
Enzymatic Synthesis of Biologically Active H-Phosphinic Analogue of α-Ketoglutarate

 , , , , and
, , , , and
Abstract
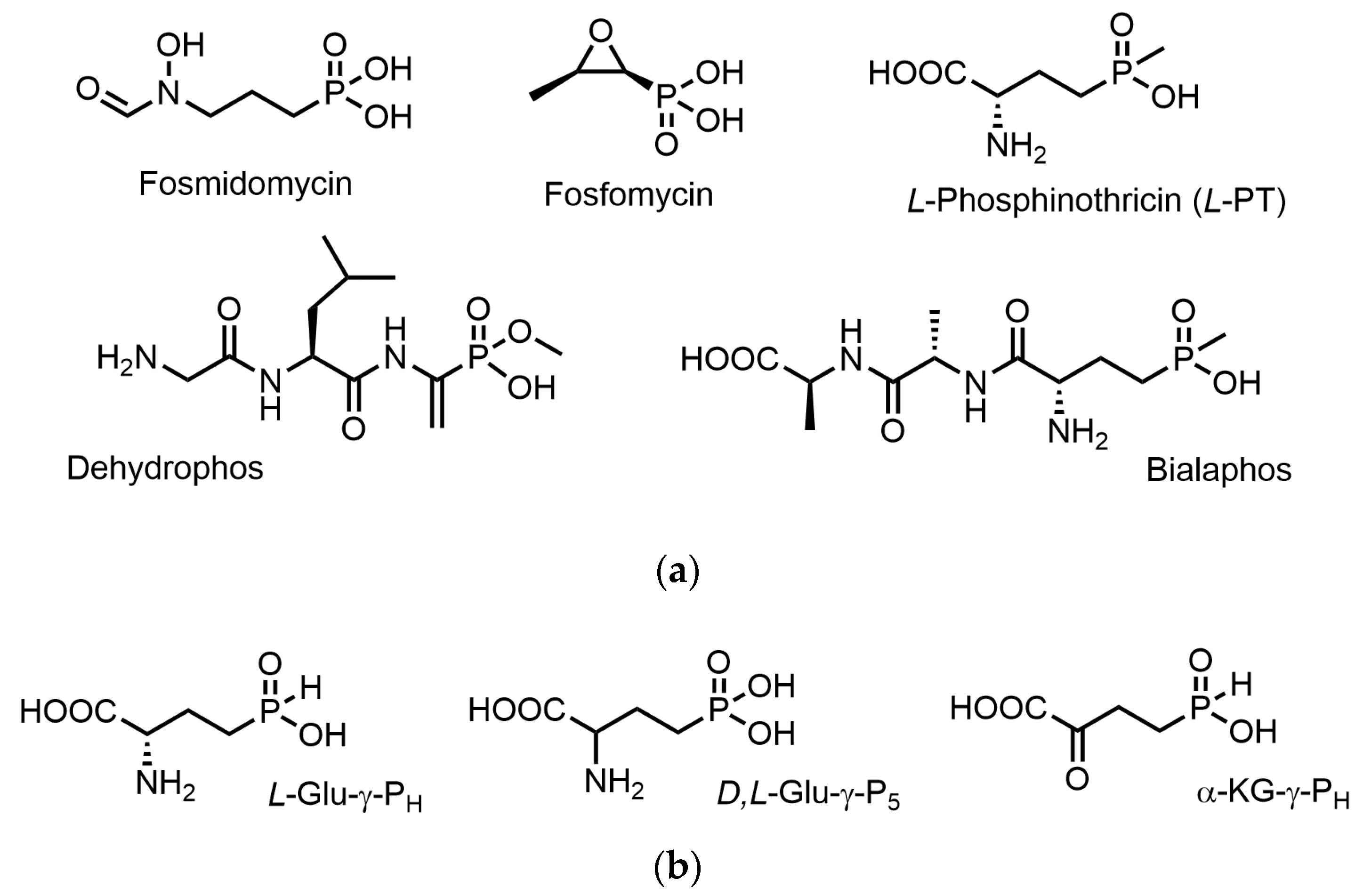
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Enzymatic Activity Assay and Kinetic Parameter Calculation
2.3. The Extent of L-Glu or L-Glu-γ-PH Conversion into α-KG or α-KG-γ-PH, Respectively
2.4. Preparative Synthesis of α-KG-γ-PH
3. Results
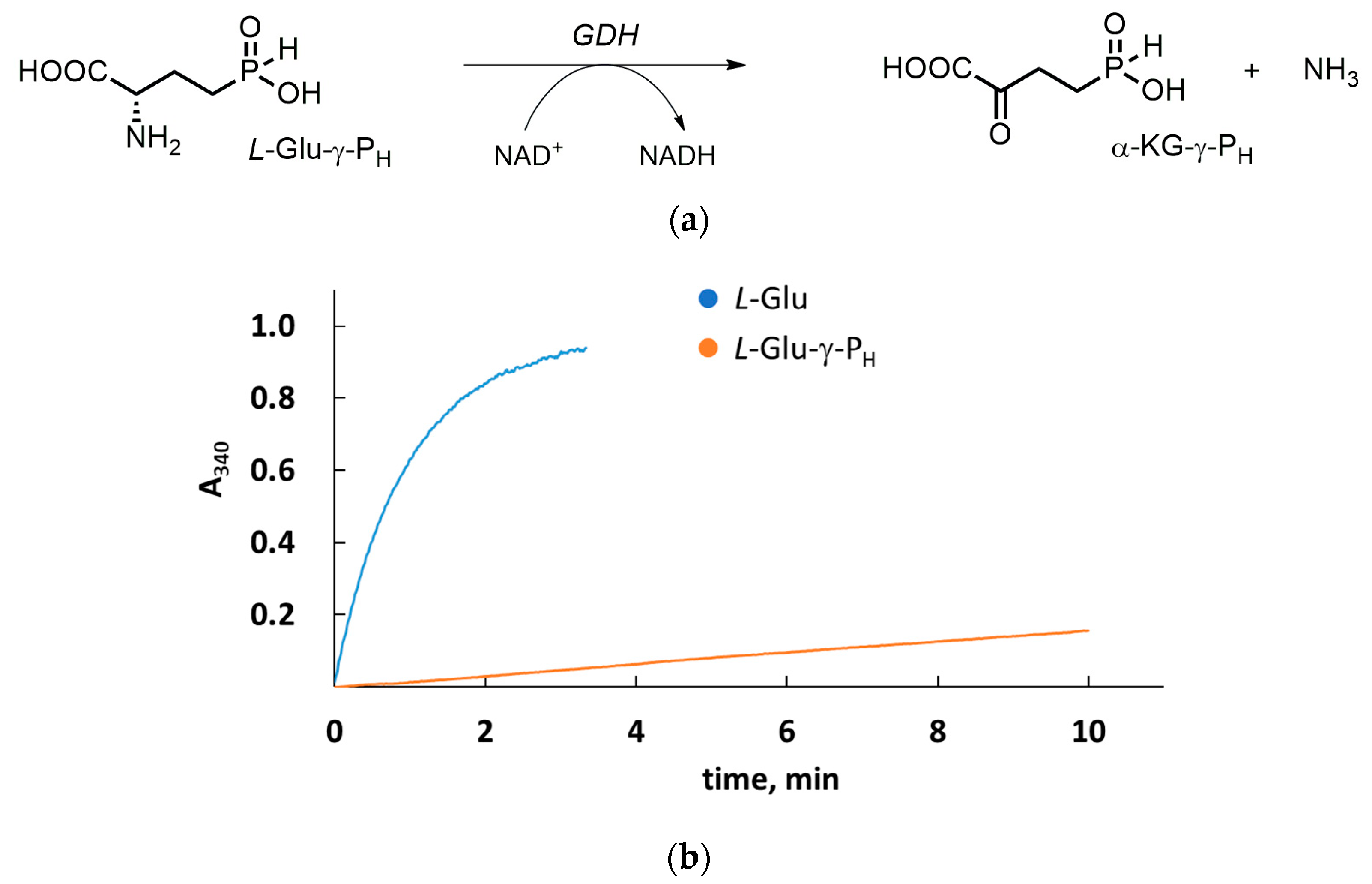
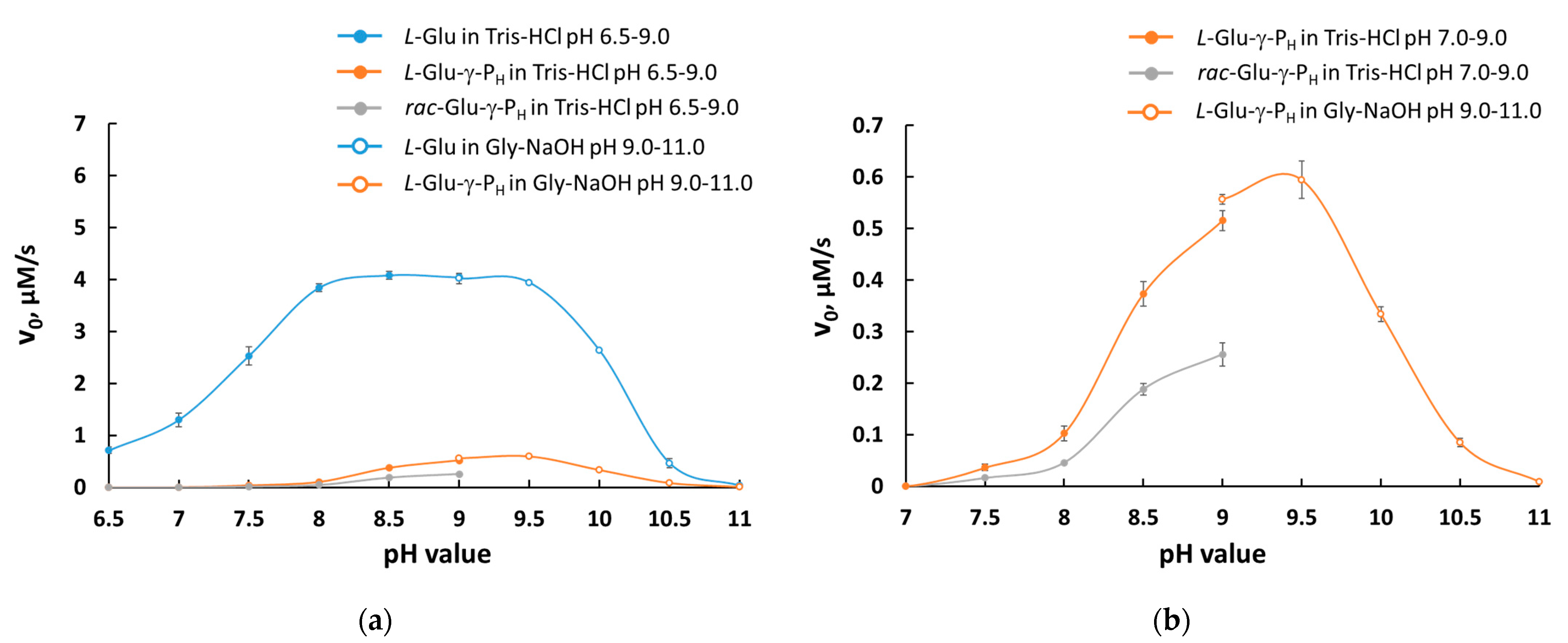
3.1. L-Glu-γ-PH But Not D-Glu-γ-PH Is a Substrate of GDH
3.2. Glu-γ-P5 and PT Are Neither Substrates Nor Inhibitors of GDH
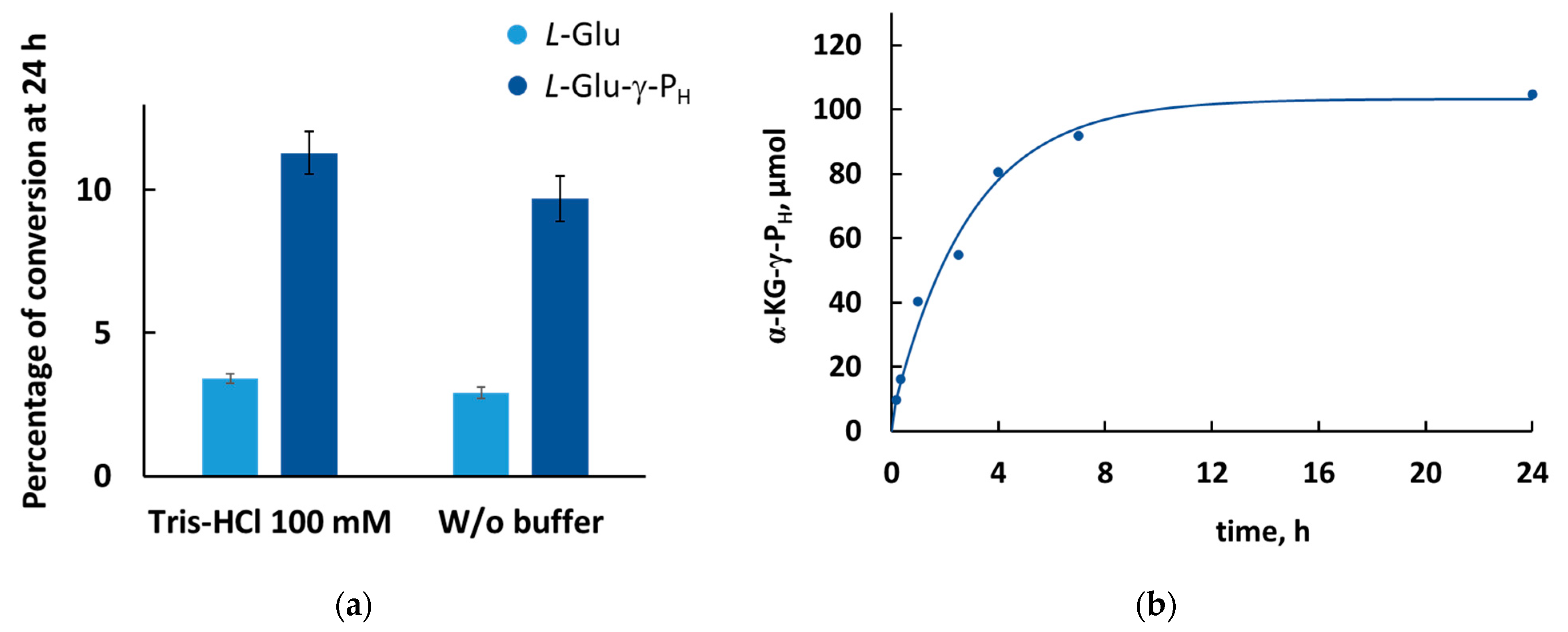
3.3. Comparison of the Percentage of Conversion of the Substrates Involved in GDH Reaction
3.4. Enzymatic Synthesis of H-Phosphinic Analogue of α-Ketoglutarate
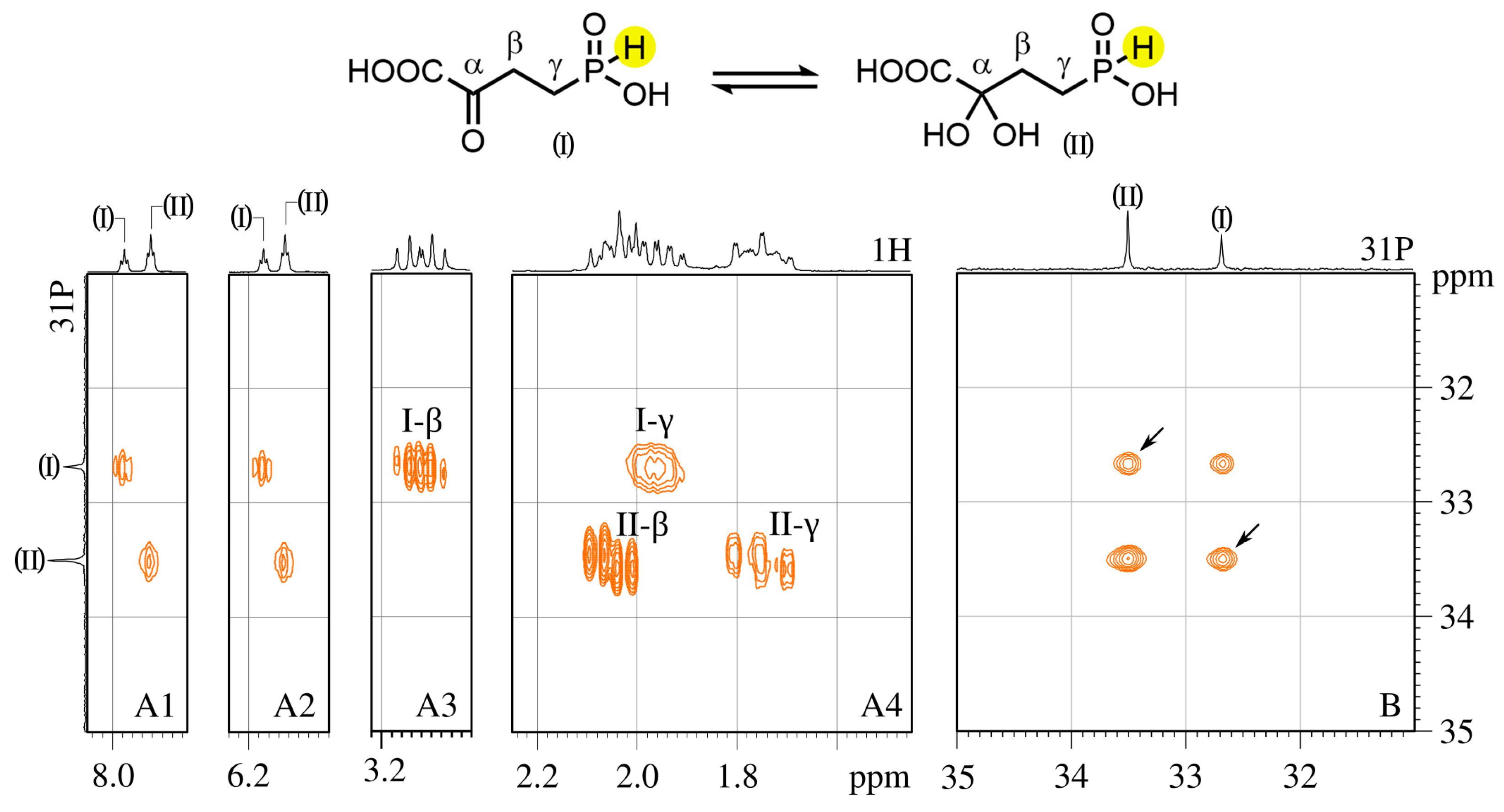
3.5. NMR Analysis of the Structure of α-KG-γ-PH
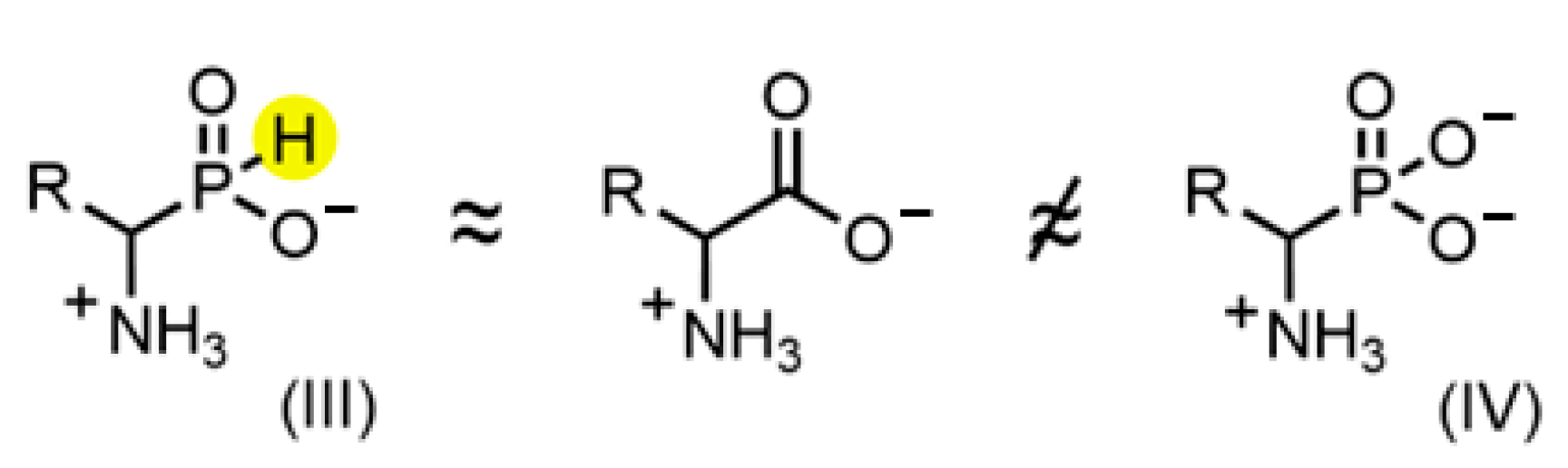
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Horsman, G.P.; Zechel, D.L. Phosphonate biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Doroghazi, J.R.; Janga, S.C.; Zhang, J.K.; Circello, B.; Grifiin, B.M.; Labeda, D.P.; Metcalf, W.W. Diversity and abundance of phosphonate biosynthetic genes in nature. Proc. Natl. Acad. Sci. USA 2013, 110, 20759–20764. