
Inspiring Tactics with the Improvement of Mitophagy and Redox Balance for the Development of Innovative Treatment against Polycystic Kidney Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
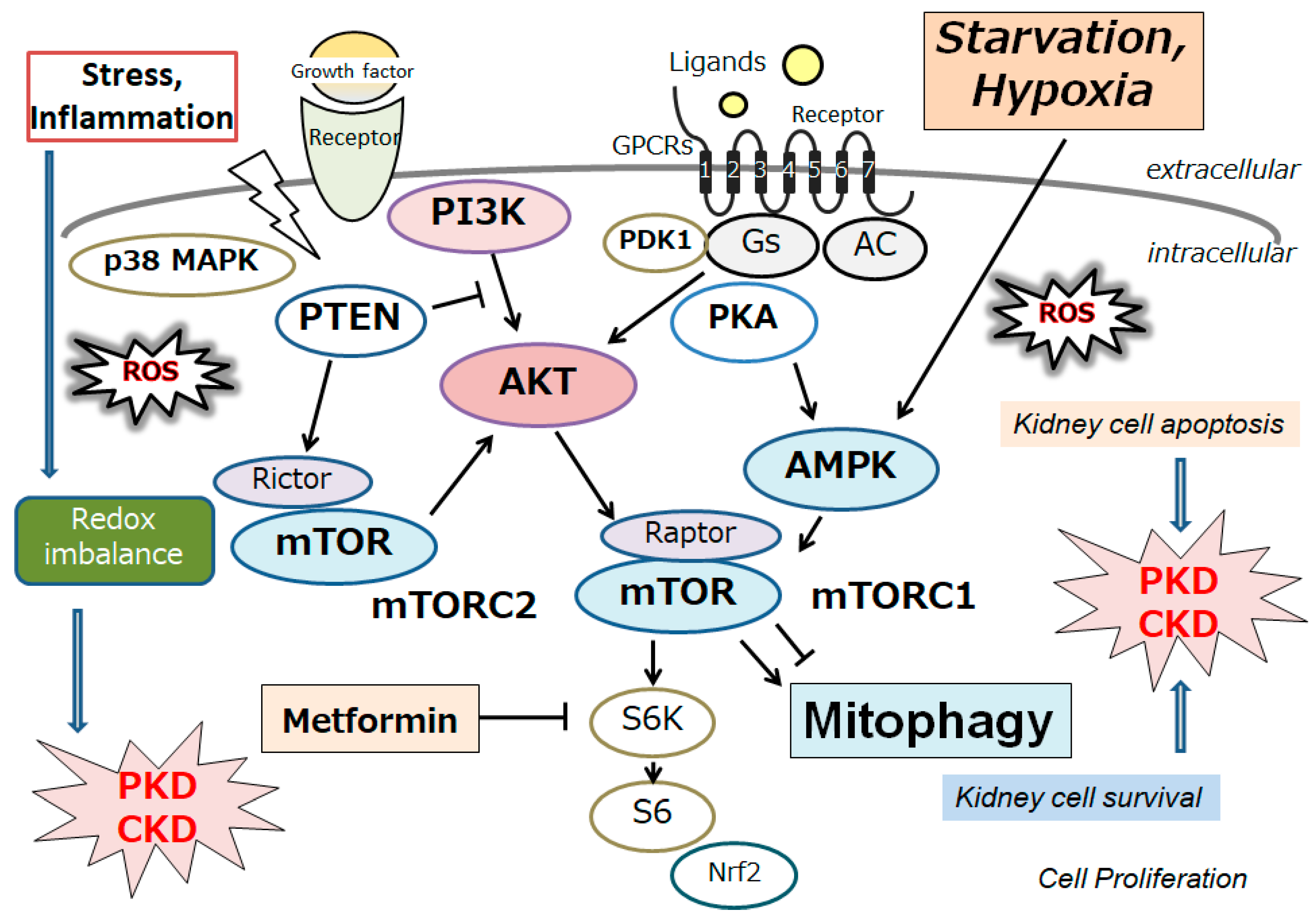
:1. Introduction
2. Autophagy/Mitophagy and Redox Imbalance in the Homeostasis of Kidney Cells
3. Autophagy/Mitophagy Involved in the Pathogenesis of Several Kidney Diseases, including Polycystic Kidney
4. Autophagy/Mitophagy as a Target of Treatment against Polycystic Kidney Disease
5. Another Tactic Regarding the Alteration of Gut Microbiome for the Treatment of Polycystic Kidney
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKI | Acute kidney injury |
| AMPK | adenosine monophosphate-activated protein kinase |
| APOL1 | apolipoprotein L1 |
| ATG13 | autophagy related 13 |
| CKD | chronic kidney disease |
| 4E-BP1 | eukaryotic translation initiation factor 4E-binding protein 1 |
| eIF3 | eukaryotic initiation factor 3 |
| FMT | fecal microbiota transplantation |
| FOXO1 | forkhead box protein O1 |
| FOXO3a | forkhead box protein O3a |
| HIF-1α | hypoxia inducible factor-1α |
| HO-1 | heme-oxygenase 1 |
| LC3 | microtubule-associated protein 1 light chain 3 |
| mRNAs | messenger RNAs |
| mTOR | mechanistic/mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| NAD+ | nicotinamide adenine dinucleotide |
| NDP52 | nuclear dot protein 52 kDa |
| NLRP3 | NLR family pyrin domain containing 3 |
| Nrf2 | nuclear factor (erythroid-derived 2)-like 2 |
| ORF | open reading framework |
| p70S6K | p70 ribosomal protein S6 kinase |
| PC1 | polycystin-1 |
| PC2 | polycystin-2 |
| PINK1 | PTEN-induced putative kinase 1 |
| PKD | Polycystic kidney disease |
| PPAR-α | peroxisome proliferator-activated receptor-α |
| PPARs | Peroxisome proliferator-activated receptors |
| SIRT1 | Sirtuin-1 |
| TGFβ | tumor growth factor β |
| ULK1 | unc-51-like autophagy activating kinase 1 |
| ULK2 | unc-51-like autophagy activating kinase 2 |
References
- Torres, V.E.; Harris, P.C. Progress in the understanding of polycystic kidney disease. Nat. Rev. Nephrol. 2019, 15, 70–72. [Google Scholar] [CrossRef]
- Harris, P.C.; Rossetti, S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2010, 6, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1α (HIF-1α) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol. Renal Physiol. 2011, 300, F1235–F1243. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Edelstein, C.L. Apoptosis and autophagy in polycystic kidney disease (PKD). Cell. Signal. 2020, 68, 109518. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Fan, S.; Xu, Y.; Meng, J.; Shen, X.; Mao, J.; Zhang, L.; Zhang, X.; Moeckel, G.; Wu, D.; et al. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J. Cell. Mol. Med. 2017, 21, 1619–1635. [Google Scholar] [CrossRef] [PubMed]
- Kou, P.; Wei, S.; Xiong, F. Recent advances of mTOR inhibitors use in autosomal dominant polycystic kidney disease: Is the road still open? Curr. Med. Chem. 2019, 26, 2962–2973. [Google Scholar] [CrossRef] [PubMed]
- Zeier, M.; Jones, E.; Ritz, E. Autosomal dominant polycystic kidney disease—The patient on renal replacement therapy. Nephrol. Dial. Transplant. 1996, 11 (Suppl. S6), 18–20. [Google Scholar] [CrossRef] [PubMed]
