
Variations of the NodB Architecture Are Attuned to Functional Specificities into and beyond the Carbohydrate Esterase Family 4

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Construction
2.2. Clustering Analysis
2.3. Topology Analysis
2.4. Correspondence Analysis
2.5. Function-Related Sequence Conservations Mapped on the NodB Fold
3. Results
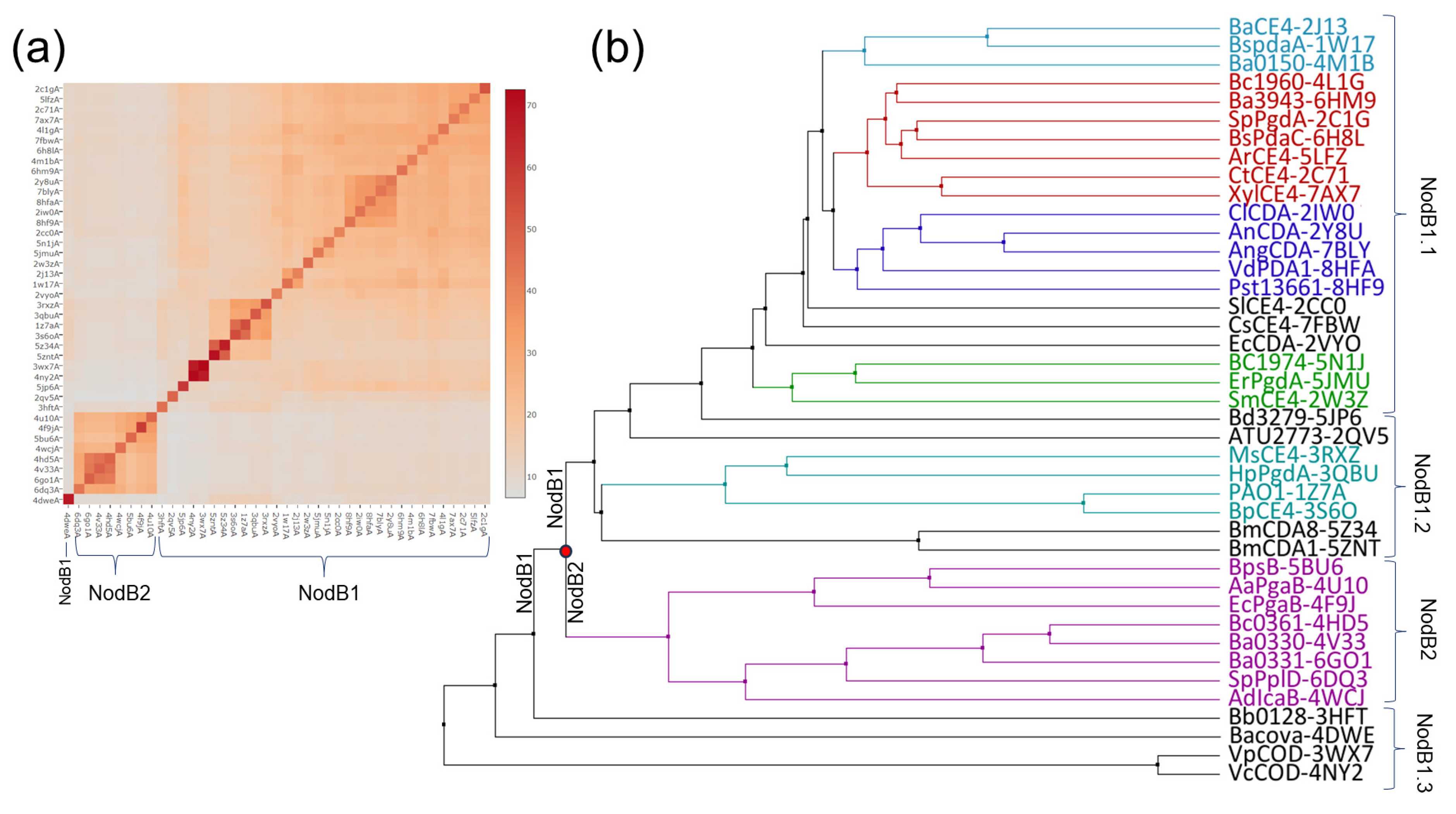
3.1. Structural Classification and Clustering Analysis of the NodB Domain
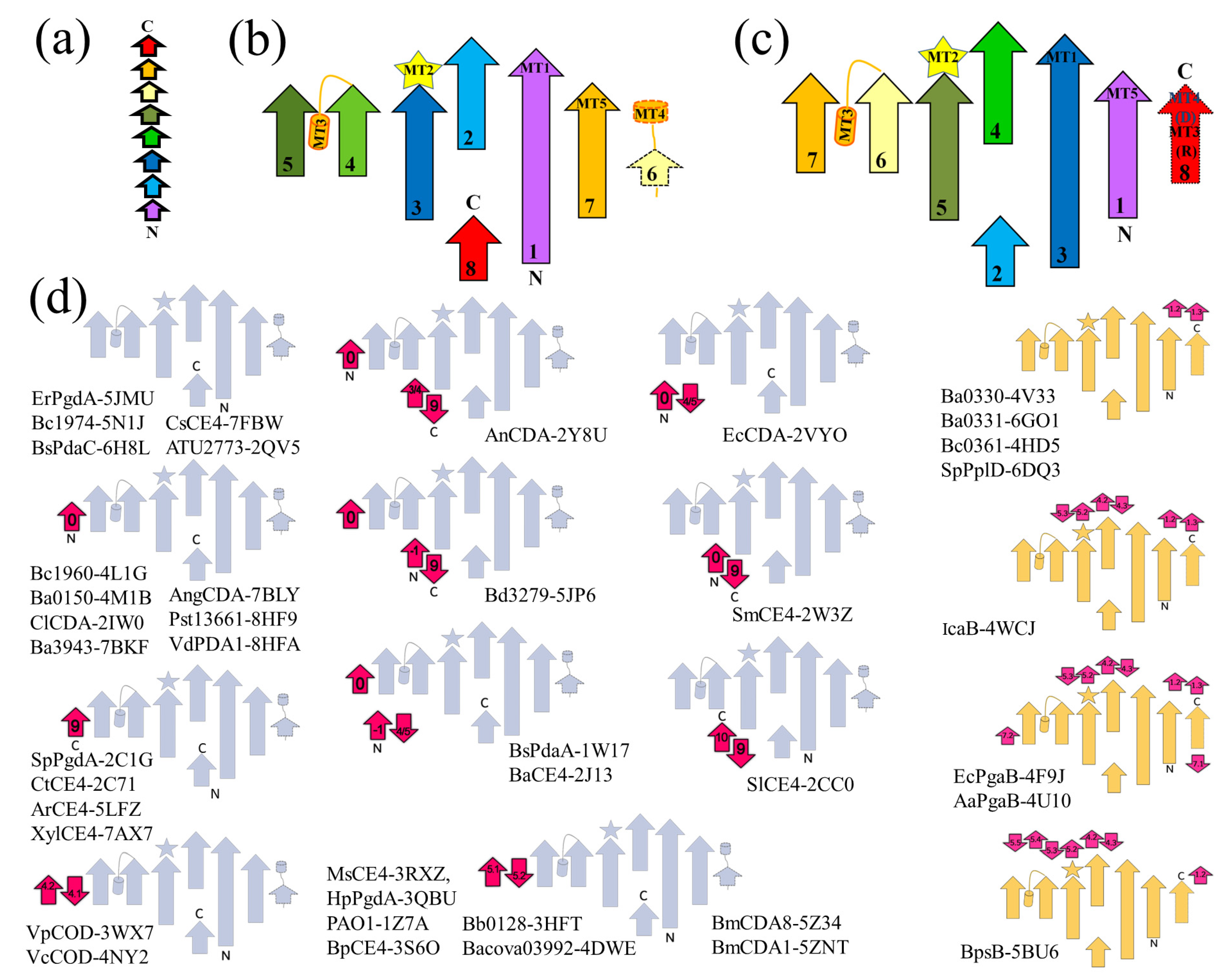
3.2. Topology Analysis
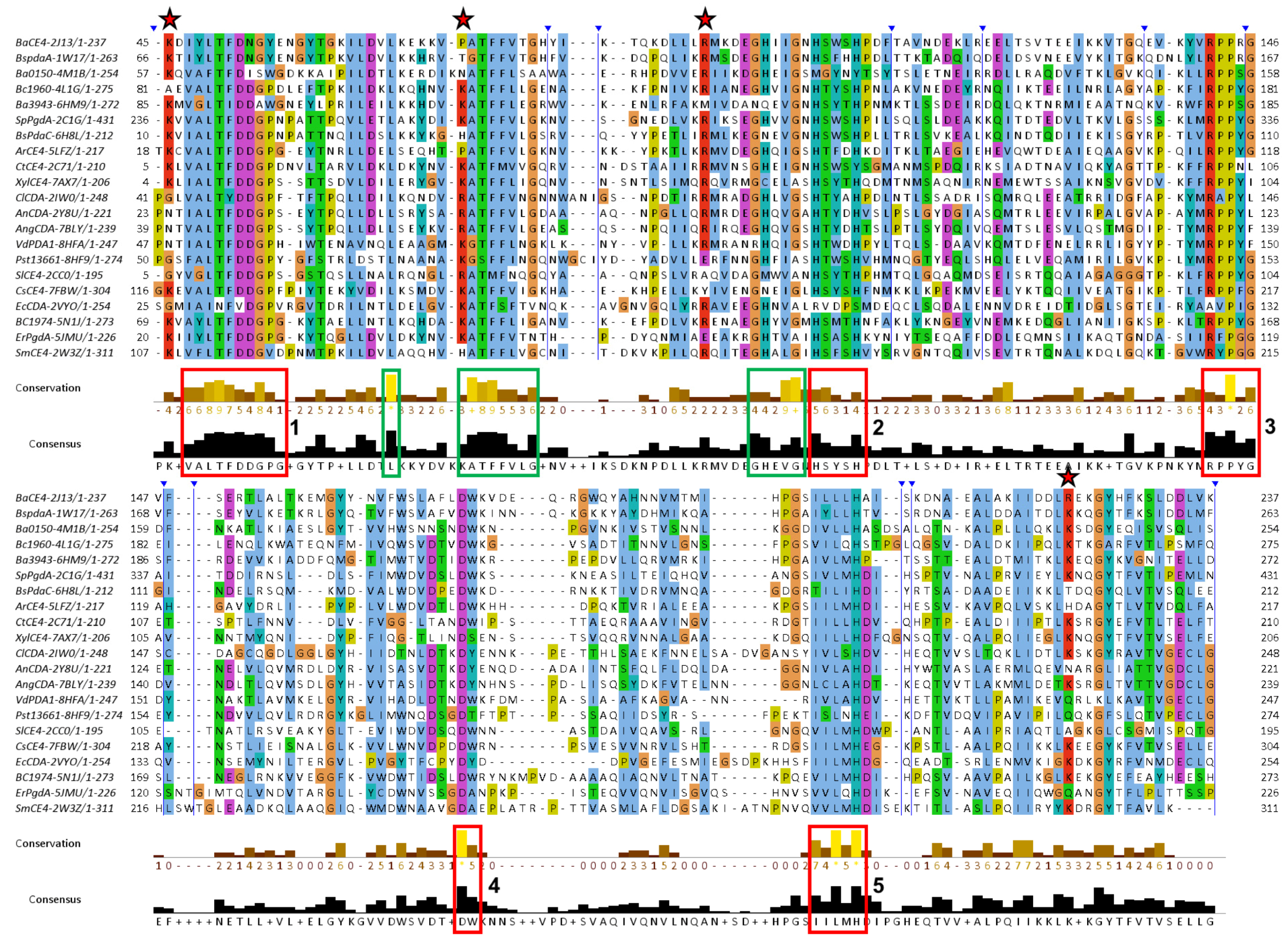
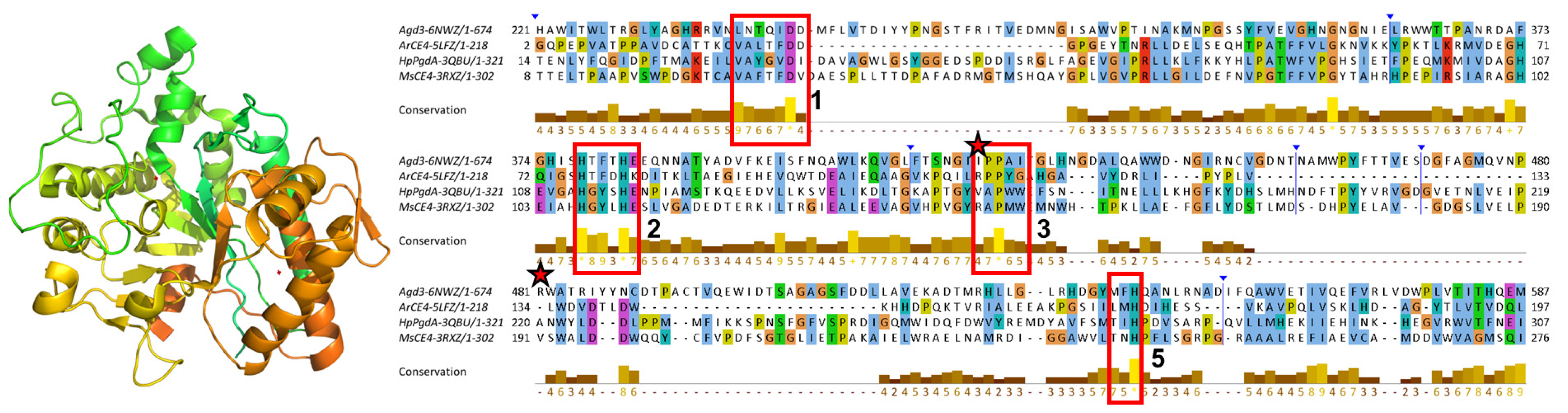
3.3. Sequence Conservation Analysis
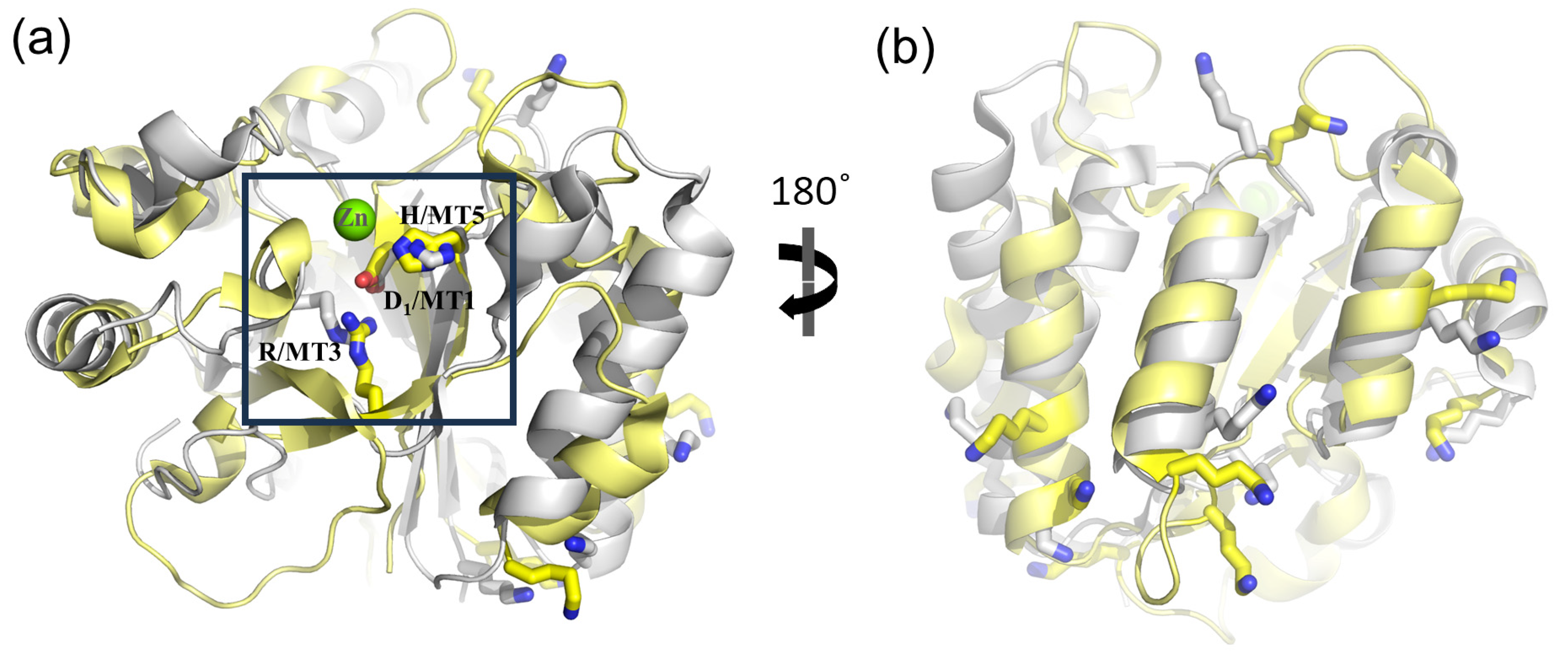
3.4. Function-Related, Structure-Distributed Sequence Conservation
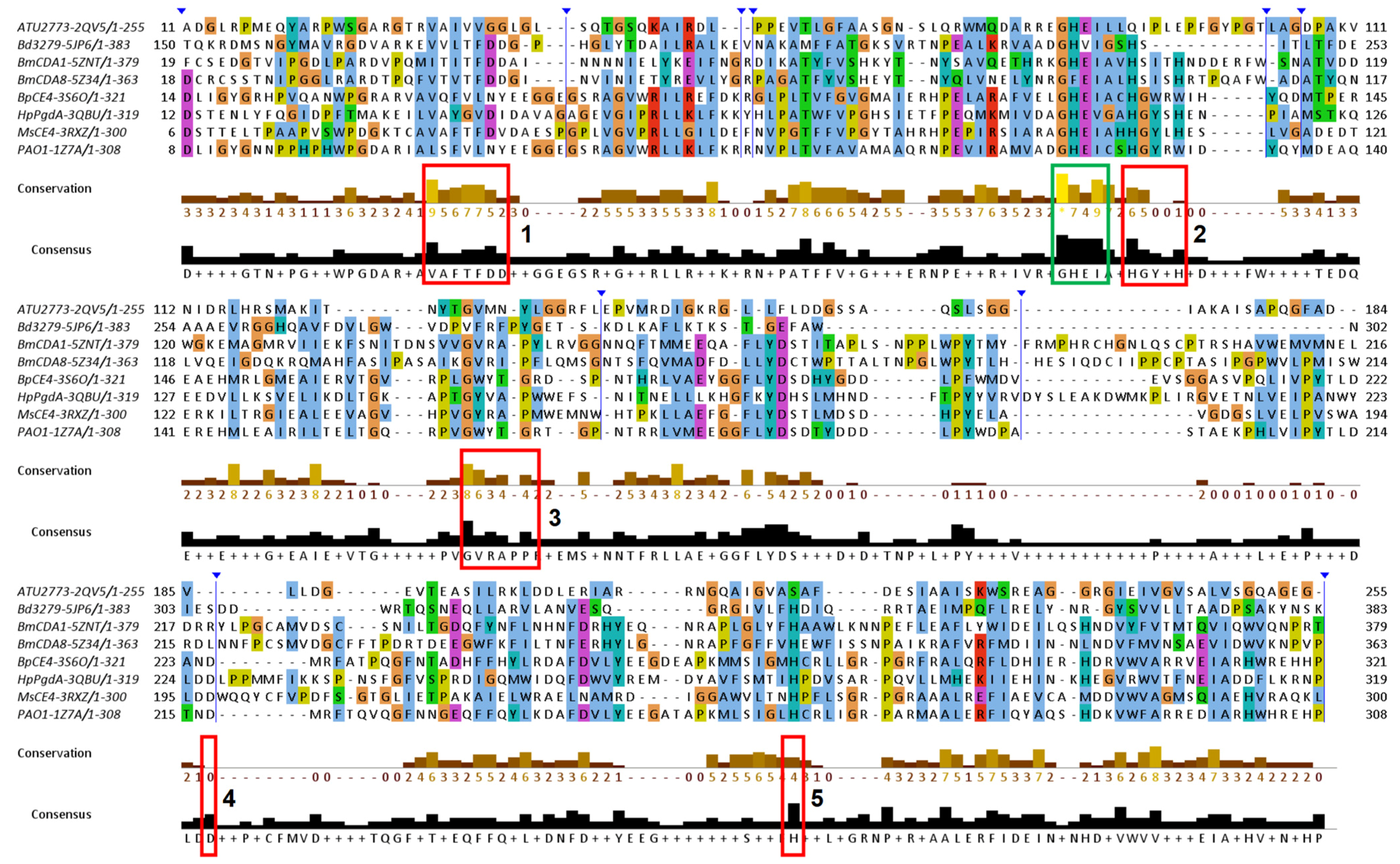
3.5. NodB Fold beyond the CE4 Family
3.5.1. NodB in PuuE Allantoinases
3.5.2. NodB in the Wider Family of Carbohydrate Esterases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branden, C.-I. The TIM Barrel—The Most Frequently Occurring Folding Motif in Proteins. Curr. Opin. Struct. Biol. 1991, 1, 978–983. [Google Scholar] [CrossRef]
- Nagano, N.; Gail Hutchinson, E.; Thornton, J.M. Barrel Structures in Proteins: Automatic Identification and Classification Including a Sequence Analysis of TIM Barrels. Protein Sci. 1999, 8, 2072–2084. [Google Scholar] [CrossRef] [PubMed]
