
Structures of Toxic Advanced Glycation End-Products Derived from Glyceraldehyde, A Sugar Metabolite

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
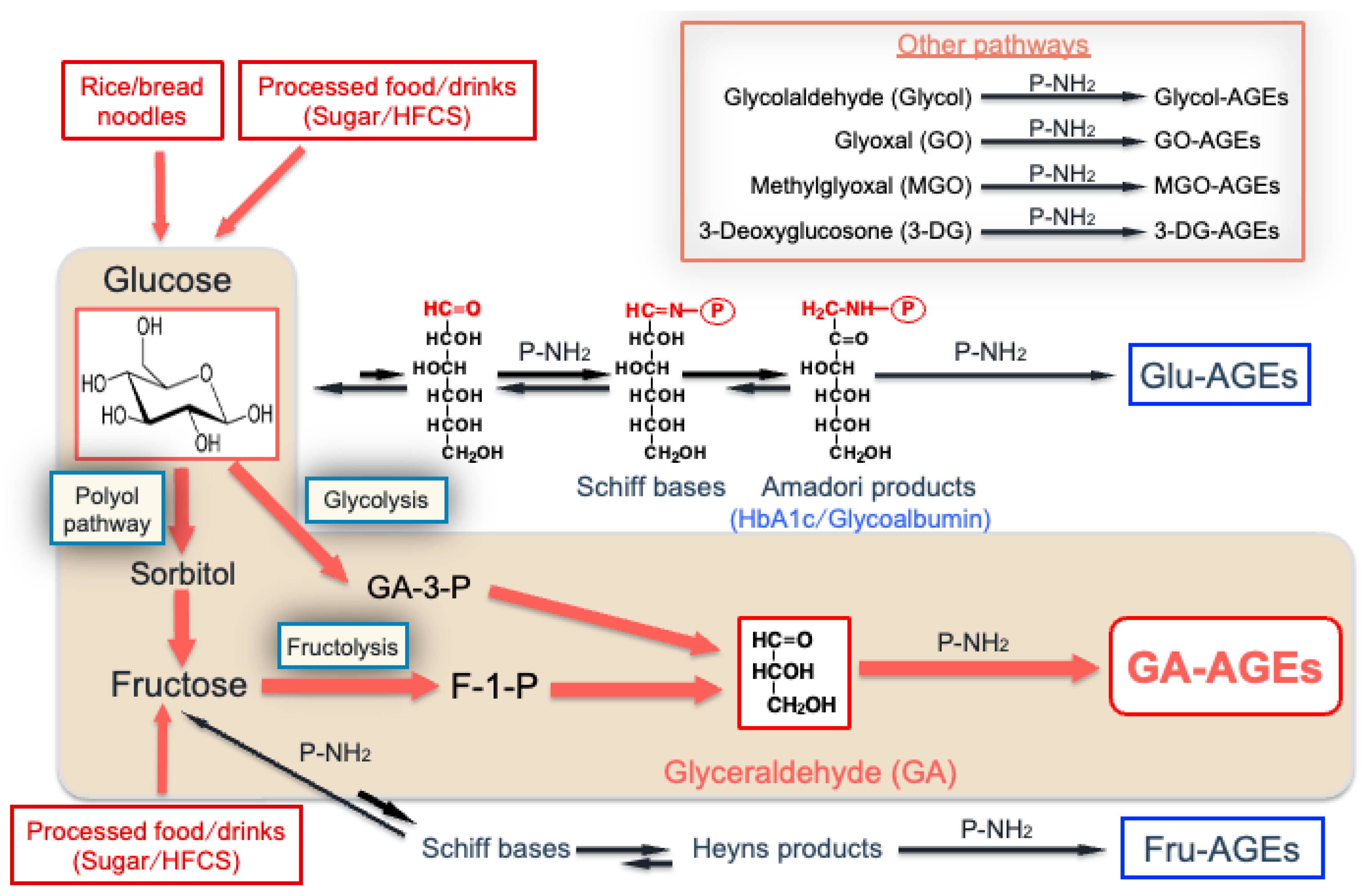
2. Routes by Which Various AGEs Are Produced in the Human Body
- Fluorescent and cross-linked products (i.e., PPGs, pentosidine, crossline, and vespelysine);
- Fluorescent and non-cross-linked products (i.e., ArgP);
- Non-fluorescent and cross-linked products (i.e., MGO-Lys-dimer, GO-Lys-dimer, and glucosepane);
- Non-fluorescent and non-cross-linked products (i.e., trihydroxy-triosidine, GLAP, MG-H1, Nε-(carboxyethyl)Lys, Nε-(carboxymethyl)Lys (CML), and pyrraline).