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Vouloumanou, K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Oh, E.J.; Bassalo, M.C.; Choudhury, A.; Garst, A.D.; Fankhauser, R.G.; Eckert, C.A.; Gill, R.T. Genomic deoxyxylulose phosphate reductoisomerase (DXR) mutations conferring resistance to the antimalarial drug fosmidomycin in E. coli. ACS Synth. Biol. 2018, 7, 2824–2832. [Google Scholar] [CrossRef]
- Jawaid, S.; Seidle, H.; Zhou, W.; Abdirahman, H.; Abadeer, M.; Hix, J.H.; van Hoek, M.L.; Couch, R.D. Kinetic characterization and phosphoregulation of the Francisella tularensis 1-deoxy-D-xylulose 5-phosphate reductoisomerase (MEP synthase). PLoS ONE 2009, 4, e8288. [Google Scholar] [CrossRef]
- Wicke, D.; Schulz, L.M.; Lentes, S.; Scholz, P.; Poehlein, A.; Gibhardt, J.; Daniel, R.; Ischebeck, T.; Commichau, F.M. Identification of the first glyphosate transporter by genomic adaptation. Environ. Microbiol. 2019, 21, 1287–1305. [Google Scholar] [CrossRef]
- Gill, H.S.; Eisenberg, D. The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry 2001, 40, 1903–1912. [Google Scholar] [CrossRef]
- Circello, B.T.; Miller, C.G.; Lee, J.-H.; van der Donk, W.A.; Metcalf, W.W. The antibiotic dehydrophos is converted to a toxic pyruvate analogue by peptide bond cleavage in Salmonella enterica. Antimicrob. Agents Chemother. 2011, 55, 3357–3362. [Google Scholar] [CrossRef]