- Nagano, N.; Orengo, C.A.; Thornton, J.M. One Fold with Many Functions: The Evolutionary Relationships between TIM Barrel Families Based on Their Sequences, Structures and Functions. J. Mol. Biol. 2002, 321, 741–765. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, R.K. The TIM-Barrel Fold: A Versatile Framework for Efficient Enzymes. FEBS Lett. 2001, 492, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.K.; Brenner, S.E.; Chandonia, J.M. SCOPe: Structural Classification of Proteins—Extended, Integrating SCOP and ASTRAL Data and Classification of New Structures. Nucleic Acids Res. 2014, 42, D304–D309. [Google Scholar] [CrossRef]
- Chandonia, J.M.; Guan, L.; Lin, S.; Yu, C.; Fox, N.K.; Brenner, S.E. SCOPe: Improvements to the Structural Classification of Proteins-Extended Database to Facilitate Variant Interpretation and Machine Learning. Nucleic Acids Res. 2022, 50, D553–D559. [Google Scholar] [CrossRef]
- Blair, D.E.; Schuttelkopf, A.W.; MacRae, J.I.; van Aalten, D.M. Structure and Metal-Dependent Mechanism of Pep-tidoglycan Deacetylase, a Streptococcal Virulence Factor. Proc. Natl. Acad. Sci. USA 2005, 102, 15429–15434. [Google Scholar] [CrossRef]
- Balomenou Arnaouteli, S.; Koutsioulis, D.; Fadouloglou, V.E.; Vassilis Bouriotis, S. Polysaccharide Deacety-lases: New Antibacterial Drug Targets. Front. Anti-Infect. Drug Discov. 2015, 4, 68–130. [Google Scholar] [CrossRef]
