


Understanding the Pathophysiology of Ischemic Stroke: The Basis of Current Therapies and Opportunity for New Ones

Abstract
1. Introduction
2. Etiology and Pathophysiology of Ischemic Stroke
2.1. Excitotoxicity
2.2. Oxidative Stress
2.3. Neuroinflammation
2.4. Apoptosis
2.4.1. Ferroptosis
2.4.2. Necroptosis
2.4.3. Pyroptosis
2.4.4. Parthanatos
2.4.5. Phagoptosis
2.5. Autophagy
3. Current Therapies for Ischemic Stroke and Their Targets
3.1. Thrombolytics/Thrombolytic Agents
3.2. Adjunctive Therapies
3.2.1. Anti-Thrombotic Agents
3.2.2. Antiplatelet Therapy
3.2.3. Fibrinogen-Depleting Agents
3.3. Cellular Therapies for Ischemic Stroke: A Paradigm Approach
Limitations of Stem Cell Therapy and Way Forward
4. Emerging Neuroprotective Agents for Ischemic Stroke: Pathophysiology-Targeted Therapies
5. Future Perspectives: Drug Repurposing and Re-Designing
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef]
- Külkens, S.; Hacke, W. Thrombolysis with alteplase for acute ischemic stroke: Review of SITS-MOST and other Phase IV studies. Expert Rev. Neurother. 2007, 7, 783–788. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar]
- Berge, E.; Whiteley, W.; Audebert, H.; De Marchis, G.M.; Fonseca, A.C.; Padiglioni, C.; Pérez de la Ossa, N.; Strbian, D.; Tsivgoulis, G.; Turc, G. European Stroke Organisation (ESO) guidelines on intravenous thrombolysis for acute ischaemic stroke. Eur. Stroke J. 2021, 6, I–LXII. [Google Scholar] [CrossRef]
- Palmer, S.J. Identification, care and prevention of stroke is possible. Br. J. Healthc. Assist. 2023, 17, 236–239. [Google Scholar] [CrossRef]
- Salaudeen, M.A.; Allan, S.; Pinteaux, E. Hypoxia and interleukin-1-primed mesenchymal stem/stromal cells as novel therapy for stroke. Hum. Cell 2023, 37, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.-L.; Brassard, P.; Rickards, C.A.; Nogueira, R.C.; Nasr, N.; McBryde, F.D.; Fisher, J.P.; Tzeng, Y.-C. Integrative cerebral blood flow regulation in ischemic stroke. J. Cereb. Blood Flow Metab. 2022, 42, 387–403. [Google Scholar] [CrossRef]
- van Putten, M.J.; Fahlke, C.; Kafitz, K.W.; Hofmeijer, J.; Rose, C.R. Dysregulation of astrocyte ion homeostasis and its relevance for stroke-induced brain damage. Int. J. Mol. Sci. 2021, 22, 5679. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Ziemens, D.; Verkhratsky, A. On the special role of NCX in astrocytes: Translating Na+-transients into intracellular Ca2+ signals. Cell Calcium 2020, 86, 102154. [Google Scholar] [CrossRef]
- Ludhiadch, A.; Sharma, R.; Muriki, A.; Munshi, A. Role of calcium homeostasis in ischemic stroke: A review. CNS Neurol. Disord.-Drug Targets Former. Curr. Drug Targets-CNS Neurol. Disord. 2022, 21, 52–61. [Google Scholar] [CrossRef]
- Feske, S.K. Ischemic stroke. Am. J. Med. 2021, 134, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xiang, M.; Chen, C.; Ding, F.; Wang, Y.; Shang, C.; Xin, L.; Zhang, Y.; Cui, X. Glutamate excitotoxicity: Potential therapeutic target for ischemic stroke. Biomed. Pharmacother. 2022, 151, 113125. [Google Scholar] [CrossRef] [PubMed]
- Pawluk, H.; Kołodziejska, R.; Grześk, G.; Woźniak, A.; Kozakiewicz, M.; Kosinska, A.; Pawluk, M.; Grzechowiak, E.; Wojtasik, J.; Kozera, G. Increased Oxidative Stress Markers in Acute Ischemic Stroke Patients Treated with Thrombolytics. Int. J. Mol. Sci. 2022, 23, 15625. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.; Chaurasia, B.; Garg, K.; Deora, H.; Umana, G.E.; Palmisciano, P.; Scalia, G.; Lu, B. Molecular mechanisms of oxidative stress in stroke and cancer. Brain Disord. 2022, 5, 100029. [Google Scholar] [CrossRef]
- Cheng, Z.; Wang, L.; Qu, M.; Liang, H.; Li, W.; Li, Y.; Deng, L.; Zhang, Z.; Yang, G.-Y. Mesenchymal stem cells attenuate blood-brain barrier leakage after cerebral ischemia in mice. J. Neuroinflamm. 2018, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, X.; Guo, X.; Chen, D.; Jiang, L.; Qi, Y.; Shao, J.; Tao, L.; Hang, J.; Lu, G. The clinical value of serum xanthine oxidase levels in patients with acute ischemic stroke. Redox Biol. 2023, 60, 102623. [Google Scholar] [CrossRef]
- Burrage, E.N.; Coblentz, T.; Prabhu, S.S.; Childers, R.; Bryner, R.W.; Lewis, S.E.; DeVallance, E.; Kelley, E.E.; Chantler, P.D. Xanthine oxidase mediates chronic stress-induced cerebrovascular dysfunction and cognitive impairment. J. Cereb. Blood Flow. Metab. 2023, 43, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.; Fieschi, F. NADPH oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.-P.; Szanto, I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 80–91. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Seun, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Shaheryar, Z.A.; Khan, M.A.; Adnan, C.S.; Zaidi, A.A.; Hänggi, D.; Muhammad, S. Neuroinflammatory triangle presenting novel pharmacological targets for ischemic brain injury. Front. Immunol. 2021, 12, 748663. [Google Scholar] [CrossRef]