- Bunik, V.I.; Artiukhov, A.; Kazantsev, A.; Goncalves, R.; Daloso, D.; Oppermann, H.; Kulakovskaya, E.; Lukashev, N.; Fernie, A.; Brand, M.; et al. Specific inhibition by synthetic analogues of pyruvate reveals that the pyruvate dehydrogenase reaction is essential for metabolism and viability of glioblastoma cells. Oncotarget 2015, 6, 40036–40052. [Google Scholar] [CrossRef]
- Kluger, R.; Tittmann, K. Thiamin diphosphate catalysis: Enzymic and nonenzymic covalent intermediates. Chem. Rev. 2008, 108, 1797–1833. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, W.W.; van der Donk, W.A. Biosynthesis of phosphonic and phosphinic acid natural products. Annu. Rev. Biochem. 2009, 78, 65–94. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; Van Dien, S.J.; Rabinowitz, J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Seto, H.; Sasaki, T.; Imai, S.; Tsuruoka, T.; Ogawa, H.; Satoh, A.; Inouye, S.; Niida, T.; Otake, N. Studies on the biosynthesis of bialaphos (SF-1293). 2. Isolation of the first natural products with a C-P-H bond and their involvement in the C-P-C bond formation. J. Antibiot. 1983, 36, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Blodgett, J.A.; Thomas, P.M.; Li, G.; Velasquez, J.E.; van der Donk, W.A.; Kelleher, N.L.; Metcalf, W.W. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat. Chem. Biol. 2007, 3, 480–485. [Google Scholar] [CrossRef]