- Fadouloglou, V.E.; Kapanidou, M.; Agiomirgianaki, A.; Arnaouteli, S.; Bouriotis, V.; Glykos, N.M.; Kokkinidis, M. Structure Determination through Homology Modelling and Torsion-Angle Simulated Annealing: Application to a Polysaccharide Deacetylase from Bacillus Cereus. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 276–283. [Google Scholar] [CrossRef]
- Fadouloglou, V.E.; Balomenou, S.; Aivaliotis, M.; Kotsifaki, D.; Arnaouteli, S.; Tomatsidou, A.; Efstathiou, G.; Kountourakis, N.; Miliara, S.; Griniezaki, M.; et al. Unusual Alpha-Carbon Hydroxylation of Proline Promotes Active-Site Maturation. J. Am. Chem. Soc. 2017, 139, 5330–5337. [Google Scholar] [CrossRef]
- Prejanò, M.; Romeo, I.; Sgrizzi, L.; Russo, N.; Marino, T. Why Hydroxy-Proline Improves the Catalytic Power of the Peptidoglycan: N-Deacetylase Enzyme: Insight from Theory. Phys. Chem. Chem. Phys. 2019, 21, 23338–23345. [Google Scholar] [CrossRef]
- Nakamura Nascimento, A.S.; Polikarpov, I.A.M. Structural Diversity of Carbohydrate Esterase. Biotechnol. Res. Innov. 2017, 1, 35–51. [Google Scholar] [CrossRef]
- Caufrier, F.; Martinou, A.; Dupont, C.; Bouriotis, V. Carbohydrate Esterase Family 4 Enzymes: Substrate Specificity. Carbohydr. Res. 2003, 338, 687–692. [Google Scholar] [CrossRef]
- John, M.; Rohrig, H.; Schmidt, J.; Wieneke, U.; Schell, J. Rhizobium Nodb Protein Involved in Nodulation Signal Synthesis Is a Chitooligosaccharide Deacetylase. Proc. Natl. Acad. Sci. USA 1993, 90, 625–629. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The Carbohydrate-Active Enzymes Database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Taylor, E.J.; Gloster, T.M.; Turkenburg, J.P.; Vincent, F.; Brzozowski, A.M.; Dupont, C.; Shareck, F.; Centeno, M.S.; Prates, J.A.; Puchart, V.; et al. Structure and Activity of Two Metal Ion-Dependent Acetylxylan Esterases Involved in Plant Cell Wall Degradation Reveals a Close Similarity to Peptidoglycan Deacetylases. J. Biol. Chem. 2006, 281, 10968–10975. [Google Scholar] [CrossRef]
- Andres, E.; Albesa-Jove, D.; Biarnes, X.; Moerschbacher, B.M.; Guerin, M.E.; Planas, A. Structural Basis of Chitin Oligosaccharide Deacetylation. Angew. Chem. Int. Ed. 2014, 53, 6882–6887. [Google Scholar] [CrossRef] [PubMed]
- Aragunde, H.; Biarnes, X.; Planas, A. Substrate Recognition and Specificity of Chitin Deacetylases and Related Family 4 Carbohydrate Esterases. Int. J. Mol. Sci. 2018, 19, 412. [Google Scholar] [CrossRef] [PubMed]
- Andreou, A.; Giastas, P.; Christoforides, E.; Eliopoulos, E.E. Structural and Evolutionary Insights within the Pol-ysaccharide Deacetylase Gene Family of Bacillus Anthracis and Bacillus Cereus. Genes 2018, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Grifoll-Romero, L.; Pascual, S.; Aragunde, H.; Biarnés, X.; Planas, A. Chitin Deacetylases: Structures, Specificities, and Biotech Applications. Polymers 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Bamford, N.C.; Le