- Lian, L.; Zhang, Y.; Liu, L.; Yang, L.; Cai, Y.; Zhang, J.; Xu, S. Neuroinflammation in ischemic stroke: Focus on MicroRNA-mediated polarization of microglia. Front. Mol. Neurosci. 2021, 13, 612439. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Nie, D.; Wang, L.-J.; Qiu, H.-C.; Ma, L.; Dong, M.-X.; Tu, W.-J.; Zhao, J. Microglial polarization: Novel therapeutic strategy against ischemic stroke. Aging Dis. 2021, 12, 466. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, stroke, blood-brain barrier dysfunction, and imaging modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Tuo, Q.Z.; Zhang, S.T.; Lei, P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med. Res. Rev. 2022, 42, 259–305. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163, 144–171. [Google Scholar] [CrossRef]
- Xu, W.; Jin, W.; Zhang, X.; Chen, J.; Ren, C. Remote limb preconditioning generates a neuroprotective effect by modulating the extrinsic apoptotic pathway and TRAIL-receptors expression. Cell. Mol. Neurobiol. 2017, 37, 169–182. [Google Scholar] [CrossRef]
- Wu, J.-R.; Tuo, Q.-Z.; Lei, P. Ferroptosis, a recent defined form of critical cell death in neurological disorders. J. Mol. Neurosci. 2018, 66, 197–206. [Google Scholar] [CrossRef]
- Yan, H.-F.; Zou, T.; Tuo, Q.-Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, H. Tau as a potential therapeutic target for ischemic stroke. Aging 2019, 11, 12827. [Google Scholar] [CrossRef]
- Jin, Y.; Zhuang, Y.; Liu, M.; Che, J.; Dong, X. Inhibiting ferroptosis: A novel approach for stroke therapeutics. Drug Discov. Today 2021, 26, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jiang, N.; Su, W.; Zhuo, Y. Necroptosis: A novel pathway in neuroinflammation. Front. Pharmacol. 2021, 12, 701564. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, F.; Duo, K.; Liu, Y.; Yu, J.; Wu, Q.; Cai, Z. The Role of Necroptosis in Cerebral Ischemic Stroke. Mol. Neurobiol. 2023, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hribljan, V.; Lisjak, D.; Petrović, D.J.; Mitrečić, D. Necroptosis is one of the modalities of cell death accompanying ischemic brain stroke: From pathogenesis to therapeutic possibilities. Croat. Med. J. 2019, 60, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.-Q.; Fang, Z.; Chen, X.-L.; Yang, S.; Zhou, Y.-F.; Mao, L.; Xia, Y.-P.; Jin, H.-J.; Li, Y.-N.; You, M.-F. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain–barrier disruption after ischemic stroke. Cell Death Dis. 2019, 10, 487. [Google Scholar] [CrossRef]
- Zhou, Y.; Liao, J.; Mei, Z.; Liu, X.; Ge, J. Insight into crosstalk between ferroptosis and necroptosis: Novel therapeutics in ischemic stroke. Oxidative Med. Cell. Longev. 2021, 2021, 9991001. [Google Scholar] [CrossRef]
- Liao, S.; Apaijai, N.; Chattipakorn, N.; Chattipakorn, S.C. The possible roles of necroptosis during cerebral ischemia and ischemia/reperfusion injury. Arch. Biochem. Biophys. 2020, 695, 108629. [Google Scholar] [CrossRef]
- Naito, M.G.; Xu, D.; Amin, P.; Lee, J.; Wang, H.; Li, W.; Kelliher, M.; Pasparakis, M.; Yuan, J. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl. Acad. Sci. USA 2020, 117, 4959–4970. [Google Scholar] [CrossRef]
- Ye, X.; Song, G.; Huang, S.; Liang, Q.; Fang, Y.; Lian, L.; Zhu, S. Caspase-1: A Promising Target for Preserving Blood–Brain Barrier Integrity in Acute Stroke. Front. Mol. Neurosci. 2022, 15, 856372. [Google Scholar] [CrossRef]
- Moltrasio, C.; Romagnuolo, M.; Marzano, A.V. NLRP3 inflammasome and NLRP3-related autoinflammatory diseases: From cryopyrin function to targeted therapies. Front. Immunol. 2022, 13, 1007705. [Google Scholar] [CrossRef]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The role of the inflammasome in neurodegenerative diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Sun, Y.; Liu, S.; Yang, S.; Chen, C.; Zhang, Z.; Chu, S.; Yang, Y.; Pei, G.; Lin, M. Targeting pyroptosis as a preventive and therapeutic approach for stroke. Cell Death Discov. 2023, 9, 155. [Google Scholar] [CrossRef]
- Koehler, R.C.; Dawson, V.L.; Dawson, T.M. Targeting parthanatos in ischemic stroke. Front. Neurol. 2021, 12, 662034. [Google Scholar] [CrossRef] [PubMed]
- Prokhorova, E.A.; Egorshina, A.Y.; Zhivotovsky, B.; Kopeina, G.S. The DNA-damage response and nuclear events as regulators of nonapoptotic forms of cell death. Oncogene 2020, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kumar, A.; Mir, K.U.I.; Yadav, V.; Chauhan, S.S. Pleiotropic role of PARP1: An overview. 3 Biotech 2022, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Luo, W.; Wang, Y. Emerging role of PARP-1 and PARthanatos in ischemic stroke. J. Neurochem. 2022, 160, 74–87. [Google Scholar] [CrossRef]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Merelli, A.; Repetto, M.; Lazarowski, A.; Auzmendi, J. Hypoxia, oxidative stress, and inflammation: Three faces of neurodegenerative diseases. J. Alzheimer’s Dis. 2021, 82, S109–S126. [Google Scholar] [CrossRef]
- Fricker, M.; Neher, J.J.; Zhao, J.-W.; Théry, C.; Tolkovsky, A.M.; Brown, G.C. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J. Neurosci. 2012, 32, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Théry, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef]
- Kanduła, Z.; Lewandowski, K. Calreticulin—A multifaced protein. Postęp. Hig. Med. Dośw. 2021, 75, 328–336. [Google Scholar] [CrossRef]
