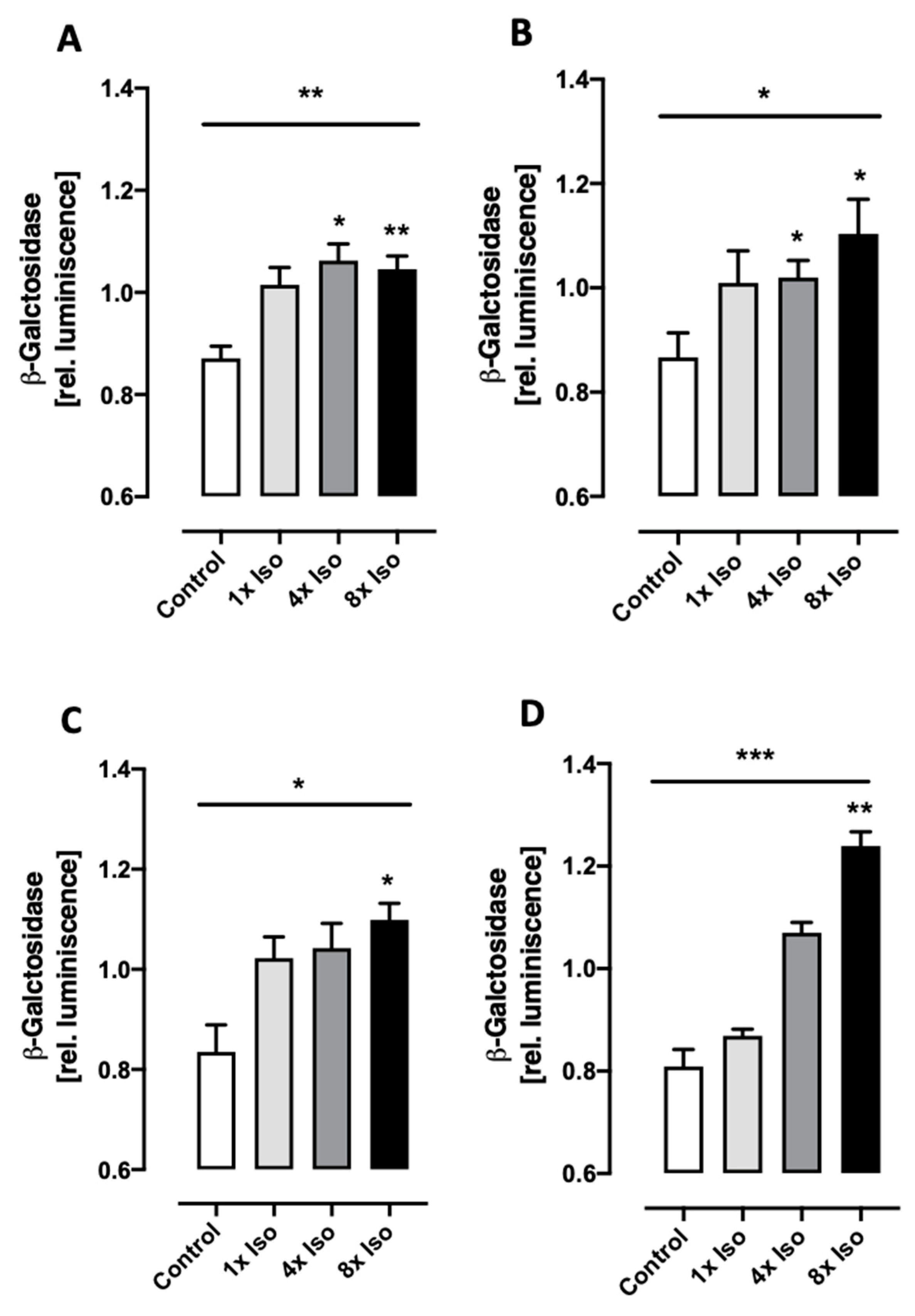
3.5. Isoproterenol Induces Senescence-Associated β-Galactosidase Activity
4. Discussion
5. Conclusions
6. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Godoy, L.D.; Rossignoli, M.T.; Delfino-Pereira, P.; Garcia-Cairasco, N.; de Lima Umeoka, E.H. A Comprehensive Overview on Stress Neurobiology: Basic Concepts and Clinical Implications. Front. Behav. Neurosci. 2018, 12, 127. [Google Scholar] [CrossRef]
- Goldstein, D.S. Adrenal responses to stress. Cell Mol. Neurobiol. 2010, 30, 1433–1440. [Google Scholar] [CrossRef]
- Tank, A.W.; Lee Wong, D. Peripheral and central effects of circulating catecholamines. Compr. Physiol. 2015, 5, 1–15. [Google Scholar] [CrossRef]
- Antonini, S.R.; Stecchini, M.F.; Ramalho, F.S. Development and Function of the Adrenal Cortex and Medulla in the Fetus and Neonate. In Maternal-Fetal and Neonatal Endocrinology Physiology, Pathophysiology, and Clinical Management; Deal, C.S.K.a.C.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 611–623. [Google Scholar]
- Crary, B.; Hauser, S.L.; Borysenko, M.; Kutz, I.; Hoban, C.; Ault, K.A.; Weiner, H.L.; Benson, H. Epinephrine-induced changes in the distribution of lymphocyte subsets in peripheral blood of humans. J. Immunol. 1983, 131, 1178–1181. [Google Scholar] [CrossRef]
- Dougherty, T.F.; Frank, J.A. The quantitative and qualitative responses of blood lymphocytes to stress stimuli. J. Lab. Clin. Med. 1953, 42, 530–537. [Google Scholar] [PubMed]
- Oberbeck, R. Catecholamines: Physiological immunomodulators during health and illness. Curr. Med. Chem. 2006, 13, 1979–1989. [Google Scholar] [CrossRef]
- Yu, D.T.; Clements, P.J. Human lymphocyte subpopulations effect of epinephrine. Clin. Exp. Immunol. 1976, 25, 472–479. [Google Scholar] [PubMed]
- Chu, B.; Marwaha, K.; Sanvictores, T.; Awosika, A.O.; Ayers, D. Physiology, Stress Reaction; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Dhabhar, F.S. Effects of stress on immune function: The good, the bad, and the beautiful. Immunol. Res. 2014, 58, 193–210. [Google Scholar] [CrossRef]
- Shin, E.; Ko, K.S.; Rhee, B.D.; Han, J.; Kim, N. Different effects of prolonged beta-adrenergic stimulation on heart and cerebral artery. Integr. Med. Res. 2014, 3, 204–210. [Google Scholar] [CrossRef]
- Bradley, P.B. Introduction to Neuropharmacology; Elsevier Ltd.: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Maddison, J.E.; Page, S.W.; Church, D.B. Small Animal Clinical Pharmacology; Elsevier Ltd.: Amsterdam, The Netherlands, 2008. [Google Scholar]
- MacGregor, D.A.; Prielipp, R.C.; Butterworth, J.F.t.; James, R.L.; Royster, R.L. Relative efficacy and potency of beta-adrenoceptor agonists for generating cAMP in human lymphocytes. Chest 1996, 109, 194–200. [Google Scholar] [CrossRef]
- Bai, L.; Kee, H.J.; Han, X.; Zhao, T.; Kee, S.J.; Jeong, M.H. Protocatechuic acid attenuates isoproterenol-induced cardiac hypertrophy via downregulation of ROCK1-Sp1-PKCgamma axis. Sci. Rep. 2021, 11, 17343. [Google Scholar] [CrossRef] [PubMed]
- Nanda, D.; Pant, P.; Machha, P.; Sowpati, D.T.; Kumarswamy, R. Transcriptional changes during isoproterenol-induced cardiac fibrosis in mice. Front. Mol. Biosci. 2023, 10, 1263913. [Google Scholar] [CrossRef] [PubMed]