Mauff, F.; Van Loon, J.C.; Ostapska, H.; Snarr, B.D.; Zhang, Y.; Kitova, E.N.; Klassen, J.S.; Codée, J.D.C.; Sheppard, D.C.; et al. Structural and Biochemical Characterization of the Exopolysaccharide Deacetylase Agd3 Required for Aspergillus Fumigatus Biofilm Formation. Nat. Commun. 2020, 11, 2450. [Google Scholar] [CrossRef] [PubMed]
- Ramazzina, I.; Cendron, L.; Folli, C.; Berni, R.; Monteverdi, D.; Zanotti, G.; Percudani, R. Logical Identification of an Allantoinase Analog (PuuE) Recruited from Polysaccharide Deacetylases. J. Biol. Chem. 2008, 283, 23295–23304. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Little, D.J.; Milek, S.; Bamford, N.C.; Ganguly, T.; DiFrancesco, B.R.; Nitz, M.; Deora, R.; Howell, P.L. The Protein BpsB Is a Poly-β-1,6-N-Acetyl-d-Glucosamine Deacetylase Required for Biofilm Formation in Bordetella Bron-chiseptica. J. Biol. Chem. 2015, 290, 22827–22840. [Google Scholar] [CrossRef]
- Andreou, A.; Giastas, P.; Arnaouteli, S.; Tzanodaskalaki, M.; Tzartos, S.J.; Bethanis, K.; Bouriotis, V.; Eliopoulos, E.E. The Putative Polysaccharide Deacetylase Ba0331: Cloning, Expression, Crystallization and Structure Determination. Acta Crystallogr. F Struct. Biol. Commun. 2019, 75, 312–320. [Google Scholar] [CrossRef]
- Rush, J.S.; Parajuli, P.; Ruda, A.; Li, J.; Pohane, A.A.; Zamakhaeva, S.; Rahman, M.M.; Chang, J.C.; Gogos, A.; Kenner, C.W.; et al. PplD Is a De-N-Acetylase of the Cell Wall Linkage Unit of Streptococcal Rhamnopolysaccha-rides. Nat. Commun. 2022, 13, 590. [Google Scholar] [CrossRef]