- Dery, M.C.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540. [Google Scholar] [CrossRef]
- De Mendonça, R.P.; Balbinot, K.M.; Martins, B.V.; da Silva Kataoka, M.S.; Mesquita, R.A.; de Jesus Viana Pinheiro, J.; de Melo Alves Júnior, S. Hypoxia and proangiogenic proteins in human ameloblastoma. Sci. Rep. 2020, 10, 17567. [Google Scholar] [CrossRef]
- Pereira-Prado, V.; Vigil-Bastitta, G.; Sánchez-Romero, C.; Arocena, M.; Molina-Frechero, N.; González-González, R.; Meleti, M.; Bologna-Molina, R. Immunoexpression of galectin-3 and its potential relation to hypoxia-inducible factor-1α in ameloblastomas. Biotech. Histochem. 2021, 96, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, N.M.M.; de Siqueira, A.S.; Ribeiro, A.L.R.; da Silva Kataoka, M.S.; Jaeger, R.G.; de Alves-Júnior, S.M.; Smith, A.M.; de Jesus Viana Pinheiro, J. Role of HIF-1α and CASPASE-3 in cystogenesis of odontogenic cysts and tumors. Clin. Oral. Investig. 2018, 22, 141–149. [Google Scholar] [CrossRef]
- Buchholz, B.; Eckardt, K.U. Role of oxygen and the HIF-pathway in polycystic kidney disease. Cell. Signal. 2020, 69, 109524. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.; Schley, G.; Faria, D.; Kroening, S.; Willam, C.; Schreiber, R.; Klanke, B.; Burzlaff, N.; Jantsch, J.; Kunzelmann, K.; et al. Hypoxia-inducible factor-1α causes renal cyst expansion through calcium-activated chloride secretion. J. Am. Soc. Nephrol. 2014, 25, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Ding, L.; Ma, R.X.; Zhang, Y.; Zhang, Y.L.; Ni, W.J.; Tang, T.T.; Wang, G.H.; Wang, B.; Lv, L.L.; et al. Activation of HIF-1α C-terminal transactivation domain protects against hypoxia-induced kidney injury through hexokinase 2-mediated mitophagy. Cell Death Dis. 2023, 14, 339. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liu, J.; Cui, J.; Su, J.; Xu, W.; Zhang, F.; Ding, Y. Ferroptosis plays a crucial role in lung cell damage caused by ventilation stretch. Free Radic. Biol. Med. 2023, 209 Pt 1, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.X.; Ding, H.; Torres, V.E.; Yu, C.; Li, X. Ferroptosis Promotes Cyst Growth in Autosomal Dominant Polycystic Kidney Disease Mouse Models. J. Am. Soc. Nephrol. 2021, 32, 2759–2776. [Google Scholar] [CrossRef]
- Falchook, G.S.; Wheler, J.J.; Naing, A.; Jackson, E.F.; Janku, F.; Hong, D.; Ng, C.S.; Tannir, N.M.; Lawhorn, K.N.; Huang, M.; et al. Targeting hypoxia-inducible factor-1α (HIF-1α) in combination with antiangiogenic therapy: A phase I trial of bortezomib plus bevacizumab. Oncotarget 2014, 5, 10280–10292. [Google Scholar] [CrossRef]
- Gregory, A.V.; Chebib, F.T.; Poudyal, B.; Holmes, H.L.; Yu, A.S.L.; Landsittel, D.P.; Bae, K.T.; Chapman, A.B.; Frederic, R.O.; Mrug, M.; et al. Utility of new image-derived biomarkers for autosomal dominant polycystic kidney disease prognosis using automated instance cyst segmentation. Kidney Int. 2023, 104, 334–342. [Google Scholar] [CrossRef]
- Kim, Y.; Tao, C.; Kim, H.; Oh, G.Y.; Ko, J.; Bae, K.T. A Deep Learning Approach for Automated Segmentation of Kidneys and Exophytic Cysts in Individuals with Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Tollens, F.; Hansen, L.; Golla, A.K.; Schad, L.R.; Nörenberg, D.; Zöllner, F.G. Deep Learning-Based Total Kidney Volume Segmentation in Autosomal Dominant Polycystic Kidney Disease Using Attention, Cosine Loss, and Sharpness Aware Minimization. Diagnostics 2022, 12, 1159. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1α-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef]