- Martinsson, A.; Lindvall, K.; Melcher, A.; Hjemdahl, P. Beta-adrenergic receptor responsiveness to isoprenaline in humans: Concentration-effect, as compared with dose-effect evaluation and influence of autonomic reflexes. Br. J. Clin. Pharmacol. 1989, 28, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kamat, A.; Singh, B.B. Isoproterenol-Dependent Activation of TRPM7 Protects Against Neurotoxin-Induced Loss of Neuroblastoma Cells. Front. Physiol. 2020, 11, 305. [Google Scholar] [CrossRef] [PubMed]
- Gan, M.; Zheng, T.; Shen, L.; Tan, Y.; Fan, Y.; Shuai, S.; Bai, L.; Li, X.; Wang, J.; Zhang, S.; et al. Genistein reverses isoproterenol-induced cardiac hypertrophy by regulating miR-451/TIMP2. Biomed. Pharmacother. 2019, 112, 108618. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.R.; Polizzi, S.M.; Harris, T.; Wilson, N.; Michel, M.C.; Maisel, A.S. Prolonged isoproterenol treatment alters immunoregulatory cell traffic and function in the rat. Brain Behav. Immun. 1993, 7, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Villanueva, M.; Morath, J.; Vanhooren, V.; Elbert, T.; Kolassa, S.; Libert, C.; Burkle, A.; Kolassa, I.T. N-glycosylation profiling of plasma provides evidence for accelerated physiological aging in post-traumatic stress disorder. Transl. Psychiatry 2013, 3, e320. [Google Scholar] [CrossRef]
- O’Donovan, A.; Epel, E.; Lin, J.; Wolkowitz, O.; Cohen, B.; Maguen, S.; Metzler, T.; Lenoci, M.; Blackburn, E.; Neylan, T.C. Childhood trauma associated with short leukocyte telomere length in posttraumatic stress disorder. Biol. Psychiatry 2011, 70, 465–471. [Google Scholar] [CrossRef]
- Morath, J.; Moreno-Villanueva, M.; Hamuni, G.; Kolassa, S.; Ruf-Leuschner, M.; Schauer, M.; Elbert, T.; Burkle, A.; Kolassa, I.T. Effects of psychotherapy on DNA strand break accumulation originating from traumatic stress. Psychother. Psychosom. 2014, 83, 289–297. [Google Scholar] [CrossRef]
- Bhatia-Dey, N.; Kanherkar, R.R.; Stair, S.E.; Makarev, E.O.; Csoka, A.B. Cellular Senescence as the Causal Nexus of Aging. Front. Genet. 2016, 7, 13. [Google Scholar] [CrossRef]
- Kipling, D. Telomeres, replicative senescence and human ageing. Maturitas 2001, 38, 25–37; discussion 37–28. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Rossiello, F.; Herbig, U.; Longhese, M.P.; Fumagalli, M.; d’Adda di Fagagna, F. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 2014, 26, 89–95. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Robetorye, R.S.; Pereira-Smith, O.M.; Smith, J.R. Senescent cells fail to express cdc2, cycA, and cycB in response to mitogen stimulation. Proc. Natl. Acad. Sci. USA 1991, 88, 11012–11016. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes. Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes. Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef]
- Varela-Eirin, M.; Demaria, M. Cellular senescence. Curr. Biol. 2022, 32, R448–R452. [Google Scholar] [CrossRef]
- Gonzalez-Gualda, E.; Baker, A.G.; Fruk, L.; Munoz-Espin, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Kudlova, N.; De Sanctis, J.B.; Hajduch, M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. Int. J. Mol. Sci. 2022, 23, 4168. [Google Scholar] [CrossRef]