- Little, D.J.; Bamford, N.C.; Podrovskaya, V.; Robinson, H.; Nitz, M.; Howell, P.L. Structural Basis for the De-N-Acetylation of Poly-Beta-1,6-N-Acetyl-D-Glucosamine in Gram-Positive Bacteria. J. Biol. Chem. 2014, 289, 35907–35917. [Google Scholar] [CrossRef]
- Nishiyama, T.; Noguchi, H.; Yoshida, H.; Park, S.Y.; Tame, J.R.H. The Structure of the Deacetylase Domain of Escherichia Coli PgaB, an Enzyme Required for Biofilm Formation: A Circularly Permuted Member of the Carbohydrate Esterase 4 Family. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2013, 69, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Arnaouteli, S.; Giastas, P.; Andreou, A.; Tzanodaskalaki, M.; Aldridge, C.; Tzartos, S.J.; Vollmer, W.; Eliopoulos, E.; Bouriotis, V. Two Putative Polysaccharide Deacetylases Are Required for Osmotic Stability and Cell Shape Maintenance in Bacillus Anthracis. J. Biol. Chem. 2015, 290, 13465–13478. [Google Scholar] [CrossRef]
- Oberbarnscheidt, L.; Taylor, E.J.; Davies, G.J.; Gloster, T.M. Structure of a Carbohydrate Esterase from Bacillus Anthracis. Proteins 2007, 66, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Blair, D.E.; van Aalten, D.M. Structures of Bacillus Subtilis PdaA, a Family 4 Carbohydrate Esterase, and a Com-plex with N-Acetyl-Glucosamine. FEBS