- Chen, M.; Wang, W.; Fu, X.; Yi, Y.; Wang, K.; Wang, M. Role of Pink1-mediated mitophagy in adenomyosis. PeerJ 2023, 11, e16497. [Google Scholar] [CrossRef]
- Ji, Y.; Leng, Y.; Lei, S.; Qiu, Z.; Ming, H.; Zhang, Y.; Zhang, A.; Wu, Y.; Xia, Z. The mitochondria-targeted antioxidant MitoQ ameliorates myocardial ischemia-reperfusion injury by enhancing PINK1/Parkin-mediated mitophagy in type 2 diabetic rats. Cell Stress Chaperones 2022, 27, 353–367. [Google Scholar] [CrossRef]
- Feng, J.; Lu, C.; Dai, Q.; Sheng, J.; Xu, M. SIRT3 Facilitates Amniotic Fluid Stem Cells to Repair Diabetic Nephropathy through Protecting Mitochondrial Homeostasis by Modulation of Mitophagy. Cell. Physiol. Biochem. 2018, 46, 1508–1524. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Chen, K.; Chen, J.; Wang, L.; Yang, J.; Xiao, F.; Wang, X.; Yuan, J.; Wang, L.; He, Y. Parkin Ubiquitinates GATA4 and Attenuates the GATA4/GAS1 Signaling and Detrimental Effects on Diabetic Nephropathy. FASEB J. 2020, 34, 8858–8875. [Google Scholar] [CrossRef]
- E, Y.; Lin, Y.; Yan, G.; Yang, J.; Jiao, L.; Wu, R.; Yan, Q.; Chen, Y.; Chen, Y.; Yan, X.; et al. Exogenous H2S alleviates senescence of glomerular mesangial cells through up-regulating mitophagy by activation of AMPK-ULK1-PINK1-parkin pathway in mice. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119568. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Olsvik, H.; Williams, G.L.; Oh, S.; Evjen, G.; Sjøttem, E.; Mandell, M.A. TBK1 is ubiquitinated by TRIM5α to assemble mitophagy machinery. bioRxiv 2023. [Google Scholar] [CrossRef]
- Gao, X.; Liu, Y.; Wang, L.; Sai, N.; Liu, Y.; Ni, J. Morroniside Inhibits H2O2-Induced Podocyte Apoptosis by Down-Regulating NOX4 Expression Controlled by Autophagy In Vitro. Front. Pharmacol. 2020, 11, 533809. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Forbes, M.S.; Thornhill, B.A.; Chevalier, R.L. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: A new look at an old model. Am. J. Physiol. Renal Physiol. 2011, 301, F110–F117. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023, 19, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhang, J.; Wang, J.; Wang, X.; Cao, E.; Yang, C.; Sun, Q.; Sivakumar, R.; Peng, Z. Pannexin 1 targets mitophagy to mediate renal ischemia/reperfusion injury. Commun. Biol. 2023, 6, 889. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in acute kidney injury and kidney repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef]
- Bhatia, D.; Choi, M.E. The emerging role of mitophagy in kidney diseases. J. Life Sci. 2019, 1, 13–22. [Google Scholar] [CrossRef]