- Imai, S.; Seto, H.; Sasaki, T.; Tsuruoka, T.; Ogawa, H.; Satoh, A.; Inouye, S.; Niida, T.; Otake, N. Studies on the biosynthesis of bialaphos (SF-1293). 6. Production of N-acetyl-demethylphosphinothricin and N-acetylbialaphos by blocked mutants of Streptomyces hygroscopicus SF-1293 and their roles in the biosynthesis of bialaphos. J. Antibiot. 1985, 38, 687–690. [Google Scholar] [CrossRef]
- De Biase, D.; Cappadocio, F.; Pennacchietti, E.; Giovannercole, F.; Coluccia, A.; Vepsalainen, J.; Khomutov, A. Enzymatic kinetic resolution of desmethylphosphinothricin indicates that phosphinic group is a bioisostere of carboxyl group. Commun. Chem. 2020, 3, 121. [Google Scholar] [CrossRef]
- Khomutov, M.A.; Giovannercole, F.; Onillon, L.; Demiankova, M.V.; Vasilieva, B.F.; Salikhov, A.I.; Kochetkov, S.N.; Efremenkova, O.V.; Khomutov, A.R.; De Biase, D. A desmethylphosphinothricin dipeptide derivative effectively inhibits Escherichia coli and Bacillus subtilis growth. Biomolecules 2023, 13, 1451. [Google Scholar] [CrossRef]