Lett. 2004, 570, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Sugiyama, K.; Sakaki, Y.; Hakamata, W.; Park, S.Y.; Nishio, T. Structure-Based Analysis of Domain Function of Chitin Oligosaccharide Deacetylase from Vibrio Parahaemolyticus. FEBS Lett. 2015, 589, 145–151. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, Y.; Qu, M.; Qiu, Y.; Guo, X.; Zhang, Y.; Liu, T.; Yang, J.; Yang, Q. Structural and Biochemical In-sights into the Catalytic Mechanisms of Two Insect Chitin Deacetylases of the Carbohydrate Esterase 4 Family. J. Biol. Chem. 2019, 294, 5774–5783. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Yang, J.; Yang, Q. An Overall Look at Insect Chitin Deacetylases: Promising Molecular Targets for Developing Green Pesticides. J. Pestic. Sci. 2021, 46, 43–52. [Google Scholar] [CrossRef]
- Grifoll-Romero, L.; Sainz-Polo, M.A.; Albesa-Jové, D.; Guerin, M.E.; Biarnés, X.; Planas, A. Structure-Function Re-lationships Underlying the Dual N-Acetylmuramic and N-Acetylglucosamine Specificities of the Bacterial Pep-tidoglycan Deacetylase PdaC. J. Biol. Chem. 2019, 294, 19066–19080. [Google Scholar] [CrossRef]
- Giastas, P.; Andreou, A.; Papakyriakou, A.; Koutsioulis, D.; Balomenou, S.; Tzartos, S.J.; Bouriotis, V.; Eliopoulos, E.E. Structures of the Peptidoglycan N-Acetylglucosamine Deacetylase Bc1974 and Its Complexes with Zinc Metalloenzyme Inhibitors. Biochemistry 2018, 57, 753–763. [Google Scholar] [CrossRef]