- Baisantry, A.; Bhayana, S.; Rong, S.; Ermeling, E.; Wrede, C.; Hegermann, J.; Pennekamp, P.; Sörensen-Zender, I.; Haller, H.; Melk, A.; et al. Autophagy induces prosenescent changes in proximal tubular S3 segments. J. Am. Soc. Nephrol. 2016, 27, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.A.; Kim, W.Y.; Kim, J.W.; Kang, M.G.; Park, S.H.; Lee, M.S.; Kim, H.W.; Yang, C.W.; Kim, J.; Kim, Y.K. Autophagy in FOXD1 stroma-derived cells regulates renal fibrosis through TGF-beta and NLRP3 inflammasome pathway. Biochem. Biophys. Res. Commun. 2019, 508, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, C.; Liu, Y.Y.; Guo, Z.A. Mitophagy in renal interstitial fibrosis. Int. Urol. Nephrol. 2023, 56, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Yu, B.; Liu, G.; Nie, W.; Wang, J.; Chen, J.; Xiao, L.; Xia, H.; Han, F.; Yang, Y. Mitophagy induced by UMI-77 preserves mitochondrial fitness in renal tubular epithelial cells and alleviates renal fibrosis. FASEB J. 2022, 36, e22342. [Google Scholar] [CrossRef]
- 48 Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- Kumar, V.; Ayasolla, K.; Jha, A.; Mishra, A.; Vashistha, H.; Lan, X.; Qayyum, M.; Chinnapaka, S.; Purohit, R.; Mikulak, J.; et al. Disrupted apolipoprotein L1-miR193a axis dedifferentiates podocytes through autophagy blockade in an APOL1 risk milieu. Am. J. Physiol. Cell Physiol. 2019, 317, C209–C225. [Google Scholar] [CrossRef]
- Asanuma, K.; Tanida, I.; Shirato, I.; Ueno, T.; Takahara, H.; Nishitani, T.; Kominami, E.; Tomino, Y. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J. 2003, 17, 1165–1167. [Google Scholar] [CrossRef]
- Zeng, C.; Fan, Y.; Wu, J.; Shi, S.; Chen, Z.; Zhong, Y.; Zhang, C.; Zen, K.; Liu, Z. Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J. Pathol. 2014, 234, 203–213. [Google Scholar] [CrossRef]
- Zschiedrich, S.; Bork, T.; Liang, W.; Wanner, N.; Eulenbruch, K.; Munder, S.; Hartleben, B.; Kretz, O.; Gerber, S.; Simons, M.; et al. Targeting mTOR signaling can prevent the progression of FSGS. J. Am. Soc. Nephrol. 2017, 28, 2144–2157. [Google Scholar] [CrossRef]
- Ma, Z.; Li, L.; Livingston, M.J.; Zhang, D.; Mi, Q.; Zhang, M.; Ding, H.F.; Huo, Y.; Mei, C.; Dong, Z. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J. Clin. Investig. 2020, 130, 5011–5026. [Google Scholar] [CrossRef]
- Chen, K.; Dai, H.; Yuan, J.; Chen, J.; Lin, L.; Zhang, W.; Wang, L.; Zhang, J.; Li, K.; He, Y. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Li, C.; Gu, C.; Fang, Y.; Tycksen, E.; Puri, A.; Pietka, T.A.; Sivapackiam, J.; Kidd, K.; Park, S.J.; et al. MANF stimulates autophagy and restores mitochondrial homeostasis to treat autosomal dominant tubulointerstitial kidney disease in mice. Nat. Commun. 2023, 14, 6493. [Google Scholar] [CrossRef] [PubMed]
- Pampliega, O.; Cuervo, A.M. Autophagy and primary cilia: Dual interplay. Curr. Opin. Cell Biol. 2016, 39, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Sagredo, A.; Quiroga, C.; Garrido-Moreno, V.; López-Crisosto, C.; Leiva-Navarrete, S.; Norambuena-Soto, I.; Ortiz-Quintero, J.; Díaz-Vesga, M.C.; Perez, W.; Hendrickson, T.; et al. Polycystin-1 regulates cardiomyocyte mitophagy. FASEB J. 2021, 35, e21796. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Piva, F.; Cimadamore, A.; Giulietti, M.; Battelli, N.; Montironi, R.; Cosmai, L.; Porta, C. Exploring the Spectrum of Kidney Ciliopathies. Diagnostics 2020, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Lea, W.A.; Winklhofer, T.; Zelenchuk, L.; Sharma, M.; Rossol-Allison, J.; Fields, T.A.; Reif, G.; Calvet, J.P.; Bakeberg, J.L.; Wallace, D.P.; et al. Polycystin-1 Interacting Protein-1 (CU062) Interacts with the Ectodomain of Polycystin-1 (PC1). Cells 2023, 12, 2166. [Google Scholar] [CrossRef] [PubMed]