- Limas, C.J.; Limas, C. Rapid recovery of cardiac beta-adrenergic receptors after isoproterenol-induced “down”-regulation. Circ. Res. 1984, 55, 524–531. [Google Scholar] [CrossRef]
- O’Callaghan, N.J.; Fenech, M. A quantitative PCR method for measuring absolute telomere length. Biol. Proced. Online 2011, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Palombo, P.; Burkle, A.; Moreno-Villanueva, M. Culture medium-dependent isoproterenol stability and its impact on DNA strand breaks formation and repair. Chem. Biol. Interact. 2022, 357, 109877. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Palombo, P.; Schuhmacher, T.; von Scheven, G.; Bazylianska, V.; Salzwedel, J.; Schafer, N.; Burkle, A.; Moreno-Villanueva, M. Impaired PARP activity in response to the beta-adrenergic receptor agonist isoproterenol. Toxicol. Vitr. 2018, 50, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Rey-Millet, M.; Pousse, M.; Soithong, C.; Ye, J.; Mendez-Bermudez, A.; Gilson, E. Senescence-associated transcriptional derepression in subtelomeres is determined in a chromosome-end-specific manner. Aging Cell 2023, 22, e13804. [Google Scholar] [CrossRef]
- Karlseder, J.; Smogorzewska, A.; de Lange, T. Senescence induced by altered telomere state, not telomere loss. Science 2002, 295, 2446–2449. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Zhao, J.; Cao, D.; Dang, X.; Wang, L.; Lian, J.; Zhang, Y.; Jia, Z.; Wu, X.Y.; Morrison, Z.; et al. Inhibition of TRF2 accelerates telomere attrition and DNA damage in naive CD4 T cells during HCV infection. Cell Death Dis. 2018, 9, 900. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.E.; Aisner, D.L.; Shay, J.W.; Wright, W.E. Lack of cell cycle regulation of telomerase activity in human cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10687–10692. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.P.; Hathcock, K.S.; Hodes, R.J. Regulation of telomere length and telomerase in T and B cells: A mechanism for maintaining replicative potential. Immunity 1998, 9, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Causes and consequences of DNA single-strand breaks. Trends Biochem. Sci. 2024, 49, 68–78. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef]
- Rieckher, M.; Garinis, G.A.; Schumacher, B. Molecular pathology of rare progeroid diseases. Trends Mol. Med. 2021, 27, 907–922. [Google Scholar] [CrossRef]
- Collin, G.; Huna, A.; Warnier, M.; Flaman, J.M.; Bernard, D. Transcriptional repression of DNA repair genes is a hallmark and a cause of cellular senescence. Cell Death Dis. 2018, 9, 259. [Google Scholar] [CrossRef]
- Hu, L.; Li, H.; Zi, M.; Li, W.; Liu, J.; Yang, Y.; Zhou, D.; Kong, Q.P.; Zhang, Y.; He, Y. Why Senescent Cells Are Resistant to Apoptosis: An Insight for Senolytic Development. Front. Cell Dev. Biol. 2022, 10, 822816. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.S.; Cook, J.G. Cell cycle exits and U-turns: Quiescence as multiple reversible forms of arrest. Fac. Rev. 2023, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.S.; Weinberg, R.A. Control of the cell cycle and apoptosis. Eur. J. Cancer 1999, 35, 1886–1894. [Google Scholar] [CrossRef]