- Konishi, H.; Koizumi, S.; Kiyama, H. Phagocytic astrocytes: Emerging from the shadows of microglia. Glia 2022, 70, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, J.; Zhang, Y.; Huang, Y.; Chen, D.; Shi, Z.; Smith, A.D.; Li, W.; Gao, Y. Central nervous system diseases related to pathological microglial phagocytosis. CNS Neurosci. Ther. 2021, 27, 528–539. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef]
- Adhami, F.; Liao, G.; Morozov, Y.M.; Schloemer, A.; Schmithorst, V.J.; Lorenz, J.N.; Dunn, R.S.; Vorhees, C.V.; Wills-Karp, M.; Degen, J.L. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. 2006, 169, 566–583. [Google Scholar] [CrossRef]
- Hwang, J.-Y.; Gertner, M.; Pontarelli, F.; Court-Vazquez, B.; Bennett, M.V.L.; Ofengeim, D.; Zukin, R.S. Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 2017, 24, 317–329. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Wang, B.; Wang, L.; Abraham, N.; Tao, K.; Huang, L.; Shi, W.; Dong, Y.; Qu, Y. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE 1 signalings. J. Pineal Res. 2017, 62, e12395. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Mahato, R.I.; Lee, M. Hypoxia-specific gene expression for ischemic disease gene therapy. Adv. Drug Deliv. Rev. 2009, 61, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Geng, X.; Warren, J.; Cosky, E.E.P.; Kaura, S.; Stone, C.; Li, F.; Ding, Y. Hypoxia inducible factor-1α (HIF-1α) mediates NLRP3 inflammasome-dependent-pyroptotic and apoptotic cell death following ischemic stroke. Neuroscience 2020, 448, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Hu, G.; Yao, Q.; Wu, J.; He, Z.; Law, B.Y.-K.; Hu, G.; Zhou, X.; Du, J.; Wu, A. Microglia autophagy in ischemic stroke: A double-edged sword. Front. Immunol. 2022, 13, 6825. [Google Scholar] [CrossRef] [PubMed]
- Tsivgoulis, G.; Katsanos, A.H.; Sandset, E.C.; Turc, G.; Nguyen, T.N.; Bivard, A.; Fischer, U.; Khatri, P. Thrombolysis for acute ischaemic stroke: Current status and future perspectives. Lancet Neurol. 2023, 22, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Barreto, A.D. Intravenous thrombolytics for ischemic stroke. Neurotherapeutics 2011, 8, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, Y.; Ma, Q.; Chen, H.; Liu, B.; Yang, Y.; Zhu, J.; Zhao, S.; Jin, X.; Li, Y. Efficacy and safety of recombinant human prourokinase in acute ischemic stroke: A phase IIa randomized clinical trial. Transl. Stroke Res. 2022, 13, 995–1004. [Google Scholar] [CrossRef]
- Menon, B.K.; Buck, B.H.; Singh, N.; Deschaintre, Y.; Almekhlafi, M.A.; Coutts, S.B.; Thirunavukkarasu, S.; Khosravani, H.; Appireddy, R.; Moreau, F. Intravenous tenecteplase compared with alteplase for acute ischaemic stroke in Canada (AcT): A pragmatic, multicentre, open-label, registry-linked, randomised, controlled, non-inferiority trial. Lancet 2022, 400, 161–169. [Google Scholar] [CrossRef]
- Gusev, E.I.; Martynov, M.Y.; Nikonov, A.A.; Shamalov, N.A.; Semenov, M.P.; Gerasimets, E.A.; Yarovaya, E.B.; Semenov, A.M.; Archakov, A.I.; Markin, S.S. Non-immunogenic recombinant staphylokinase versus alteplase for patients with acute ischaemic stroke 4·5 h after symptom onset in Russia (FRIDA): A randomised, open label, multicentre, parallel-group, non-inferiority trial. Lancet Neurol. 2021, 20, 721–728. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, Y.; Zhang, L.; Zhang, Y.; Treurniet, K.M.; Chen, W.; Peng, Y.; Han, H.; Wang, J.; Wang, S. Endovascular thrombectomy with or without intravenous alteplase in acute stroke. N. Engl. J. Med. 2020, 382, 1981–1993. [Google Scholar] [CrossRef]
- Zi, W.; Qiu, Z.; Li, F.; Sang, H.; Wu, D.; Luo, W.; Liu, S.; Yuan, J.; Song, J.; Shi, Z. Effect of endovascular treatment alone vs intravenous alteplase plus endovascular treatment on functional independence in patients with acute ischemic stroke: The DEVT randomized clinical trial. JAMA 2021, 325, 234–243. [Google Scholar] [CrossRef]
- Collen, D.; Lijnen, H.R. Thrombolytic agents. Thromb. Haemost. 2005, 93, 627–630. [Google Scholar] [CrossRef]
- Warner, J.J.; Harrington, R.A.; Sacco, R.L.; Elkind, M.S. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke. Stroke 2019, 50, 3331–3332. [Google Scholar] [CrossRef]
- Goyal, M.; Menon, B.K.; Van Zwam, W.H.; Dippel, D.W.; Mitchell, P.J.; Demchuk, A.M.; Dávalos, A.; Charles, B.L.M.M.; Aad, V.D.L.; Maria, A.D.M.; et al. Endovascular thrombectomy after large-vessel ischaemic stroke: A meta-analysis of individual patient data from five randomised trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef]