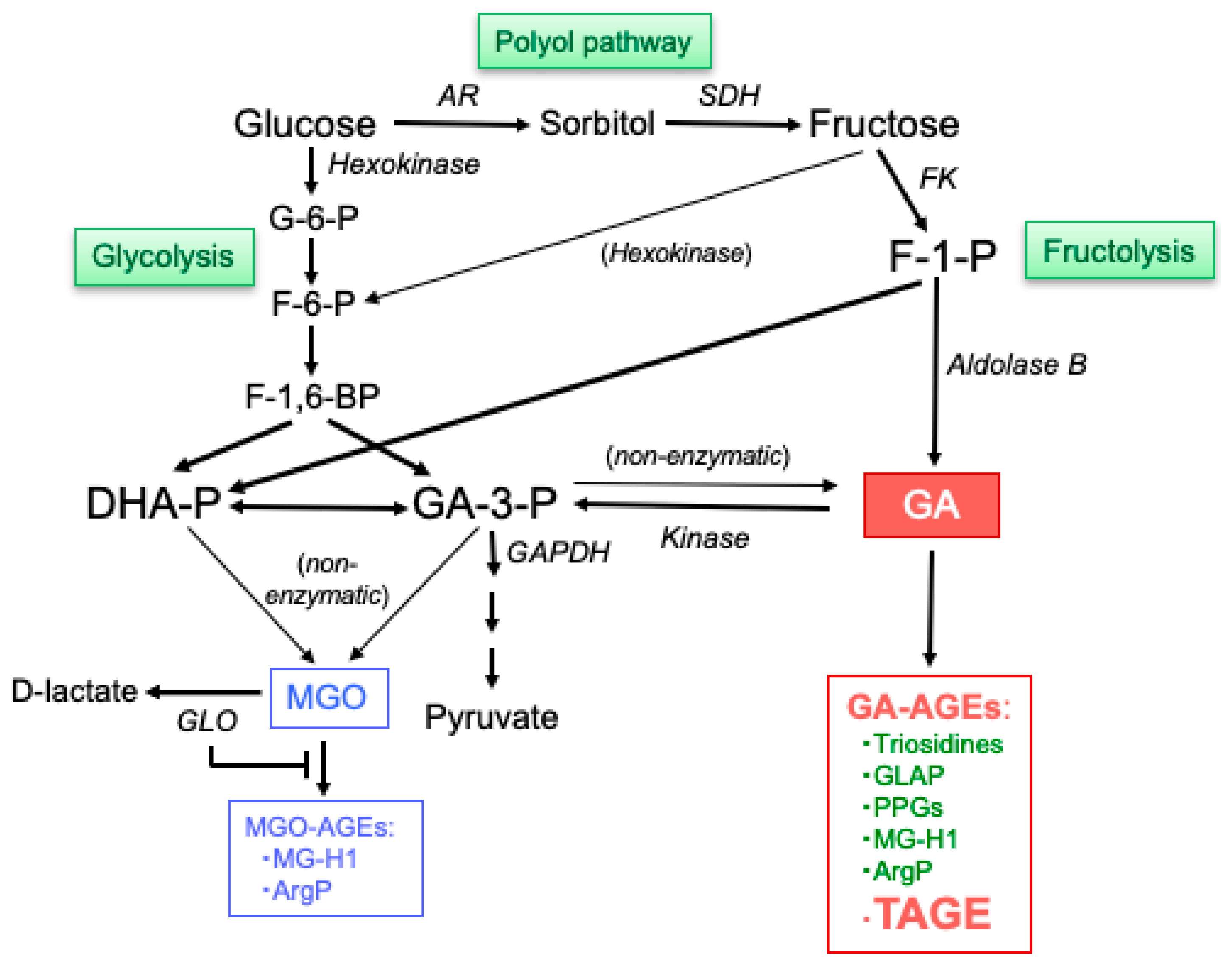
3. Generation of GA
3.1. Fructolysis
3.2. Glycolysis
3.3. Polyol Pathway
4. Generation of MGO
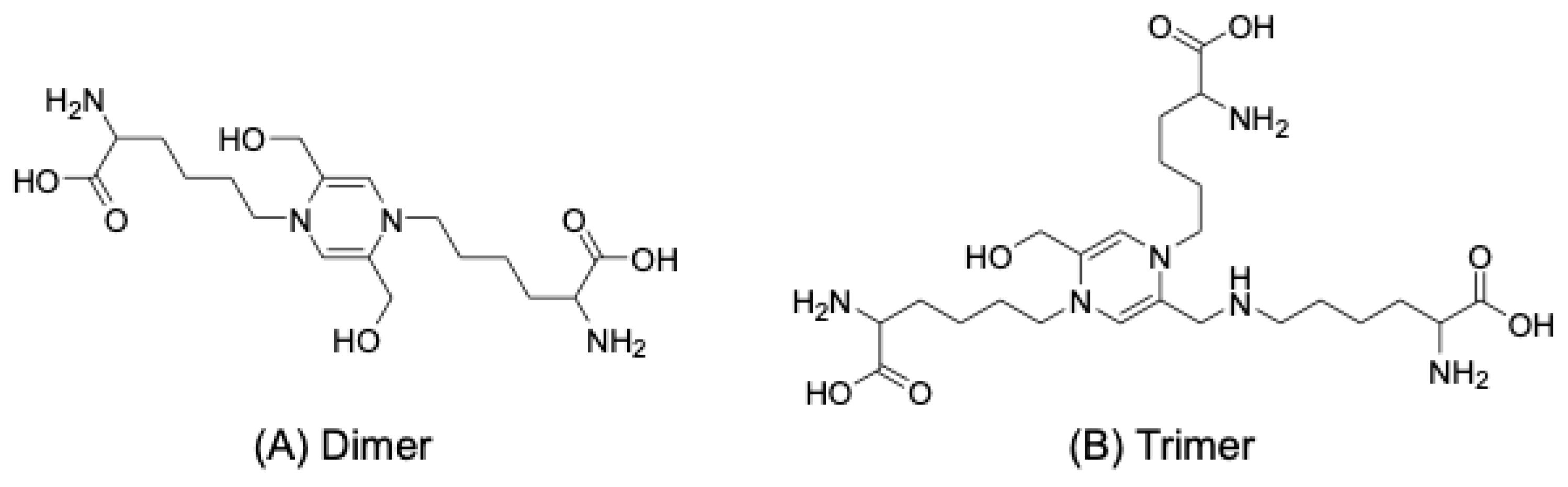
5. Structures of GA-AGEs
5.1. Structures of GA-AGEs with Lys Modifications
5.1.1. Triosidines
5.1.2. GLAP
5.1.3. PPGs
5.2. Structures of GA-AGEs with Arg Modifications
5.2.1. MG-H1
5.2.2. ArgP
6. Structures of TAGE
6.1. Proposed TAGE Structures
6.2. New Concept in LSRDs: The TAGE Theory
7. Cytotoxicity of TAGE
7.1. Accumulation of Intracellular TAGE and Cell Damage
7.1.1. Intracellular TAGE and Cell Death
TAGE and Hepatocytes
TAGE and Neurons
TAGE, Cardiomyocytes, and CFs
7.1.2. Mechanisms Underlying Intracellular TAGE Degradation
7.1.3. Extracellular TAGE and the TAGE–RAGE Axis
7.2. Involvement of Circulating TAGE in LSRDs
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bucala, R.; Cerami, A. Advanced glycosylation: Chemistry, biology, and implications for diabetes and aging. Adv. Pharmacol. 1992, 23, 1–34. [Google Scholar] [CrossRef]
- Vlassara, H.; Bucala, R.; Striker, L. Pathogenic effects of advanced glycosylation: Biochemical, biologic, and clinical implications for diabetes and aging. Lab. Investig. 1994, 70, 138–151. [Google Scholar]
- Brownlee, M. Advanced protein glycosylation in diabetes and aging. Ann. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. Recent progress in advanced glycation end products and diabetic complications. Diabetes 1997, 46, S19–S25. [Google Scholar] [CrossRef] [PubMed]
- Schroter, D.; Hohn, A. Role of advanced glycation end products in carcinogenesis and their therapeutic implications. Curr. Pharm. Des. 2018, 24, 5245–5251. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Lin, X.; Bu, C.; Zhang, X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr. Metab. 2018, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.Y.; Lu, C.H.; Wu, C.H.; Li, K.J.; Kuo, Y.M.; Hsieh, S.C.; Yu, C.L. The development of Maillard reaction, and advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling inhibitors as novel therapeutic strategies for patients with AGE-related diseases. Molecules 2020, 25, 5591. [Google Scholar] [CrossRef] [PubMed]
- Wells-Knecht, K.J.; Zyzak, D.V.; Litchfield, J.E.; Thorpe, S.R.; Baynes, J.W. Mechanism of autoxidative glycosylation: Identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry 1995, 34, 3702–3709. [Google Scholar] [CrossRef] [PubMed]
- Glomb, M.A.; Monnier, V.M. Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J. Biol. Chem. 1995, 270, 10017–10026. [Google Scholar] [CrossRef]
- Hayase, F.; Konishi, Y.; Kato, H. Identification of the modified structure of arginine residues in proteins with 3-deoxyglucosone, a Maillard reaction intermediate. Biosci. Biotech. Biochem. 1995, 59, 1407–1411. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344 Pt 1, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol. Med. 2000, 6, 114–125. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yanase, Y.; Matsuura, N.; Yamagishi, S.; Kameda, Y.; Bucala, R.; Makita, Z. Immunological detection of a novel advanced glycation end-product. Mol. Med. 2001, 7, 783–791. [Google Scholar] [CrossRef]