- Peintner, L.; Venkatraman, A.; Waeldin, A.; Hofherr, A.; Busch, T.; Voronov, A.; Viau, A.; Kuehn, E.W.; Köttgen, M.; Borner, C. Loss of PKD1/Polycystin-1 Impairs Lysosomal Activity in a CAPN (Calpain)-Dependent Manner. Autophagy 2021, 17, 2384–2400. [Google Scholar] [CrossRef]
- Peña-Oyarzun, D.; Rodriguez-Peña, M.; Burgos-Bravo, F.; Vergara, A.; Kretschmar, C.; Sotomayor-Flores, C.; Ramirez-Sarmiento, C.A.; De Smedt, H.; Reyes, M.; Perez, W.; et al. PKD2/Polycystin-2 Induces Autophagy by Forming a Complex with BECN1. Autophagy 2021, 17, 1714–1728. [Google Scholar] [CrossRef]
- Pan, F.; Bu, L.; Wu, K.; Wang, A.; Xu, X. PKD2/polycystin-2 inhibits LPS-induced acute lung injury in vitro and in vivo by activating autophagy. BMC Pulm. Med. 2023, 23, 171. [Google Scholar] [CrossRef]
- Criollo, A.; Altamirano, F.; Pedrozo, Z.; Schiattarella, G.G.; Li, D.L.; Rivera-Mejías, P.; Sotomayor-Flores, C.; Parra, V.; Villalobos, E.; Battiprolu, P.K.; et al. Polycystin-2-Dependent Control of Cardiomyocyte Autophagy. J. Mol. Cell. Cardiol. 2018, 118, 110–121. [Google Scholar] [CrossRef]
- Decuypere, J.-P.; Ceulemans, L.J.; Agostinis, P.; Monbaliu, D.; Naesens, M.; Pirenne, J.; Jochmans, I. Autophagy and the Kidney: Implications for Ischemia-Reperfusion Injury and Therapy. Am. J. Kidney Dis. 2015, 66, 699–709. [Google Scholar] [CrossRef]
- Yuajit, C.; Muanprasat, C.; Homvisasevongsa, S.; Chatsudthipong, V. Steviol Stabilizes Polycystin 1 Expression and Promotes Lysosomal Degradation of CFTR and β-Catenin Proteins in Renal Epithelial Cells. Biomed. Pharmacother. 2017, 94, 820–826. [Google Scholar] [CrossRef]
- Mei, Y.; Hu, H.; Deng, L.; Sun, X.; Tan, W. Therapeutic effects of isosteviol sodium on non-alcoholic fatty liver disease by regulating autophagy via Sirt1/AMPK pathway. Sci. Rep. 2022, 12, 12857. [Google Scholar] [CrossRef]
- Collins, J.; Robinson, C.; Danhof, H.; Knetsch, C.W.; van Leeuwen, H.C.; Lawley, T.D.; Auchtung, J.M.; Britton, R.A. Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature 2018, 553, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Haddad, G.G. Role of trehalose phosphate synthase and trehalose during hypoxia: From flies to mammals. J. Exp. Biol. 2004, 207 Pt 18, 3125–3129. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, R.E.; Hill, S.D.; Bispham, N.Z.; Santos-Parker, J.R.; Nowlan, M.J.; Snyder, L.L.; Chonchol, M.; LaRocca, T.J.; McQueen, M.B.; Seals, D.R. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging 2016, 8, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.L.; Saleem, M.A.; Chan, K.W.; Yung, B.Y.; Law, H.K. Trehalose, an mTOR independent autophagy inducer, alleviates human podocyte injury after puromycin aminonucleoside treatment. PLoS ONE 2014, 9, e113520. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Yang, H.; Wang, M.G.; Yang, D.B.; Wang, Z.Y.; Wang, L. Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis. 2017, 8, e3099. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.; Choi, H.I.; Park, S.H.; Ahn, J.; Jang, Y.J.; Ha, T.Y.; Lee, D.H.; Seo, H.D.; Jung, C.H. The unc-51 like autophagy activating kinase 1-autophagy related 13 complex has distinct functions in tunicamycin-treated cells. Biochem. Biophys. Res. Commun. 2020, 524, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell 2015, 57, 456–466. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE 2010, 5, e9199. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef]