- Dalkara, T.; Arsava, E.M. Can restoring incomplete microcirculatory reperfusion improve stroke outcome after thrombolysis? J. Cereb. Blood Flow Metab. 2012, 32, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Sun, X.; Cheng, H.; Burgin, W.S.; Luo, W.; Jia, W.; Liu, Y.; He, W.; Geng, X.; Zhu, L. Combined Approach to Eptifibatide and Thrombectomy in Acute Ischemic Stroke Because of Large Vessel Occlusion: A matched-control analysis. Stroke 2022, 53, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Barreto, A.D.; Ford, G.A.; Shen, L.; Pedroza, C.; Tyson, J.; Cai, C.; Rahbar, M.H.; Grotta, J.C. Randomized, multicenter trial of ARTSS-2 (argatroban with recombinant tissue plasminogen activator for acute stroke). Stroke 2017, 48, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M. Safety and tolerability, pharmacokinetics, and pharmacodynamics of ACT017, an antiplatelet GPVI (glycoprotein VI) Fab: First-in-human healthy volunteer trial. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Shuai, J.; Gong, Z.; Huang, L.; Liu, J.; Tang, K.; Duan, Z.; Ni, H.; Liu, Y.; Xie, F.; Yan, D. Effect of intravenous tirofiban vs placebo before endovascular thrombectomy on functional outcomes in large vessel occlusion stroke: The RESCUE BT randomized clinical trial. JAMA 2022, 328, 543–553. [Google Scholar]
- Hackam, D.G.; David, S.J. Antiplatelet therapy in ischemic stroke and transient ischemic attack: An overview of major trials and meta-analyses. Stroke 2019, 50, 773–778. [Google Scholar] [CrossRef]
- Stringberg, A.; Ryan, C.; Kathryn, Q.; Syed, N.H. Update on dual antiplatelet therapy for secondary stroke prevention. Mo. Med. 2019, 116, 303. [Google Scholar]
- Mehta, S.R.; Yusuf, S.; Peters, R.J.; Bertrand, M.E.; Lewis, B.S.; Natarajan, M.K.; Malmberg, K.; Rupprecht, H.-J.; Zhao, F.; Chrolavicius, S. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001, 358, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.J.; Mehta, S.R.; Fox, K.A.; Zhao, F.; Lewis, B.S.; Kopecky, S.L.; Diaz, R.; Commerford, P.J.; Valentin, V.; Yusuf, S. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: Observations from the Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) study. Circulation 2003, 108, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S. Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators: Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N. Engl. J. Med. 2001, 345, 494–502. [Google Scholar] [PubMed]
- Diener, H.-C.; Julien, B.; Lawrence, B.M.; Claudio, C.; Laszlo, C.; Markku, K.; Didier, L.; Jordi, M.-G.; Hans-Jürgen, R. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): Randomised, double-blind, placebo-controlled trial. Lancet 2004, 364, 331–337. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Fox, K.A.; Hacke, W.; Berger, P.B.; Black, H.R.; Boden, W.E.; Cacoub, P.; Cohen, E.A.; Creager, M.A.; Easton, D.J.; et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N. Engl. J. Med. 2006, 354, 1706–1717. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Topol, E.J.T.; CHARISMA, Executive Committee. Clopidogrel added to aspirin versus aspirin alone in secondary prevention and high-risk primary prevention: Rationale and design of the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial. Am. Heart J. 2004, 148, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Turaj, W.; Słowik, A.; Dziedzic, T.; Pułyk, R.; Adamski, M.; Strojny, J.; Szczudlik, A. Increased plasma fibrinogen predicts one-year mortality in patients with acute ischemic stroke. J. Neurol. Sci. 2006, 246, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J.; Levy, D.E.; Wasiewski, W.W.; Pancioli, A.M.; Demchuk, A.M.; Trammel, J.; Demaerschalk, B.M.; Kaste, M.; Albers, G.W.; Ringelstein, E.B. Hyperfibrinogenemia and functional outcome from acute ischemic stroke. Stroke 2009, 40, 1687–1691. [Google Scholar] [CrossRef]
- Sherman, D.G.; Atkinson, R.P.; Chippendale, T.; Levin, K.A.; Ng, K.; Futrell, N.; Hsu, C.Y.; Levy, D.E. Intravenous ancrod for treatment of acute ischemic stroke: The STAT study: A randomized controlled trial. JAMA 2000, 283, 2395–2403. [Google Scholar] [CrossRef]
- Hennerici, M.G.; Kay, R.; Bogousslavsky, J.; Lenzi, G.L.; Verstraete, M.; Orgogozo, J.M. Intravenous ancrod for acute ischaemic stroke in the European Stroke Treatment with Ancrod Trial: A randomised controlled trial. Lancet 2006, 368, 1871–1878. [Google Scholar] [CrossRef]
- Woodward, M.; Lowe, G.D.; Campbell, D.J.; Colman, S.; Rumley, A.; Chalmers, J.; Neal, B.C.; Patel, A.; Jenkins, A.J.; Kemp, B.E. Associations of inflammatory and hemostatic variables with the risk of recurrent stroke. Stroke 2005, 36, 2143–2147. [Google Scholar] [CrossRef]
- Chen, J.; Dalong, S.; Mingli, L.; Shufan, Z.; Chuancheng, R. Defibrinogen therapy for acute ischemic stroke: 1332 consecutive cases. Sci. Rep. 2018, 8, 9489. [Google Scholar] [CrossRef]
- Hao, Z.; Ming, L.; Carl, C.; Joanna, W.M.; Sen, L.; Xiaoling, Z. Fibrinogen depleting agents for acute ischaemic stroke. Cochrane Database Syst. Rev. 2012, 6, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; James, T.; Warren, W.W.; Team, A.S.P.A.S. Ancrod for acute ischemic stroke: A new dosing regimen derived from analysis of prior ancrod stroke studies. J. Stroke Cerebrovasc. Dis. 2009, 18, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Theodore, K.k.; Jeffrey, J.R.; Rodrigo, M.; Alba, R.I.; Stephen, P.G.; Thomas, F.B.; McKeown, P.; Cahill, D.W.; Nishino, H.; et al. Neural transplantation as an experimental treatment modality for cerebral ischemia. Neurosci. Biobehav. Rev. 1997, 21, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Kuniyasu, N.; Maxim, M.; Mari, D.; Cesar, B.V. Cell-based therapy for stroke: Musing with muse cells. Stroke 2020, 51, 2854–2862. [Google Scholar] [CrossRef]