- Tauer, A.; Zhang, X.; Schaub, T.P.; Zimmeck, T.; Niwa, T.; Passlick-Deetjen, J.; Pischetsrieder, M. Formation of advanced glycation end products during CAPD. Am. J. Kidney Dis. 2003, 41 (Suppl. 1), S57–S60. [Google Scholar] [CrossRef]
- Takeuchi, M.; Iwaki, M.; Takino, J.; Shirai, H.; Kawakami, M.; Bucala, R.; Yamagishi, S. Immunological detection of fructose-derived advanced glycation end-products. Lab. Investig. 2010, 90, 1117–1127. [Google Scholar] [CrossRef]
- Gugliucci, A. Formation of fructose-mediated advanced glycation end products and their roles in metabolic and inflammatory diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef]
- Bunn, H.F.; Higgins, P.J. Reaction of monosaccharides with proteins: Possible evolutionary significance. Science 1981, 213, 222–224. [Google Scholar] [CrossRef]
- Suárez, G.; Rajaram, R.; Oronsky, A.L.; Gawinowicz, M.A. Nonenzymatic glycation of bovine serum albumin by fructose (fructation). Comparison with the Maillard reaction initiated by glucose. J. Biol. Chem. 1989, 264, 3674–3679. [Google Scholar] [CrossRef]
- Giardino, I.; Edelstein, D.; Brownlee, M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J. Clin. Investig. 1994, 94, 110–117. [Google Scholar] [CrossRef]
- Yokota, M.; Sekita, M.; Okano, Y.; Masaki, H.; Takeuchi, M.; Tokudome, Y. Glyceraldehyde-derived advanced glycation end products accumulate faster than Nε-(carboxymethyl) lysine. Ann. Dermatol. 2017, 29, 508–511. [Google Scholar] [CrossRef]
- Takeuchi, M. Toxic AGEs (TAGE) theory: A new concept for preventing the development of diseases related to lifestyle. Diabetol. Metab. Syndr. 2020, 12, 105. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y. Effects of toxic AGEs (TAGE) on human health. Cells 2022, 11, 2178. [Google Scholar] [CrossRef]
- Takino, J.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J. Gastroenterol. 2010, 45, 646–655. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products promote the production of reactive oxygen species in HepG2 cells. Int. J. Mol. Sci. 2020, 21, 4861. [Google Scholar] [CrossRef]
- Kikuchi, C.; Sakasai-Sakai, A.; Okimura, R.; Tanaka, H.; Takata, T.; Takeuchi, M.; Matsunaga, T. Accumulation of toxic advanced glycation end-products induces cytotoxicity and inflammation in hepatocyte-like cells differentiated from human induced pluripotent stem cells. Biol. Pharm. Bull. 2021, 44, 1399–1402. [Google Scholar] [CrossRef]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer’s disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci. Rep. 2015, 5, 13313. [Google Scholar] [CrossRef]
- Nasu, R.; Furukawa, A.; Suzuki, K.; Takeuchi, M.; Koriyama, Y. The effect of glyceraldehyde-derived advanced glycation end-products on β-tubulin-inhibited neurite outgrowth in SH-SY5Y human neuroblastoma cells. Nutrients 2020, 12, 2958. [Google Scholar] [CrossRef]
- Ooi, H.; Nasu, R.; Furukawa, A.; Takeuchi, M.; Koriyama, Y. Pyridoxamine and aminoguanidine attenuate the abnormal aggregation of β-tubulin and suppression of neurite outgrowth by glyceraldehyde-derived toxic advanced glycation end-products. Front. Pharmacol. 2022, 13, 921611. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Ueda, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci. Rep. 2019, 9, 2121. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular toxic advanced glycation end-products may induce cell death and suppress cardiac fibroblasts. Metabolites 2022, 12, 615. [Google Scholar] [CrossRef]