- Giovannercole, F.; Gafeira Gonçalves, L.; Armengaud, J.; Varela Coelho, A.; Khomutov, A.; De Biase, D. Integrated multi-omics unveil the impact of H-phosphinic analogues of glutamate and α-ketoglutarate on Escherichia coli metabolism. J. Biol. Chem. 2024, 300, 107803. [Google Scholar] [CrossRef]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef]
- Rogers, K.S.; Yusko, S.C. Sodium dodecyl sulfate inactivation of bovine liver glutamate dehydrogenase. J. Biol. Chem. 1969, 244, 6690–6695. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.L.; Shaka, A.J. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Li, M.; Smith, C.J.; Walker, M.T.; Smith, T.J. Novel inhibitors complexed with glutamate dehydrogenase. Allosteric regulation by control of protein dynamics. J. Biol. Chem. 2009, 284, 22988–23000. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Piszkiewicz, D. Bovine glutamate dehydrogenase. The pH dependence of native and nitrated enzyme in the presence of allosteric modifiers. J. Biol. Chem. 1973, 248, 3089–3092. [Google Scholar] [CrossRef]
- Fisher, H.F. L-Glutamate dehydrogenase from bovine liver. Methods Enzymol. 1985, 113, 16–27. [Google Scholar]
- Schulz, A.; Taggeselle, P.; Tripier, D.; Bartsch, K. Stereospecific production of the herbicide phosphinothricin (glufosinate) by transamination: Isolation and characterization of a phosphinothricin-specific transaminase from Escherichia coli. Appl. Environ. Microbiol. 1990, 56, 1–6. [Google Scholar] [CrossRef]