- Chapman, N.M.; Chi, H. Hallmarks of T-cell Exit from Quiescence. Cancer Immunol. Res. 2018, 6, 502–508. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, M.R.; Johns, S.; Wilcox, P. The effect of PHA stimulation on lymphocyte sub-populations in whole-blood cultures. Mutagenesis 1995, 10, 371–374. [Google Scholar] [CrossRef]
- Lossaint, G.; Horvat, A.; Gire, V.; Bacevic, K.; Mrouj, K.; Charrier-Savournin, F.; Georget, V.; Fisher, D.; Dulic, V. Reciprocal regulation of p21 and Chk1 controls the cyclin D1-RB pathway to mediate senescence onset after G2 arrest. J. Cell Sci. 2022, 135, jcs259114. [Google Scholar] [CrossRef]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef]
- Zhang, X.; Min, K.W.; Wimalasena, J.; Baek, S.J. Cyclin D1 degradation and p21 induction contribute to growth inhibition of colorectal cancer cells induced by epigallocatechin-3-gallate. J. Cancer Res. Clin. Oncol. 2012, 138, 2051–2060. [Google Scholar] [CrossRef]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef]
- Wolfson, M.; Budovsky, A.; Tacutu, R.; Fraifeld, V. The signaling hubs at the crossroad of longevity and age-related disease networks. Int. J. Biochem. Cell Biol. 2009, 41, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican-A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Kang, I.; Merrilees, M.J. Versican and the control of inflammation. Matrix Biol. 2014, 35, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113 Pt 20, 3613–3622. [Google Scholar] [CrossRef]
- Bulbiankova, D.; Diaz-Puertas, R.; Alvarez-Martinez, F.J.; Herranz-Lopez, M.; Barrajon-Catalan, E.; Micol, V. Hallmarks and Biomarkers of Skin Senescence: An Updated Review of Skin Senotherapeutics. Antioxidants 2023, 12, 444. [Google Scholar] [CrossRef]
- Beausejour, C. Bone marrow-derived cells: The influence of aging and cellular senescence. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 67–88. [Google Scholar] [CrossRef]
- Behmoaras, J.; Gil, J. Similarities and interplay between senescent cells and macrophages. J. Cell Biol. 2021, 220, e202010162. [Google Scholar] [CrossRef]
- Vasudevan, N.T.; Mohan, M.L.; Goswami, S.K.; Naga Prasad, S.V. Regulation of beta-adrenergic receptor function: An emphasis on receptor resensitization. Cell Cycle 2011, 10, 3684–3691. [Google Scholar] [CrossRef]
- Hara, M.R.; Kovacs, J.J.; Whalen, E.J.; Rajagopal, S.; Strachan, R.T.; Grant, W.; Towers, A.J.; Williams, B.; Lam, C.M.; Xiao, K.; et al. A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature 2011, 477, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Santos-Otte, P.; Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems and Their Role in Cellular Senescence. Comput. Struct. Biotechnol. J. 2019, 17, 1265–1277. [Google Scholar] [CrossRef]
- Sun, R.; Zhu, B.; Xiong, K.; Sun, Y.; Shi, D.; Chen, L.; Zhang, Y.; Li, Z.; Xue, L. Senescence as a novel mechanism involved in beta-adrenergic receptor mediated cardiac hypertrophy. PLoS ONE 2017, 12, e0182668. [Google Scholar] [CrossRef]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Brand, T. Length doesn’t matter-telomere damage triggers cellular senescence in the ageing heart. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Victorelli, S.; Passos, J.F. Telomeres: Beacons of autocrine and paracrine DNA damage during skin aging. Cell Cycle 2020, 19, 532–540. [Google Scholar] [CrossRef]