- Tabassum, S.; Misrani, A.; Huang, H.X.; Zhang, Z.Y.; Li, Q.W.; Long, C. Resveratrol Attenuates Chronic Unpredictable Mild Stress-Induced Alterations in the SIRT1/PGC1α/SIRT3 Pathway and Associated Mitochondrial Dysfunction in Mice. Mol. Neurobiol. 2023, 60, 5102–5116. [Google Scholar] [CrossRef]
- El Ters, M.; Zhou, X.; Lepping, R.J.; Lu, P.; Karcher, R.T.; Mahnken, J.D.; Brooks, W.M.; Winklhofer, F.T.; Li, X.; Alan, S.L. Biological Efficacy and Safety of Niacinamide in Patients with ADPKD. Kidney Int. Rep. 2020, 5, 1271–1279. [Google Scholar] [CrossRef]
- Lempiäinen, J.; Finckenberg, P.; Mervaala, E.E.; Sankari, S.; Levijoki, J.; Mervaala, E.M. Caloric restriction ameliorates kidney ischaemia/reperfusion injury through PGC-1α-eNOS pathway and enhanced autophagy. Acta Physiol. 2013, 208, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rubio, M.; Burón, M.I.; López-Lluch, G.; Navas, P.; de Cabo, R.; Ramsey, J.J.; Villalba, J.M.; González-Reyes, J.A. Dietary fat composition influences glomerular and proximal convoluted tubule cell structure and autophagic processes in kidneys from calorie-restricted mice. Aging Cell. 2016, 15, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Duca, F.A.; Côté, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K. Metformin activates a duodenal Ampk–dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2015, 21, 506–511. [Google Scholar] [CrossRef]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: Potential role in atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef]
- Musi, N.; Hirshman, M.F.; Nygren, J.; Svanfeldt, M.; Bavenholm, P.; Rooyackers, O.; Zhou, G.; Williamson, J.M.; Ljunqvist, O.; Efendic, S.; et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 2002, 51, 2074–2081. [Google Scholar] [CrossRef]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J. Bioenerg. Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Chowdhury, S.; Bhattacharyya, M. Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients. Diabetes Res. Clin. Pract. 2011, 93, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.H.; Ding, W.; Zhang, N.; Zhou, Z.Y.; Ling, Z.; Li, W.J.; Chen, S.; Tang, Q.Z. Activation of AMPKα2 attenuated doxorubicin-induced cardiotoxicity via inhibiting lipid peroxidation associated ferroptosis. Free Radic. Biol. Med. 2023, 205, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Padovano, V.; Podrini, C.; Boletta, A.; Caplan, M.J. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat. Rev. Nephrol. 2018, 14, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Lakhia, R.; Yheskel, M.; Flaten, A.; Quittner-Strom, E.B.; Holland, W.L.; Patel, V. PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Renal Physiol. 2018, 314, F122–F131. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Duszka, K.; Wahli, W. PPARs and Microbiota in Skeletal Muscle Health and Wasting. Int. J. Mol. Sci. 2020, 21, 8056. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, W.D.; Wang, Y.D. Nuclear receptors: A bridge linking the gut microbiome and the host. Mol. Med. 2021, 27, 144. [Google Scholar] [CrossRef]