- Aizman, I.; Ciara, T.C.; Michael, M.; Casey, C.C. Extracellular matrix produced by bone marrow stromal cells and by their derivative, SB623 cells, supports neural cell growth. J. Neurosci. Res. 2009, 87, 3198–3206. [Google Scholar] [CrossRef]
- Sun, J.; Huang, Y.; Gong, J.; Wang, J.; Fan, Y.; Cai, J.; Wang, Y.; Qiu, Y.; Wei, Y.; Xiong, C.; et al. Transplantation of hPSC-derived pericyte-like cells promotes functional recovery in ischemic stroke mice. Nat. Commun. 2020, 11, 5196. [Google Scholar] [CrossRef]
- Redondo-Castro, E.; Cunningham, C.; Miller, J.; Martuscelli, L.; Aoulad-Ali, S.; Rothwell, N.J.; Kielty, C.M.; Allan, S.M.; Pinteaux, E. Interleukin-1 primes human mesenchymal stem cells towards an anti-inflammatory and pro-trophic phenotype in vitro. Stem Cell Res. Ther. 2017, 8, 79. [Google Scholar] [CrossRef]
- Solari, C.; Petrone, M.V.; Vazquez Echegaray, C.; Cosentino, M.S.; Waisman, A.; Francia, M.; Barañao, L.; Miriuka, S.; Guberman, A. Superoxide dismutase 1 expression is modulated by the core pluripotency transcription factors Oct4, Sox2 and Nanog in embryonic stem cells. Mech. Dev. 2018, 154, 116–121. [Google Scholar] [CrossRef]
- Chopp, M.; Yi, L.; Zheng Gang, Z. Mechanisms underlying improved recovery of neurological function after stroke in the rodent after treatment with neurorestorative cell-based therapies. Stroke 2009, 40 (Suppl. S1), S143–S145. [Google Scholar] [CrossRef]
- Prasad, K.; Sharma, A.; Garg, A.; Mohanty, S.; Bhatnagar, S.; Johri, S.; Singh, K.K.; Nair, V.; Sarkar, R.S.; Gorthi, S.P.; et al. Intravenous autologous bone marrow mononuclear stem cell therapy for ischemic stroke: A multicentric, randomized trial. Stroke 2014, 45, 3618–3624. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Hong, J.M.; Moon, G.J.; Lee, P.H.; Ahn, Y.H.; Bang, O.Y. A long-term follow-up study of intravenous autologous mesenchymal stem cell transplantation in patients with ischemic stroke. Stem Cells 2010, 28, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, W.; Jiang, R.; Qian, H.; Chen, M.; Hu, J.; Cao, W.; Han, C.; Chen, Y. Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp. Mol. Pathol. 2006, 80, 267–274. [Google Scholar] [CrossRef]
- Kawabori, M.; Shichinohe, H.; Kuroda, S.; Houkin, K. Clinical Trials of Stem Cell Therapy for Cerebral Ischemic Stroke. Int. J. Mol. Sci. 2020, 21, 7380. [Google Scholar] [CrossRef]
- Lai, R.C.; Tan, S.S.; Teh, B.J.; Sze, S.K.; Arslan, F.; de Kleijn, D.P.; Choo, A.; Lim, S.K. Proteolytic Potential of the MSC Exosome Proteome: Implications for an Exosome-Mediated Delivery of Therapeutic Proteasome. Int. J. Proteom. 2012, 2012, 971907. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Wallace, B.K.; Yuen, N.; Jenkins, D.P.; Wulff, H.; O’Donnell, M.E. Blood–brain barrier KCa3. 1 channels: Evidence for a role in brain Na uptake and edema in ischemic stroke. Stroke 2015, 46, 237–244. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Grössinger, E.M.; Horiuchi, M.; Davis, K.W.; Jin, L.W.; Maezawa, I.; Wulff, H. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017, 65, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Kolski-Andreaco, A.; Sankaranarayanan, A.; Sabatier, J.M.; Shakkottai, V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr. Med. Chem. 2007, 14, 1437–1457. [Google Scholar] [CrossRef] [PubMed]
- Angeli, F.; Paolo, V.; Reboldi, G.P.; Roberto, G.; Maurizio, B.; Jan, S.A.; Carlo, P. Calcium channel blockade to prevent stroke in hypertension: A meta-analysis of 13 studies with 103,793 subjects. Am. J. Hypertens. 2004, 17, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Infeld, B.; Davis, S.M.; Donnan, G.A.; Yasaka, M.; Lichtenstein, M.; Mitchell, P.J.; Fitt, G.J. Nimodipine and Perfusion Changes After Stroke. Stroke 1999, 30, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Segura, T.; Calleja, S.; Joaquin, J. Recommendations and treatment strategies for the management of acute ischemic stroke. Expert Opin. Pharmacother. 2008, 9, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, K.E.; Peter, S.K.; Howard, L.J. The use-dependent sodium channel blocker mexiletine is neuroprotective against global ischemic injury. Brain Res. 2001, 898, 281–287. [Google Scholar] [CrossRef]
- Woodruff, T.M.; John, T.; Sung-Chun, T.; Christopher, S.G.; Stephen, T.M.; Thiruma, A.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Zivin, J.A. Ebselen, a seleno-organic antioxidant, is neuroprotective after embolic strokes in rabbits: Synergism with low-dose tissue plasminogen activator. Stroke 2003, 34, 2013–2018. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ogawa, A.; Yoshimoto, T.; Kikuchi, H.; Sano, K.; Saito, I.; Yamaguchi, T.; Yasuhara, H. Ebselen in acute middle cerebral artery occlusion: A placebo-controlled, double-blind clinical trial. Cerebrovasc. Dis. 1999, 9, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Saito, I.; Asano, T.; Sano, K.; Takakura, K.; Abe, H.; Yoshimoto, T.; Kikuchi, H.; Ohta, T.; Ishibashi, S. Neuroprotective effect of an antioxidant, ebselen, in patients with delayed neurological deficits after aneurysmal subarachnoid hemorrhage. Neurosurgery 1998, 42, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in acute ischemic stroke: A placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Aoki, J.; Sakamoto, Y.; Kobayashi, K.; Sakai, K.; Inoue, T.; Iguchi, Y.; Shibazaki, K. Administration of edaravone, a free radical scavenger, during t-PA infusion can enhance early recanalization in acute stroke patients—A preliminary study. J. Neurol. Sci. 2012, 313, 132–136. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Araujo, D.M.; Song, D.; Wei, J.; Zivin, J.A. Neuroprotective effects of the spin trap agent disodium-[(tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (generic NXY-059) in a rabbit small clot embolic stroke model: Combination studies with the thrombolytic tissue plasminogen activator. Stroke 2002, 33, 1411–1415. [Google Scholar] [CrossRef]
- Lees, K.R.; Zivin, J.A.; Ashwood, T.; Davalos, A.; Davis, S.M.; Diener, H.C.; Grotta, J.; Lyden, P.; Shuaib, A.; Hårdemark, H.G.; et al. NXY-059 for acute ischemic stroke. N. Engl. J. Med. 2006, 354, 588–600. [Google Scholar] [CrossRef]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [CrossRef]
- Zaman, K.; Ryu, H.; Hall, D.; O’Donovan, K.; Lin, K.-I.; Miller, M.P.; Marquis, J.C.; Baraban, J.M.; Semenza, G.L.; Ratan, R.R. Protection from oxidative stress–induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced dna binding of hypoxia-inducible factor-1 and atf-1/creb and increased expression of glycolytic enzymes, p21waf1/cip1, and erythropoietin. J. Neurosci. 1999, 19, 9821–9830. [Google Scholar]
- Regan, R.F.; Panter, S. Hemoglobin potentiates excitotoxic injury in cortical cell culture. J. Neurotrauma 1996, 13, 223–231. [Google Scholar] [CrossRef]
- Selim, M. Deferoxamine Mesylate. Stroke 2009, 40, S90–S91. [Google Scholar] [CrossRef]
- Majid, A. Neuroprotection in stroke: Past, present, and future. ISRN Neurol. 2014, 2014, 515716. [Google Scholar] [CrossRef]
- Antoni, D.V.; José, C.; José, A.l.-S.N.; Julio, S.J.; Joan, M.; Sonia, L.P.; Erik, C.; Steven, W.; David, S.; Wayne, C.; et al. Oral citicoline in acute ischemic stroke: An individual patient data pooling analysis of clinical trials. Stroke 2002, 33, 2850–2857. [Google Scholar]
- Cho, H.J.; Kim, Y.J. Efficacy and safety of oral citicoline in acute ischemic stroke: Drug surveillance study in 4,191 cases. Methods Find. Exp. Clin. Pharmacol. 2009, 31, 171–176. [Google Scholar] [CrossRef]
- Xu, S.-Y.; Pan, S.-Y. The failure of animal models of neuroprotection in acute ischemic stroke to translate to clinical efficacy. Med. Sci. Monit. Basic Res. 2013, 19, 37. [Google Scholar] [CrossRef]
- Westermann, B. Molecular machinery of mitochondrial fusion and fission. J. Biol. Chem. 2008, 283, 13501–13505. [Google Scholar] [CrossRef]
- Jordan, J.; Cena, V.; Prehn, J.H.M. Mitochondrial control of neuron death and its role in neurodegenerative disorders. J. Physiol. Biochem. 2003, 59, 129–144. [Google Scholar] [CrossRef]
- Muranyi, M.; Li, P.-A. Bongkrekic acid ameliorates ischemic neuronal death in the cortex by preventing cytochrome c release and inhibiting astrocyte activation. Neurosci. Lett. 2005, 384, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Forsse, A.; Nielsen, T.H.; Nygaard, K.H.; Nordström, C.-H.; Gramsbergen, J.B.; Poulsen, F.R. Cyclosporin A ameliorates cerebral oxidative metabolism and infarct size in the endothelin-1 rat model of transient cerebral ischaemia. Sci. Rep. 2019, 9, 3702. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Yu, G.; Matsukawa, N.; Xu, L.; Hess, D.C.; Sanberg, P.R.; Wang, Y. Acute functional effects of cyclosporine-A and methylprednisolone treatment in adult rats exposed to transient ischemic stroke. Life Sci. 2005, 76, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Janelidze, S.; Siesjö, B.K. The immunosuppressants cyclosporin A and FK506 equally ameliorate brain damage due to 30-min middle cerebral artery occlusion in hyperglycemic rats. Brain Res. 1999, 835, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, Y.; Furuichi, Y.; Tojo, N.; Moriguchi, A.; Maemoto, T.; Nakada, H.; Hino, M.; Matsuoka, N. Neuroprotective efficacy of FR901459, a novel derivative of cyclosporin A, in in vitro mitochondrial damage and in vivo transient cerebral ischemia models. Brain Res. 2007, 1149, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-L.; Manenko, A.; Ye, Z.; Sun, X.-J.; Hu, Q. Hypoxia therapy—A new hope for the treatment of mitochondrial dysfunctions. Med. Gas Res. 2016, 6, 172. [Google Scholar]
- Huang, P.; Galloway, C.A.; Yoon, Y. Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS ONE 2011, 6, e20655. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Gräber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Zhao, Y.X.; Cui, M.; Chen, S.F.; Dong, Q.; Liu, X.Y. Amelioration of ischemic mitochondrial injury and Bax-dependent outer membrane permeabilization by Mdivi-1. CNS Neurosci. Ther. 2014, 20, 528–538. [Google Scholar] [CrossRef]
- Grohm, J.; Kim, S.W.; Mamrak, U.; Tobaben, S.; Cassidy-Stone, A.; Nunnari, J.; Plesnila, N.; Culmsee, C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012, 19, 1446–1458. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, S.; Li, Y.; Che, L.; Zhao, Q. A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci. Lett. 2013, 535, 104–109. [Google Scholar] [CrossRef]