- Takata, T.; Ueda, T.; Sakasai-Sakai, A.; Takeuchi, M. Generation of glyceraldehyde-derived advanced glycation end-products in pancreatic cancer cells and the potential of tumor promotion. World J. Gastroenterol. 2017, 23, 4910–4919. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular toxic advanced glycation end-products 1.4E7 cell line induce death with reduction of microtubule-associated protein 1 light chain 3 and p62. Nutrients 2022, 14, 332. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Impact of intracellular toxic advanced glycation end-products (TAGE) on murine myoblast cell death. Diabetol. Metab. Syndr. 2020, 12, 54. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. The association between accumulation of toxic advanced glycation end-products and cytotoxic effect in MC3T3-E1 cells. Nutrients 2022, 14, 990. [Google Scholar] [CrossRef]
- Takeuchi, M. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker for the onset/progression of lifestyle-related diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular toxic AGEs (TAGE) triggers numerous types of cell damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef] [PubMed]
- Sakasai-Sakai, A.; Takeda, K.; Takeuchi, M. Involvement of intercellular TAGE and the TAGE-RAGE-ROS axis in the onset and progression of NAFLD/NASH. Antioxidants 2023, 12, 748. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J. Neuropathol. Exp. Neurol. 2000, 59, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Tessier, F.J.; Monnier, V.M.; Sayre, L.M.; Kornfield, J.A. Triosidines: Novel Maillard reaction products and cross-links from the reaction of triose sugars with lysine and arginine residues. Biochem. J. 2003, 369, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Hayase, F. Isolation and identification of the 3-hydroxy-5-hydroxymethyl-pyridinium compound as a novel advanced glycation end product on glyceraldehyde-related Maillard reaction. Biosci. Biotechnol. Biochem. 2003, 67, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, T.; Sakamoto, K.; Yamamoto, T. Glyceraldehyde-derived advanced glycation end-products having pyrrolopyridinium-based crosslinks. Biochem. Biophys. Rep. 2021, 26, 100963. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Watanabe, H.; Hayase, F. Isolation and identification of 5-methyl-imidazolin-4-one derivative as glyceraldehyde-derived advanced glycation end product. Biosci. Biotechnol. Biochem. 2006, 70, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Ohguchi, M.; Watanabe, H.; Hayase, F. The formation of argpyrimidine in glyceraldehyde-related glycation. Biosci. Biotechnol. Biochem. 2008, 72, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 1037. [Google Scholar] [CrossRef]
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and advanced glycation endproducts in the development of diabetic complications and targets for intervention. Int. J. Mol. Sci. 2017, 18, 984. [Google Scholar] [CrossRef]
- Jud, P.; Sourij, H. Therapeutic options to reduce advanced glycation end products in patients with diabetes mellitus: A review. Diabetes Res. Clin. Pract. 2019, 148, 54–63. [Google Scholar] [CrossRef]