- Slawinska, N.; Krupa, R. Molecular Aspects of Senescence and Organismal Ageing-DNA Damage Response, Telomeres, Inflammation and Chromatin. Int. J. Mol. Sci. 2021, 22, 590. [Google Scholar] [CrossRef]
- Sitte, N.; Saretzki, G.; von Zglinicki, T. Accelerated telomere shortening in fibroblasts after extended periods of confluency. Free Radic. Biol. Med. 1998, 24, 885–893. [Google Scholar] [CrossRef]
- Vidacek, N.S.; Cukusic, A.; Ivankovic, M.; Fulgosi, H.; Huzak, M.; Smith, J.R.; Rubelj, I. Abrupt telomere shortening in normal human fibroblasts. Exp. Gerontol. 2010, 45, 235–242. [Google Scholar] [CrossRef]
- Toussaint, O.; Medrano, E.E.; von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- Anerillas, C.; Herman, A.B.; Rossi, M.; Munk, R.; Lehrmann, E.; Martindale, J.L.; Cui, C.Y.; Abdelmohsen, K.; De, S.; Gorospe, M. Early SRC activation skews cell fate from apoptosis to senescence. Sci. Adv. 2022, 8, eabm0756. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.L.; Jia, S.H.; Parodo, J.; Plant, P.; Lodyga, M.; Charbonney, E.; Szaszi, K.; Kapus, A.; Marshall, J.C. Tyrosine Phosphorylation of Caspase-8 Abrogates Its Apoptotic Activity and Promotes Activation of c-Src. PLoS ONE 2016, 11, e0153946. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Ma, Y.C.; Benjamin, J.; Littman, D.; Chao, M.V.; Huang, X.Y. Apoptotic signaling through the beta -adrenergic receptor. A new Gs effector pathway. J. Biol. Chem. 2000, 275, 20726–20733. [Google Scholar] [CrossRef] [PubMed]
- Remondino, A.; Kwon, S.H.; Communal, C.; Pimentel, D.R.; Sawyer, D.B.; Singh, K.; Colucci, W.S. Beta-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ. Res. 2003, 92, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Hiroi, Y.; Zou, Y.; Aikawa, R.; Toko, H.; Shibasaki, F.; Yazaki, Y.; Nagai, R.; Komuro, I. beta-Adrenergic pathway induces apoptosis through calcineurin activation in cardiac myocytes. J. Biol. Chem. 2000, 275, 34528–34533. [Google Scholar] [CrossRef]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3′-kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar] [CrossRef]
- Safi, S.Z.; Saeed, L.; Shah, H.; Latif, Z.; Ali, A.; Imran, M.; Muhammad, N.; Emran, T.B.; Subramaniyan, V.; Ismail, I.S.B. Mechanisms of beta-adrenergic receptors agonists in mediating pro and anti-apoptotic pathways in hyperglycemic Muller cells. Mol. Biol. Rep. 2022, 49, 9473–9480. [Google Scholar] [CrossRef]
- Southwick, S.M.; Paige, S.; Morgan, C.A., 3rd; Bremner, J.D.; Krystal, J.H.; Charney, D.S. Neurotransmitter alterations in PTSD: Catecholamines and serotonin. Semin. Clin. Neuropsychiatry 1999, 4, 242–248. [Google Scholar]
- Applebaum, M.A.; Skol, A.D.; Bond, E.E.; Overholtzer, M.; Bond, G.L.; Onel, K. Radiation-induced apoptosis varies among individuals and is modified by sex and age. Int. J. Radiat. Biol. 2014, 90, 903–908. [Google Scholar] [CrossRef]