- Magkou, D.; Tziomalos, K. Antidiabetic treatment, stroke severity and outcome. World J. Diabetes 2014, 5, 84. [Google Scholar] [CrossRef]
- Zhao, M.; Li, X.W.; Chen, D.Z.; Hao, F.; Tao, S.X.; Yu, H.Y.; Cheng, R.; Liu, H. Neuro-protective role of metformin in patients with acute stroke and type 2 diabetes mellitus via AMPK/mammalian target of rapamycin (mTOR) signaling pathway and oxidative stress. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 2186. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Sheth, K.N.; Kimberly, W.T.; Simard, J.M. Profile of intravenous glyburide for the prevention of cerebral edema following large hemispheric infarction: Evidence to date. Drug Des. Dev. Ther. 2018, 12, 2539–2552. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Sehgal, K.; Sodnar, B.; Bhosale, N.; Sarmah, D.; Datta, A.; Chaudhary, A.; Kalia, K.; Wang, X.; Bhattacharya, P. Drug repurposing for stroke intervention. Drug Discov. Today 2022, 27, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V.A.; Bharatam, P.V. SAR and computer-aided drug design approaches in the discovery of peroxisome proliferator-activated receptor γ activators: A perspective. J. Comput. Med. 2013, 2013, 406049. [Google Scholar] [CrossRef]
- Wang, J.-G.; Li, Y.; Franklin, S.S.; Safar, M. Prevention of Stroke and Myocardial Infarction by Amlodipine and Angiotensin Receptor Blockers. Hypertension 2007, 50, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Twede, V.D.; Miljanich, G.; Olivera, B.M.; Bulaj, G. Neuroprotective and cardioprotective conopeptides: An emerging class of drug leads. Curr. Opin. Drug Discov. Devel 2009, 12, 231–239. [Google Scholar]
- Buchan, A.M.; Gertler, S.Z.; Li, H.; Xue, D.; Huang, Z.G.; Chaundy, K.E.; Barnes, K.; Lesiuk, H.J. A selective N-type Ca(2+)-channel blocker prevents CA1 injury 24 h following severe forebrain ischemia and reduces infarction following focal ischemia. J. Cereb. Blood Flow. Metab. 1994, 14, 903–910. [Google Scholar] [CrossRef]
- Colbourne, F.; Li, H.; Buchan, A.M.; Clemens, J.A. Continuing postischemic neuronal death in CA1: Influence of ischemia duration and cytoprotective doses of NBQX and SNX-111 in rats. Stroke 1999, 30, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Valentino, K.; Newcomb, R.; Gadbois, T.; Singh, T.; Bowersox, S.; Bitner, S.; Justice, A.; Yamashiro, D.; Hoffman, B.B.; Ciaranello, R.; et al. A selective N-type calcium channel antagonist protects against neuronal loss after global cerebral ischemia. Proc. Natl. Acad. Sci. USA 1993, 90, 7894–7897. [Google Scholar] [CrossRef]
- Dale, E.; Staal, R.G.; Eder, C.; Möller, T. KCa 3.1-a microglial target ready for drug repurposing? Glia 2016, 64, 1733–1741. [Google Scholar] [CrossRef]
- Staal, R.G.W.; Weinstein, J.R.; Nattini, M.; Cajina, M.; Chandresana, G.; Möller, T. Senicapoc: Repurposing a Drug to Target Microglia KCa3.1 in Stroke. Neurochem. Res. 2017, 42, 2639–2645. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Heal, C.; Vail, A.; Jeans, A.R.; Westendorp, W.F.; Nederkoorn, P.J.; Van de Beek, D.; Kalra, L.; Montaner, J.; Woodhead, M. Antibiotic class and outcome in post-stroke infections: An individual participant data pooled analysis of VISTA-Acute. Front. Neurol. 2019, 10, 504. [Google Scholar] [CrossRef] [PubMed]
- Yong, V.W.; Wells, J.; Giuliani, F.; Casha, S.; Power, C.; Metz, L.M. The promise of minocycline in neurology. Lancet Neurol. 2004, 3, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Uchikado, H.; Morioka, M.; Murai, Y.; Tanaka, E. Clinical neuroprotective drugs for treatment and prevention of stroke. Int. J. Mol. Sci. 2012, 13, 7739–7761. [Google Scholar] [CrossRef] [PubMed]
- Peyravian, N.; Dikici, E.; Deo, S.; Toborek, M.; Daunert, S. Opioid antagonists as potential therapeutics for ischemic stroke. Prog. Neurobiol. 2019, 182, 101679. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Chang, C.-Y.; Shih, K.-C.; Hung, C.-J.; Wang, Y.-Y.; Lin, S.-Y.; Chen, W.-Y.; Kuan, Y.-H.; Liao, S.-L.; Wang, W.-Y. β-Funaltrexamine displayed anti-inflammatory and neuroprotective effects in cells and rat model of stroke. Int. J. Mol. Sci. 2020, 21, 3866. [Google Scholar] [CrossRef]
- Anttila, J.E.; Albert, K.; Wires, E.S.; Mätlik, K.; Loram, L.C.; Watkins, L.R.; Rice, K.C.; Wang, Y.; Harvey, B.K.; Airavaara, M. Post-stroke Intranasal (+)-Naloxone Delivery Reduces Microglial Activation and Improves Behavioral Recovery from Ischemic Injury. eNeuro 2018, 5, ENEURO.0395-17.2018. [Google Scholar] [CrossRef]
- Wood, P.L.; Hawkinson, J.E. N-methyl-D-aspartate antagonists for stroke and head trauma. Expert Opin. Investig. Drugs 1997, 6, 389–397. [Google Scholar] [CrossRef]
- Hantson, L.; Tritsmans, L.; Crabbé, R.; Gheuens, J.; Van Rooy, P. The safety and tolerability of single intravenous doses of lubeluzole (Prosynap) in healthy volunteers. Int. J. Clin. Pharmacol. Ther. 1997, 35, 491–495. [Google Scholar] [PubMed]