- Song, Q.; Liu, J.; Dong, L.; Wang, X.; Zhang, X. Novel advances in inhibiting advanced glycation end product formation using natural compounds. Biomed. Pharmacother. 2021, 140, 111750. [Google Scholar] [CrossRef]
- Twarda-Clapa, A.; Olczak, A.; Białkowska, A.M.; Koziołkiewicz, M. Advanced glycation end-products (AGEs): Formation, chemistry, classification, receptors, and diseases related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef]
- Li, L.; Zhuang, Y.; Zou, X.; Chen, M.; Cui, B.; Jiao, Y.; Cheng, Y. Advanced glycation end products: A comprehensive review of their detection and occurrence in food. Foods 2023, 12, 2103. [Google Scholar] [CrossRef]
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660. [Google Scholar] [CrossRef]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, 754S–765S. [Google Scholar] [CrossRef]
- Yamagishi, S.; Imaizumi, T. Diabetic vascular complications: Pathophysiology, biochemical basis and potential therapeutic strategy. Curr. Pharm. Des. 2005, 11, 2279–2299. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Yamagishi, S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: A novel therapeutic strategy. Curr. Drug Targets 2010, 11, 1468–1482. [Google Scholar] [CrossRef] [PubMed]
- Tatton, W.G.; Chalmers-Redman, R.M.; Elstner, M.; Leesch, W.; Jagodzinski, F.B.; Stupak, D.P.; Sugrue, M.M.; Tatton, N.A. Glyceraldehyde-3-phosphate dehydrogenase in neurodegeneration and apoptosis signaling. J. Neural Transm. Suppl. 2000, 60, 77–100. [Google Scholar] [CrossRef]
- Oates, P.J. Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 2002, 50, 325–392. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, K.; Tanimoto, T.; Okada, S. Gene expression of enzymes comprising the polyol pathway in various rat tissues determined by the competitive RT-PCR method. Jpn. J. Pharmacol. 2002, 88, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates: Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Methylglyoxal-induced dicarbonyl stress in aging and disease: First steps towards glyoxalase 1-based treatments. Clin. Sci. 2016, 130, 1677–1696. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Thornalley, P.J. Chromatographic assay of glycation adducts in human serum albumin glycated in vitro by derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and intrinsic fluorescence. Biochem. J. 2002, 364, 15–24. [Google Scholar] [CrossRef]
- Ahmed, N.; Thornalley, P.J. Peptide mapping of human serum albumin modified minimally by methylglyoxal in vitro and in vivo. Ann. N. Y. Acad. Sci. 2005, 1043, 260–266. [Google Scholar] [CrossRef]
- Shipanova, I.N.; Glomb, M.A.; Nagaraj, R.H. Protein modification by methylglyoxal: Chemical nature and synthetic mechanism of a major fluorescent adduct. Arch. Biochem. Biophys. 1997, 344, 29–36. [Google Scholar] [CrossRef]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999, 274, 18492–18502. [Google Scholar] [CrossRef]
- Hollenbach, M. The role of glyoxalase-I (Glo-I), advanced glycation endproducts (AGEs), and their receptor (RAGE) in chronic liver disease and hepatocellular carcinoma (HCC). Int. J. Mol. Sci. 2017, 18, 2466. [Google Scholar] [CrossRef]