- Frąk, W.; Kućmierz, J.; Szlagor, M.; Młynarska, E.; Rysz, J.; Franczyk, B. New Insights into Molecular Mechanisms of Chronic Kidney Disease. Biomedicines 2022, 10, 2846. [Google Scholar] [CrossRef]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef]
- Gulati, A.; Watnick, T. Vascular Complications in Autosomal Dominant Polycystic Kidney Disease: Perspectives, Paradigms, and Current State of Play. Adv. Kidney Dis. Health 2023, 30, 429–439. [Google Scholar] [CrossRef]
- Wehedy, E.; Shatat, I.F.; Al Khodor, S. The Human Microbiome in Chronic Kidney Disease: A Double-Edged Sword. Front. Med. 2022, 8, 790783. [Google Scholar] [CrossRef] [PubMed]
- Noce, A.; Marchetti, M.; Marrone, G.; Di Renzo, L.; Di Lauro, M.; Di Daniele, F.; Albanese, M.; Di Daniele, N.; De Lorenzo, A. Link between Gut Microbiota Dysbiosis and Chronic Kidney Disease. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 2057–2074. [Google Scholar] [PubMed]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gasparini, A.; Ishigami, J.; Mzayen, K.; Su, G.; Barany, P.; Ärnlöv, J.; Lindholm, B.; Elinder, C.G.; Matsushita, K.; et al. eGFR and the risk of community-acquired infections. Clin. J. Am. Soc. Nephrol. 2017, 12, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Trevisan, M.; Ishigami, J.; Matsushita, K.; Stålsby Lundborg, C.; Carrero, J.J. Short-and long-term outcomes after incident pneumonia in adults with chronic kidney disease: A time-dependent analysis from the stockholm creatinine measurement project. Nephrol. Dial. Transplant. 2020, 35, 1894–1900. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Zhang, P.; Fang, Y.; Liu, Y.; Ding, Y.; Zhang, W. Intestinal homeostasis in the gut-lung-kidney axis: A prospective therapeutic target in immune-related chronic kidney diseases. Front. Immunol. 2023, 14, 1266792. [Google Scholar] [CrossRef]
- Cooper, T.E.; Khalid, R.; Chan, S.; Craig, J.C.; Hawley, C.M.; Howell, M.; Johnson, D.W.; Jaure, A.; Teixeira-Pinto, A.; Wong, G. Synbiotics, prebiotics and probiotics for people with chronic kidney disease. Cochrane Database Syst. Rev. 2023, 10, CD013631. [Google Scholar]
- Yacoub, R.; Nadkarni, G.N.; McSkimming, D.I.; Chaves, L.D.; Abyad, S.; Bryniarski, M.A.; Honan, A.M.; Thomas, S.A.; Gowda, M.; He, J.C.; et al. Fecal microbiota analysis of polycystic kidney disease patients according to renal function: A pilot study. Exp. Biol. Med. 2019, 244, 505–513. [Google Scholar] [CrossRef]
- Mancin, S.; Mazzoleni, B.; Reggiani, F.; Calatroni, M.; Alterchi, E.; Donizzetti, D.; Finazzi, S.; Soekeland, F.; Sguanci, M.; Badalamenti, S. Integrated protocol for the prevention and treatment of skin ulcers in patients with end-stage renal disease. MethodsX 2023, 11, 102482. [Google Scholar] [CrossRef]
- Du, Y.; Zhu, Y.J.; Zhou, Y.X.; Ding, J.; Liu, J.Y. Metformin in therapeutic applications in human diseases: Its mechanism of action and clinical study. Mol. Biomed. 2022, 3, 41. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Nicholas, S.B.; Norris, K.C.; Vaziri, N.D. Role of impaired Nrf2 activation in the pathogenesis of oxidative stress and inflammation in chronic tubulo-interstitial nephropathy. Nephrol. Dial. Transplant. 2013, 28, 2038–2045. [Google Scholar] [CrossRef]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim. Pol. 2019, 66, 13–21. [Google Scholar] [CrossRef]