- Schmitz, A.; Bayer, J.; Dechamps, N.; Goldin, L.; Thomas, G. Heritability of susceptibility to ionizing radiation-induced apoptosis of human lymphocyte subpopulations. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 1169–1177. [Google Scholar] [CrossRef]
- Schmitz, A.; Bayer, J.; Dechamps, N.; Thomas, G. Intrinsic susceptibility to radiation-induced apoptosis of human lymphocyte subpopulations. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Gire, V.; Dulic, V. Senescence from G2 arrest, revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Ando, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef]
- Ghiani, C.A.; Eisen, A.M.; Yuan, X.; DePinho, R.A.; McBain, C.J.; Gallo, V. Neurotransmitter receptor activation triggers p27(Kip1)and p21(CIP1) accumulation and G1 cell cycle arrest in oligodendrocyte progenitors. Development 1999, 126, 1077–1090. [Google Scholar] [CrossRef]
- Brown, N.E.; Jeselsohn, R.; Bihani, T.; Hu, M.G.; Foltopoulou, P.; Kuperwasser, C.; Hinds, P.W. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012, 72, 6477–6489. [Google Scholar] [CrossRef] [PubMed]
- Han, E.K.; Ng, S.C.; Arber, N.; Begemann, M.; Weinstein, I.B. Roles of cyclin D1 and related genes in growth inhibition, senescence and apoptosis. Apoptosis 1999, 4, 213–219. [Google Scholar] [CrossRef]
- Guan, L.; Crasta, K.C.; Maier, A.B. Assessment of cell cycle regulators in human peripheral blood cells as markers of cellular senescence. Ageing Res. Rev. 2022, 78, 101634. [Google Scholar] [CrossRef]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Mills, P.J.; Carter, S.M.; Chou, J.; Thomas, J.; Maisel, A.S. Dynamic exercise leads to an increase in circulating ICAM-1: Further evidence for adrenergic modulation of cell adhesion. Brain Behav. Immun. 1997, 11, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Enserink, J.M.; Kuiperij, H.B.; de Rooij, J.; Price, L.S.; Schwede, F.; Bos, J.L. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the beta 2-adrenergic receptor. J. Cell Biol. 2003, 160, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, M.; Honma, T.; Sato, K.; Shinohara, N.; Ito, J.; Tanaka, Y.; Tsuduki, T.; Ikeda, I. Increased monocytic adhesion by senescence in human umbilical vein endothelial cells. Biosci. Biotechnol. Biochem. 2011, 75, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, D.E.; Jeanneret, C.; Husak, M.; Lobell, M.; Jacob, H.S. Lymphocyte aggregation in response to adrenergic stimulation. Blood 1988, 71, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Barallobre-Barreiro, J.; Radovits, T.; Fava, M.; Mayr, U.; Lin, W.Y.; Ermolaeva, E.; Martinez-Lopez, D.; Lindberg, E.L.; Duregotti, E.; Daroczi, L.; et al. Extracellular Matrix in Heart Failure: Role of ADAMTS5 in Proteoglycan Remodeling. Circulation 2021, 144, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Sotoodehnejadnematalahi, F.; Burke, B. Structure, function and regulation of versican: The most abundant type of proteoglycan in the extracellular matrix. Acta Med. Iran. 2013, 51, 740–750. [Google Scholar] [PubMed]