- Bellier, J.; Nokin, M.J.; Lardé, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcène, A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663. [Google Scholar] [CrossRef] [PubMed]
- Yumnam, S.; Subedi, L.; Kim, S.Y. Glyoxalase system in the progression of skin aging and skin malignancies. Int. J. Mol. Sci. 2021, 22, 310. [Google Scholar] [CrossRef] [PubMed]
- Senavirathna, L.; Ma, C.; Chen, R.; Pan, S. Proteomic investigation of glyceraldehyde-derived intracellular AGEs and their potential influence on pancreatic ductal cells. Cells 2021, 10, 1005. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Shimohira, K.; Watanabe, H.; Hayase, F. Detection and determination of glyceraldehyde-derived pyridinium-type advanced glycation end product in streptozotocin-induced diabetic rats. Biosci. Biotechnol. Biochem. 2007, 71, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Shizuuchi, S.; Watanabe, H.; Hayase, F. Cytotoxicity and oxidative stress induced by the glyceraldehyde-related Maillard reaction products for HL-60 cells. Biosci. Biotechnol. Biochem. 2004, 68, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Fujino, T.; Hasegawa, T.; Kurachi, R.; Miura, A.; Daikoh, T.; Usui, T.; Hayase, F.; Watanabe, H. Receptor for advanced glycation end products (RAGE)-mediated cytotoxicity of 3-hydroxypyridinium derivatives. Biosci. Biotechnol. Biochem. 2018, 82, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Oda, E.; Higashimoto, Y.; Yamagishi, S. Glyceraldehyde-derived pyridinium (GLAP) evokes oxidative stress and inflammatory and thrombogenic reactions in endothelial cells via the interaction with RAGE. Cardiovasc. Diabetol. 2015, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Adaikalakoteswari, A.; Larkin, J.R.; Panagiotopoulos, S.; MacIsaac, R.J.; Yue, D.K.; Fulcher, G.R.; Roberts, M.A.; Thomas, M.; Ekinci, E.; et al. Analysis of serum advanced glycation endproducts reveals methylglyoxal-derived advanced glycation MG-H1 free adduct is a risk marker in non-diabetic and diabetic chronic kidney disease. Int. J. Mol. Sci. 2023, 24, 152. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Q.; Ren, Z.; Wang, C.; Chen, L.; Chen, I.; Li, J.; Xia, Y.; Wan, Z. The relationships between plasma advanced glycation end products level and cognitive function in middle-aged and elderly Chinese subjects. Int. J. Hyg. Environ. Health 2023, 254, 114270. [Google Scholar] [CrossRef]
- Takeuchi, M.; Suzuki, H.; Takeda, K.; Sakai-Sakasai, A. Toxic advanced glycation end-products (TAGE) are major structures of cytotoxic AGEs derived from glyceraldehyde. Med. Hypotheses 2024, 183, 111248. [Google Scholar] [CrossRef]
- Choei, H.; Sasaki, N.; Takeuchi, M.; Yoshida, T.; Ukai, W.; Yamagishi, S.; Kikuchi, S.; Saito, T. Glyceraldehyde-derived advanced glycation end products in Alzheimer’s disease. Acta Neuropathol. 2004, 108, 189–193. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yonekura, H.; Watanabe, T.; Sakurai, S.; Li, H.; Harashima, A.; Myint, K.M.; Osawa, M.; Takeuchi, A.; Takeuchi, M.; et al. Short-chain aldehyde-derived ligands for RAGE and their actions on endothelial cells. Diabetes Res. Clin. Pract. 2007, 77 (Suppl. S1), S30–S40. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takino, J.; Takeuchi, M. Evidence for toxic advanced glycation end-products generated in the normal rat liver. Nutrients 2019, 11, 1612. [Google Scholar] [CrossRef]