- Lacoste, A.M.; Mansour, S.; Cassaigne, A.; Neuzil, E. Effect of phosphonic analogues of glutamic acid on glutamate decarboxylase. Experientia 1985, 41, 643–644. [Google Scholar] [CrossRef]
- Yin, X.; Wu, J.; Yang, L. Efficient reductive amination process for enantioselective synthesis of L-phosphinothricin applying engineered glutamate dehydrogenase. Appl. Microbiol. Biotechnol. 2018, 102, 4425–4433. [Google Scholar] [CrossRef]
- Barton, J.S.; Fisher, J.R. Nonlinear kinetics of glutamate dehydrogenase. Studies with substrates-glutamate and nicotinamide-adenine dinucleotide. Biochemistry 1971, 10, 577–585. [Google Scholar]
- Laber, B.; Amrhein, N. Metabolism of 1-aminoethylphosphinate generates acetylphosphinate, a potent inhibitor of pyruvate dehydrogenase. Biochem. J. 1987, 248, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Vrtis, J.M.; White, A.K.; Metcalf, W.W.; van der Donk, W.A. Phosphite dehydrogenase: A versatile cofactor-regeneration enzyme. Angew. Chem. Int. Ed. Engl. 2002, 41, 3257–3259. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, V.A.; Bunik, V.I.; Bruch, E.M.; Bellinzoni, M. Structural basis for the binding of allosteric activators leucine and ADP to mammalian glutamate dehydrogenase. Int. J. Mol. Sci. 2022, 23, 11306. [Google Scholar] [CrossRef] [PubMed]