- Xu, D.; Li, Y.; Zhang, B.; Wang, Y.; Liu, Y.; Luo, Y.; Niu, W.; Dong, M.; Liu, M.; Dong, H.; et al. Resveratrol alleviate hypoxic pulmonary hypertension via anti-inflammation and anti-oxidant pathways in rats. Int. J. Med. Sci. 2016, 13, 942–954. [Google Scholar] [CrossRef]
- Tsuji, A.; Ikeda, Y.; Yoshikawa, S.; Taniguchi, K.; Sawamura, H.; Morikawa, S.; Nakashima, M.; Asai, T.; Matsuda, S. The Tryptophan and Kynurenine Pathway Involved in the Development of Immune-Related Diseases. Int. J. Mol. Sci. 2023, 24, 5742. [Google Scholar] [CrossRef]
- De Almeida, R.M.; Clendenon, S.G.; Richards, W.G.; Boedigheimer, M.; Damore, M.; Rossetti, S.; Harris, P.C.; Herbert, B.S.; Xu, W.M.; Wandinger-Ness, A.; et al. Transcriptome analysis reveals manifold mechanisms of cyst development in ADPKD. Hum. Genomics 2016, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Swenson-Fields, K.I.; Vivian, C.J.; Salah, S.M.; Peda, J.D.; Davis, B.M.; van Rooijen, N.; Wallace, D.P.; Fields, T.A. Macrophages promote polycystic kidney disease progression. Kidney Int. 2013, 83, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Bulley, S.; Fernández-Peña, C.; Hasan, R.; Leo, M.D.; Muralidharan, P.; Mackay, C.E.; Evanson, K.W.; Moreira-Junior, L.; Mata-Daboin, A.; Burris, S.K.; et al. Arterial smooth muscle cell PKD2 (TRPP1) channels regulate systemic blood pressure. Elife 2018, 7, e42628. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Leo, M.D.; Muralidharan, P.; Mata-Daboin, A.; Yin, W.; Bulley, S.; Fernandez-Peña, C.; MacKay, C.E.; Jaggar, J.H. SUMO1 modification of PKD2 channels regulates arterial contractility. Proc. Natl. Acad. Sci. USA 2019, 116, 27095–27104. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.A.; Whitmire, S.; Patton, S.; Clark, C.; Blanchette, C.M.; Howden, R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J. Med. Econ. 2017, 20, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. ACE inhibitors, left ventricular mass and renal cyst growth in ADPKD. Pharmacol. Res. 2016, 114, 166–168. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakashima, M.; Suga, N.; Ikeda, Y.; Yoshikawa, S.; Matsuda, S. Inspiring Tactics with the Improvement of Mitophagy and Redox Balance for the Development of Innovative Treatment against Polycystic Kidney Disease. Biomolecules 2024, 14, 207. https://doi.org/10.3390/biom14020207
Nakashima M, Suga N, Ikeda Y, Yoshikawa S, Matsuda S. Inspiring Tactics with the Improvement of Mitophagy and Redox Balance for the Development of Innovative Treatment against Polycystic Kidney Disease. Biomolecules. 2024; 14(2):207. https://doi.org/10.3390/biom14020207
Chicago/Turabian StyleNakashima, Moeka, Naoko Suga, Yuka Ikeda, Sayuri Yoshikawa, and Satoru Matsuda. 2024. "Inspiring Tactics with the Improvement of Mitophagy and Redox Balance for the Development of Innovative Treatment against Polycystic Kidney Disease" Biomolecules 14, no. 2: 207. https://doi.org/10.3390/biom14020207
APA StyleNakashima, M., Suga, N., Ikeda, Y., Yoshikawa, S., & Matsuda, S. (2024). Inspiring Tactics with the Improvement of Mitophagy and Redox Balance for the Development of Innovative Treatment against Polycystic Kidney Disease. Biomolecules, 14(2), 207. https://doi.org/10.3390/biom14020207