- Dai, Y.; Grant, S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 2010, 16, 376–383. [Google Scholar] [CrossRef]
- Takeda, K.; Sakai-Sakasai, A.; Kajinami, K.; Takeuchi, M. A novel approach: Investigating the intracellular clearance mechanism of glyceraldehyde-derived advanced glycation end-products using the artificial checkpoint kinase 1 d270KD mutant as a substrate model. Cells 2023, 12, 2838. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef]
- Bettiga, A.; Fiorio, F.; Di Marco, F.; Trevisani, F.; Romani, A.; Porrini, E.; Salonia, A.; Montorsi, F.; Vago, R. The modern western diet rich in advanced glycation end-products (AGEs): An overview of its impact on obesity and early progression of renal pathology. Nutrients 2019, 11, 1748. [Google Scholar] [CrossRef]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced glycation end products (AGEs): Biochemistry, signaling, analytical methods, and epigenetic effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef]
- Manig, F.; Hellwig, M.; Pietz, F.; Henle, T. Studies about the dietary impact on “free” glycation compounds in human saliva. Foods 2022, 11, 2112. [Google Scholar] [CrossRef]
- Kuzan, A. Toxicity of advanced glycation end products (Review). Biomed. Rep. 2021, 14, 46. [Google Scholar] [CrossRef]
- Takeuchi, M.; Takino, J.; Furuno, S.; Shirai, H.; Kawakami, M.; Muramatsu, M.; Kobayashi, Y.; Yamagishi, S. Assessment of the concentrations of various advanced glycation end-products in beverages and foods that are commonly consumed in Japan. PLoS ONE 2015, 10, e0118652. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; del Castillo, M.D.; de la Maza, M.P.; Filip, R.; Gugliucci, A.; Luevano-Contreras, C.; Macías-Cervantes, M.H.; Markowicz Bastos, D.H.; Medrano, A.; Menini, T.; et al. Dietary advanced glycation end products and their role in health and disease. Adv. Nutr. 2015, 6, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Gill, V.; Kumar, V.; Singh, K.; Kumar, A.; Kim, J.-J. Advanced glycation end products (AGEs) may be a striking link between modern diet and health. Biomolecules 2019, 9, 888. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Boulestin, H.; Campbell, F.M.; Williams, L.M. The role of dietary advanced glycation end products in metabolic dysfunction. Mol. Nutr. Food Res. 2021, 65, e1900934. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916. [Google Scholar] [CrossRef]
- Sato, T.; Wu, X.; Shimogaito, N.; Takino, J.; Yamagishi, S.; Takeuchi, M. Effects of high-AGE beverage on RAGE and VEGF expressions in the liver and kidneys. Eur. J. Nutr. 2009, 48, 6–11. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Olmesartan blocks inflammatory reactions in endothelial cells evoked by advanced glycation end products by suppressing generation of reactive oxygen species. Ophthalmic Res. 2008, 40, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Glucagon-like peptide-1 (GLP-1) inhibits advanced glycation end product (AGE)-induced up-regulation of VCAM-1 mRNA levels in endothelial cells by suppressing AGE receptor (RAGE) expression. Biochem. Biophys. Res. Commun. 2010, 391, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Maeda, S.; Higashimoto, Y.; Yamagishi, S. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc. Diabetol. 2013, 12, 125. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Ueda, S.; Fukami, K.; Yamagishi, S. Advanced glycation end products potentiate citrated plasma-evoked oxidative and inflammatory reactions in endothelial cells by up-regulating protease-activated receptor-1 expression. Cardiovasc. Diabetol. 2014, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef]