- Wight, T.N.; Kinsella, M.G.; Evanko, S.P.; Potter-Perigo, S.; Merrilees, M.J. Versican and the regulation of cell phenotype in disease. Biochim. Biophys. Acta 2014, 1840, 2441–2451. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Qin, D.; Hu, B.; Zhang, C.; Liu, S.; Wu, D.; Huang, W.; Huang, X.; Wang, L.; Chen, X.; et al. Deletion of miR-126a Promotes Hepatic Aging and Inflammation in a Mouse Model of Cholestasis. Mol. Ther. Nucleic Acids 2019, 16, 494–504. [Google Scholar] [CrossRef]
- Islam, S.; Watanabe, H. Versican: A Dynamic Regulator of the Extracellular Matrix. J. Histochem. Cytochem. 2020, 68, 763–775. [Google Scholar] [CrossRef]
- Kohli, J.; Wang, B.; Brandenburg, S.M.; Basisty, N.; Evangelou, K.; Varela-Eirin, M.; Campisi, J.; Schilling, B.; Gorgoulis, V.; Demaria, M. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat. Protoc. 2021, 16, 2471–2498. [Google Scholar] [CrossRef]
- Bursuker, I.; Rhodes, J.M.; Goldman, R. Beta-galactosidase—An indicator of the maturational stage of mouse and human mononuclear phagocytes. J. Cell Physiol. 1982, 112, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Invest. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Polet, H.; Spieker-Polet, H. Serum albumin is essential for in vitro growth of activated human lymphocytes. J. Exp. Med. 1975, 142, 949–959. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Santulli, G.; Pascale, V.; Trimarco, B.; Iaccarino, G. Adrenergic receptors and metabolism: Role in development of cardiovascular disease. Front. Physiol. 2013, 4, 265. [Google Scholar] [CrossRef]
- Jovanovic, A.; Xu, B.; Zhu, C.; Ren, D.; Wang, H.; Krause-Hauch, M.; Abel, E.D.; Li, J.; Xiang, Y.K. Characterizing Adrenergic Regulation of Glucose Transporter 4-Mediated Glucose Uptake and Metabolism in the Heart. JACC Basic. Transl. Sci. 2023, 8, 638–655. [Google Scholar] [CrossRef]
- Straat, M.E.; Hoekx, C.A.; van Velden, F.H.P.; Pereira Arias-Bouda, L.M.; Dumont, L.; Blondin, D.P.; Boon, M.R.; Martinez-Tellez, B.; Rensen, P.C.N. Stimulation of the beta-2-adrenergic receptor with salbutamol activates human brown adipose tissue. Cell Rep. Med. 2023, 4, 100942. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laumann, M.; Palombo, P.; Fieres, J.; Thomas, M.; Saretzki, G.; Bürkle, A.; Moreno-Villanueva, M. Senescence-like Phenotype After Chronic Exposure to Isoproterenol in Primary Quiescent Immune Cells. Biomolecules 2024, 14, 1528. https://doi.org/10.3390/biom14121528
Laumann M, Palombo P, Fieres J, Thomas M, Saretzki G, Bürkle A, Moreno-Villanueva M. Senescence-like Phenotype After Chronic Exposure to Isoproterenol in Primary Quiescent Immune Cells. Biomolecules. 2024; 14(12):1528. https://doi.org/10.3390/biom14121528
Chicago/Turabian StyleLaumann, Michael, Philipp Palombo, Judy Fieres, Mara Thomas, Gabriele Saretzki, Alexander Bürkle, and Maria Moreno-Villanueva. 2024. "Senescence-like Phenotype After Chronic Exposure to Isoproterenol in Primary Quiescent Immune Cells" Biomolecules 14, no. 12: 1528. https://doi.org/10.3390/biom14121528
APA StyleLaumann, M., Palombo, P., Fieres, J., Thomas, M., Saretzki, G., Bürkle, A., & Moreno-Villanueva, M. (2024). Senescence-like Phenotype After Chronic Exposure to Isoproterenol in Primary Quiescent Immune Cells. Biomolecules, 14(12), 1528. https://doi.org/10.3390/biom14121528