- Kafarski, P. Phosphonopeptides containing free phosphonic groups: Recent advances. RSC Adv. 2020, 10, 25898–25910. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, A.Y.; Mariasina, S.S.; Bolikhova, A.K.; Nikulin, M.V.; Ozhiganov, R.M.; Vasil’ev, V.G.; Ikhalaynen, Y.A.; Khandazhinskaya, A.L.; Khomutov, M.A.; Sergiev, P.V.; et al. Organophosphorus S-adenosyl-L-methionine mimetics: Synthesis, stability, and substrate properties. Front. Chem. 2024, 12, 1448747. [Google Scholar] [CrossRef]
- Filonov, V.L.; Khomutov, M.A.; Sergeev, A.V.; Khandazhinskaya, A.L.; Kochetkov, S.N.; Gromova, E.S.; Khomutov, A.R. Interaction of DNA methyltransferase Dnmt3a with phosphorus analogues of S-adenosylmethionine and S-adenosylhomocysteine. Mol. Biol. 2023, 57, 747–754. [Google Scholar] [CrossRef]
- Cheng, F.; Li, H.; Zhang, K.; Li, Q.H.; Xie, D.; Xue, Y.P.; Zheng, Y.G. Tuning amino acid dehydrogenases with featured sequences for L-phosphinothricin synthesis by reductive amination. J. Biotechnol. 2020, 312, 35–43. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Km, mM | kcat, s−1 | kcat/Km, s−1∙M−1 | |
|---|---|---|---|
| L-Glu | 0.9 ± 0.2 1 | 0.065 ± 0.003 | 70 ± 20 |