- Lee, S.Y.; Pardhe, B.D.; Oh, T.J.; Park, H.H. Crystal Structure of ChbG from Klebsiella Pneumoniae Reveals the Molecular Basis of Diacetylchitobiose Deacetylation. Commun. Biol. 2022, 5, 862. [Google Scholar] [CrossRef]
- Muñoz-Escudero, D.; Breazeale, S.D.; Lee, M.; Guan, Z.; Raetz, C.R.H.; Sousa, M.C. Structure and Function of ArnD. A Deformylase Essential for Lipid A Modification with 4-Amino-4-Deoxy-l-Arabinose and Polymyxin Resistance. Biochemistry 2023, 62, 2970–2981. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molfetas, A.S.; Boutris, N.; Tomatsidou, A.; Kokkinidis, M.; Fadouloglou, V.E. Variations of the NodB Architecture Are Attuned to Functional Specificities into and beyond the Carbohydrate Esterase Family 4. Biomolecules 2024, 14, 325. https://doi.org/10.3390/biom14030325
Molfetas AS, Boutris N, Tomatsidou A, Kokkinidis M, Fadouloglou VE. Variations of the NodB Architecture Are Attuned to Functional Specificities into and beyond the Carbohydrate Esterase Family 4. Biomolecules. 2024; 14(3):325. https://doi.org/10.3390/biom14030325
Chicago/Turabian StyleMolfetas, Alexis S., Nikiforos Boutris, Anastasia Tomatsidou, Michael Kokkinidis, and Vasiliki E. Fadouloglou. 2024. "Variations of the NodB Architecture Are Attuned to Functional Specificities into and beyond the Carbohydrate Esterase Family 4" Biomolecules 14, no. 3: 325. https://doi.org/10.3390/biom14030325