- Takino, J.; Sato, T.; Kanetaka, T.; Okihara, K.; Nagamine, K.; Takeuchi, M.; Hori, T. RasGRP2 inhibits glyceraldehyde-derived toxic advanced glycation end-products from inducing permeability in vascular endothelial cells. Sci. Rep. 2021, 11, 2959. [Google Scholar] [CrossRef]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S. RAGE-aptamer blocks the development and progression of experimental diabetic nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef]
- Nakamura, N.; Matsui, T.; Ishibashi, Y.; Sotokawauchi, A.; Fukami, K.; Higashimoto, Y.; Yamagishi, S. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Mol. Med. 2017, 23, 295–306. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamagishi, S.; Yokoro, M.; Ito, S.; Kodama, G.; Kaida, Y.; Nakayama, Y.; Ando, R.; Yamada-Obara, N.; Asanuma, K.; et al. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Sci. Rep. 2018, 8, 2686. [Google Scholar] [CrossRef]
- Nakamura, N.; Matsui, T.; Nishino, Y.; Sotokawauchi, A.; Higashimoto, Y.; Yamagishi, S. Long-term local injection of RAGE-aptamer suppresses the growth of malignant melanoma in nude mice. J. Oncol. 2019, 2019, 7387601. [Google Scholar] [CrossRef] [PubMed]
- Sotokawauchi, A.; Matsui, T.; Higashimoto, Y.; Nishino, Y.; Koga, Y.; Yagi, M.; Yamagishi, S. DNA aptamer raised against receptor for advanced glycation end products suppresses renal tubular damage and improves insulin resistance in diabetic mice. Diab. Vasc. Dis. Res. 2021, 18, 1479164121990533. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Baker, J.R.; Takeuchi, M.; Hyogo, H.; Tjønneland, A.; Eriksen, A.K.; Severi, G.; Rothwell, J.; Laouali, N.; Katzke, V.; et al. Prediagnostic serum glyceraldehyde-derived advanced glycation end products and mortality among colorectal cancer patients. Int. J. Cancer 2023, 152, 2257–2268. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakai-Sakasai, A.; Takeda, K.; Suzuki, H.; Takeuchi, M. Structures of Toxic Advanced Glycation End-Products Derived from Glyceraldehyde, A Sugar Metabolite. Biomolecules 2024, 14, 202. https://doi.org/10.3390/biom14020202
Sakai-Sakasai A, Takeda K, Suzuki H, Takeuchi M. Structures of Toxic Advanced Glycation End-Products Derived from Glyceraldehyde, A Sugar Metabolite. Biomolecules. 2024; 14(2):202. https://doi.org/10.3390/biom14020202
Chicago/Turabian StyleSakai-Sakasai, Akiko, Kenji Takeda, Hirokazu Suzuki, and Masayoshi Takeuchi. 2024. "Structures of Toxic Advanced Glycation End-Products Derived from Glyceraldehyde, A Sugar Metabolite" Biomolecules 14, no. 2: 202. https://doi.org/10.3390/biom14020202
APA StyleSakai-Sakasai, A., Takeda, K., Suzuki, H., & Takeuchi, M. (2024). Structures of Toxic Advanced Glycation End-Products Derived from Glyceraldehyde, A Sugar Metabolite. Biomolecules, 14(2), 202. https://doi.org/10.3390/biom14020202