| L-Glu-γ-PH | 52 ± 4 | 0.032 ± 0.002 | 0.6 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filonov, V.L.; Khomutov, M.A.; Tkachev, Y.V.; Udod, A.V.; Yanvarev, D.V.; Giovannercole, F.; Khurs, E.N.; Kochetkov, S.N.; De Biase, D.; Khomutov, A.R. Enzymatic Synthesis of Biologically Active H-Phosphinic Analogue of α-Ketoglutarate. Biomolecules 2024, 14, 1574. https://doi.org/10.3390/biom14121574
Filonov VL, Khomutov MA, Tkachev YV, Udod AV, Yanvarev DV, Giovannercole F, Khurs EN, Kochetkov SN, De Biase D, Khomutov AR. Enzymatic Synthesis of Biologically Active H-Phosphinic Analogue of α-Ketoglutarate. Biomolecules. 2024; 14(12):1574. https://doi.org/10.3390/biom14121574
Chicago/Turabian StyleFilonov, Vsevolod L., Maxim A. Khomutov, Yaroslav V. Tkachev, Artem V. Udod, Dmitry V. Yanvarev, Fabio Giovannercole, Elena N. Khurs, Sergei N. Kochetkov, Daniela De Biase, and Alex R. Khomutov. 2024. "Enzymatic Synthesis of Biologically Active H-Phosphinic Analogue of α-Ketoglutarate" Biomolecules 14, no. 12: 1574. https://doi.org/10.3390/biom14121574
APA StyleFilonov, V. L., Khomutov, M. A., Tkachev, Y. V., Udod, A. V., Yanvarev, D. V., Giovannercole, F., Khurs, E. N., Kochetkov, S. N., De Biase, D., & Khomutov, A. R. (2024). Enzymatic Synthesis of Biologically Active H-Phosphinic Analogue of α-Ketoglutarate. Biomolecules, 14(12), 1574. https://doi.org/10.3390/biom14121574