- Kotoda, M.; Sohei, H.; Tadahiko, I.; Kazuha, M.; Takashi, M. Amiodarone exacerbates brain injuries after hypoxic–ischemic insult in mice. BMC Neurosci. 2019, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, N.G.; Stig, B.; Anil, S.; Björn, C.; Thierry, R.; Timothy, A.; Lennart, C.; Group, C.S. The Clomethiazole Acute Stroke Study (CLASS): Efficacy results in 545 patients classified as total anterior circulation syndrome (TACS). J. Stroke Cerebrovasc. Dis. 1999, 8, 231–239. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
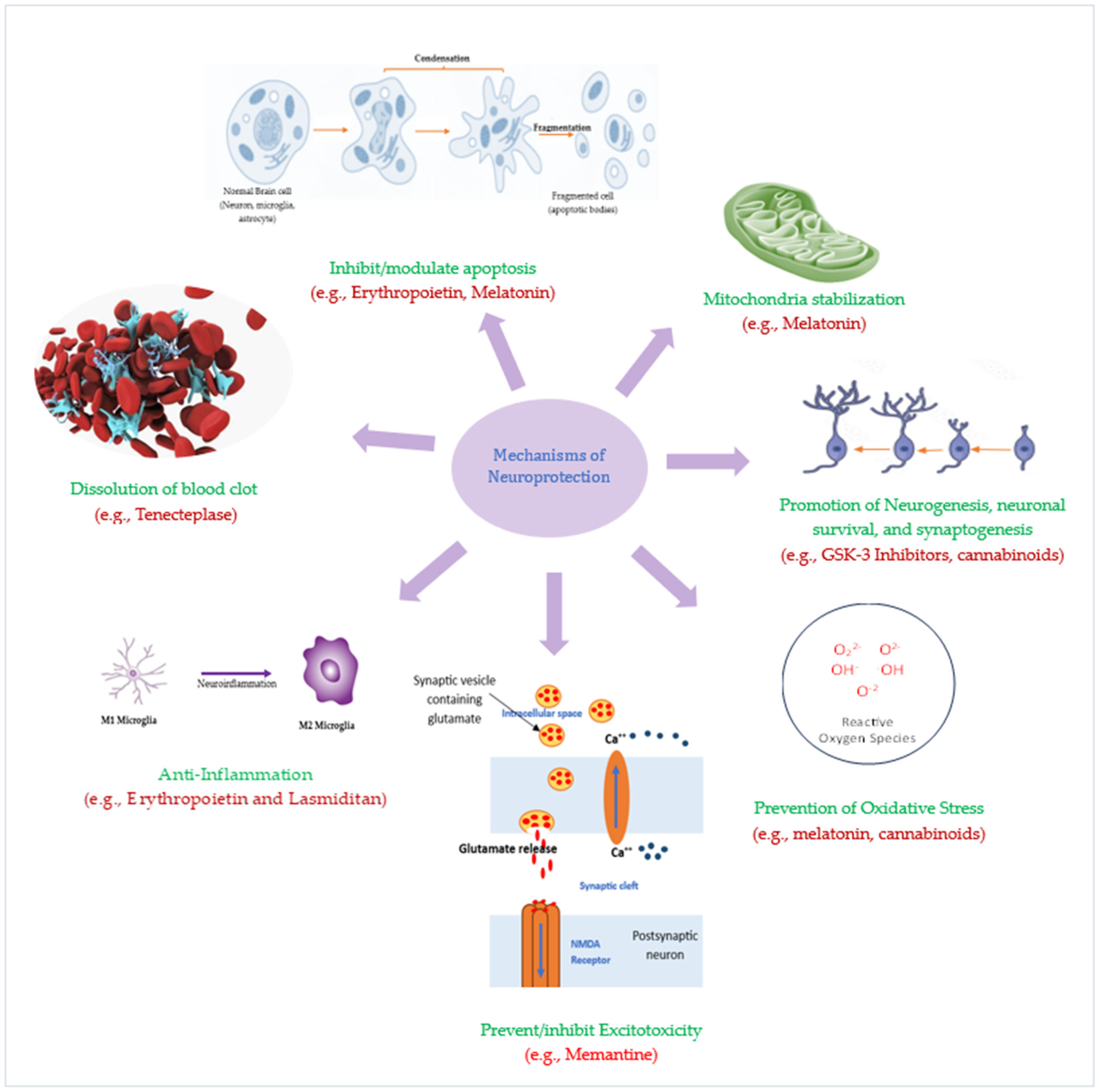{kind=link}
| S/No | Identifier | Stem Cell Type | Study Centre | Study Aim | Clinical Trial Phase | Study Status |
|---|---|---|---|---|---|---|
| 1 | NCT00875654 | Autologous mesenchymal stem cells | France, Europe | Feasibility and tolerance | Phase I | Completed |
| 2 | NCT05008588 | Umbilical cord mesenchymal stem cells (whole cell and conditioned medium) | Indonesia, Asia | Safety and Efficacy | Phase I/II | Ongoing |
| 3 | NCT02117635 | Allogeneic human neural stem cell | United Kingdom, Europe | Efficacy | Phase II | Completed |
| 4 | NCT01501773 | Autologous bone marrow stem cell | India, Asia | Safety, feasibility, and efficacy | Phase II | Completed |
| 5 | NCT04811651 | Umbilical cord-derived mesenchymal stem cells | China, Asia | Safety and efficacy | Phase II | Recruiting |
| 6 | NCT01716481 | Autologous mesenchymal stem cells | South Korea, Asia | Neuroprotection | Phase III | Completed |
| 7 | NCT04631406 | Neural stem cell | California, North America | Safety and tolerability profile | Phase I/II | Recruiting |
| 8 | NCT03356821 | Stromal cells (intranasal) | Netherlands, Europe. | Safety and feasibility | Phase I/II | Completed |
| 9 | NCT05292625 | Umbilical cord-derived MSC (intrathecal and intravenous) | Vietnam, Asia | Safety and efficacy | Phase I/II | Recruiting |
| 11 | NCT06138210 | Induced pluripotent stem cell | China, Asia | Safety and preliminary efficacy | Phase I | Starts 2024 |
| 12 | NCT00859014 | Autologous mononuclear bone marrow cells (intravenous) | Houston, North America | Safety and tolerability | Phase I | Completed |
| 13 | NCT02178657 | Autologous mononuclear bone marrow cells (intra-arterial) | Spain, Europe | Safety and neuroprotection | Phase II | Ongoing |
| 14 | NCT00950521 | CD34+ stem cell (intracerebral implantation) | Taiwan, Asia | Efficacy | Phase II | Completed |
| 15 | NCT00535197 | Autologous CD34+ subset bone marrow stem cell (intra-arterial infusion) | London, Europe | Safety and tolerability | Phase I/II | Completed |
| Drugs | Drug Class | Approved Indication(s) | Pathophysiology Target(s) | |
|---|---|---|---|---|
| Wang et al., 2007 [155] | Amlodipine | Calcium channel blocker (CCB) | Hypertension, Angina | Excitotoxicity |
| Smith et al., 2019 [162] | Azithromycin | Macrolide antibiotic | Sinusitis, conjunctivitis, community-acquired pneumonia | Neuroinflammation |
| Cho & Kim, 2009 [134] | Citicoline | Neurotropic | Parkinsonism, head injury | Apoptosis |
| Forsse et al., 2019 [139] | Cyclosporin A | Immunomodulator | Post-transplant immunosuppression | Mitochondrial dysfunction secondary to oxidative stress and excitotoxicity |
| Selim et al., 2009 [131] | Deferoxamine | Chelate | Iron poisoning | Excitotoxicity, oxidative stress, ferroptosis |
| King et al., 2018 [152] | Glyburide | Sulfonylureas antidiabetic | Diabetes mellitus | Neuroinflammation and oxidative stress |
| Zhao et al., 2019 [151] | Metformin | Biguanide antidiabetic | Diabetes mellitus, polycystic ovary syndrome | Neuroinflammation and oxidative stress |
| Kikuchi et al., 2012 [164] | Minocycline | Tetracycline antibiotic | Inflammatory acne, gonococcal infections, urinary tract infection | Excitotoxicity, neuroinflammation, apoptosis, and oxidative stress |
| Anttila et al., 2018 [167] | Naloxone | Opioid receptor antagonist | Opioid overdose | Neuroinflammation |
| Staal et al., 2017 [161] | Senicapoc | Calcium-dependent potassium channel (KCa3.1) blocker | Sickle cell anemia | Neuroinflammation |
| Twede et al., 2009 [156] | Ziconotide | Conopeptide, analgesic, N-type CCB | Chronic intractable pain | Excitotoxicity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salaudeen, M.A.; Bello, N.; Danraka, R.N.; Ammani, M.L. Understanding the Pathophysiology of Ischemic Stroke: The Basis of Current Therapies and Opportunity for New Ones. Biomolecules 2024, 14, 305. https://doi.org/10.3390/biom14030305
Salaudeen MA, Bello N, Danraka RN, Ammani ML. Understanding the Pathophysiology of Ischemic Stroke: The Basis of Current Therapies and Opportunity for New Ones. Biomolecules. 2024; 14(3):305. https://doi.org/10.3390/biom14030305
Chicago/Turabian StyleSalaudeen, Maryam A., Nura Bello, Rabiu N. Danraka, and Maryam L. Ammani. 2024. "Understanding the Pathophysiology of Ischemic Stroke: The Basis of Current Therapies and Opportunity for New Ones" Biomolecules 14, no. 3: 305. https://doi.org/10.3390/biom14030305
APA StyleSalaudeen, M. A., Bello, N., Danraka, R. N., & Ammani, M. L. (2024). Understanding the Pathophysiology of Ischemic Stroke: The Basis of Current Therapies and Opportunity for New Ones. Biomolecules, 14(3), 305. https://doi.org/10.3390/biom14030305