
Phosphatase and Tensin Homolog Inhibition in Proteolipid Protein 1-Expressing Cells Stimulates Neurogenesis and Gliogenesis in the Postnatal Enteric Nervous System


Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Ex Vivo Colonic Motility
2.3. Whole Gut Transit Time
2.4. Paraffin Embedded Colonic Sections
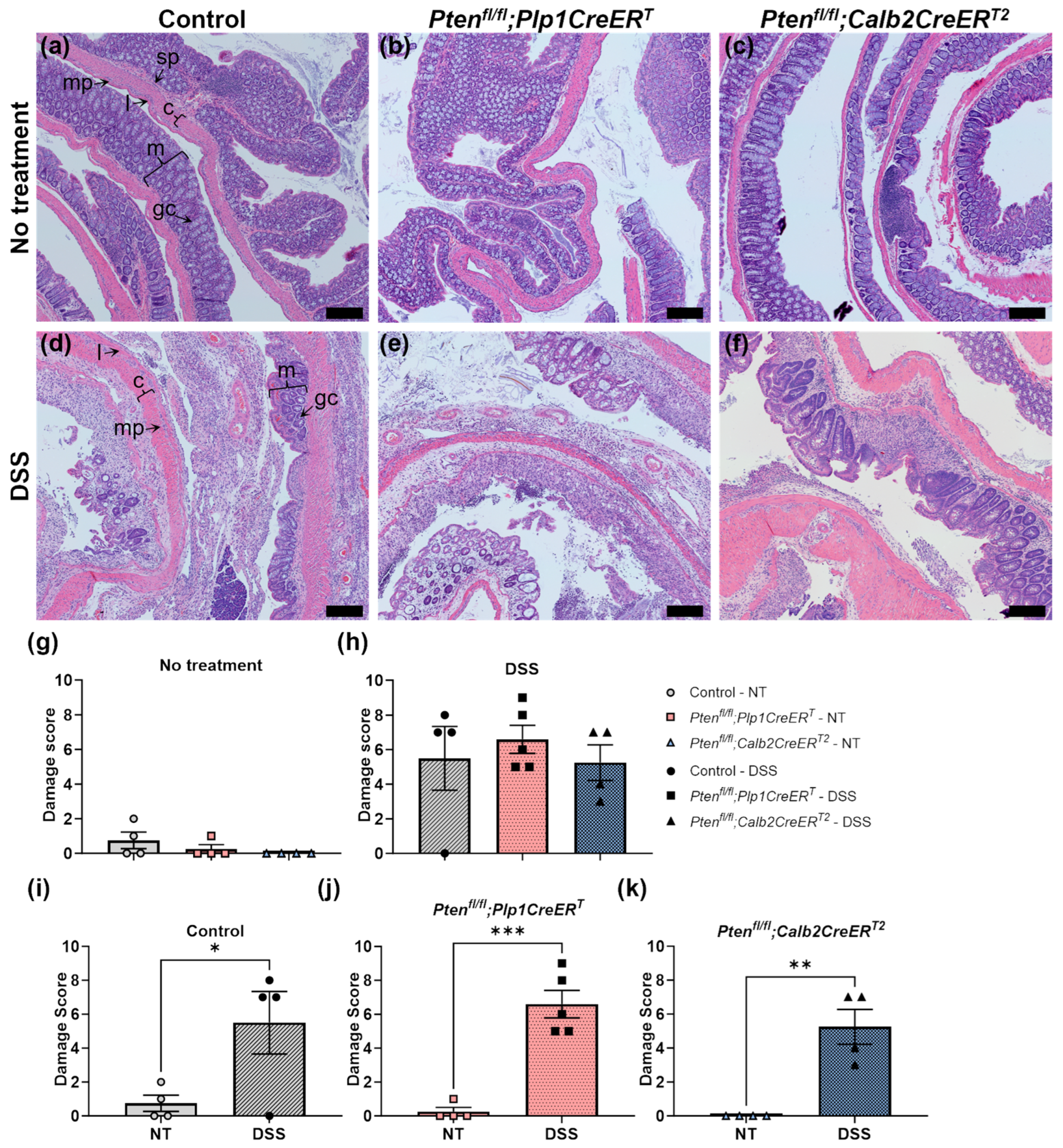
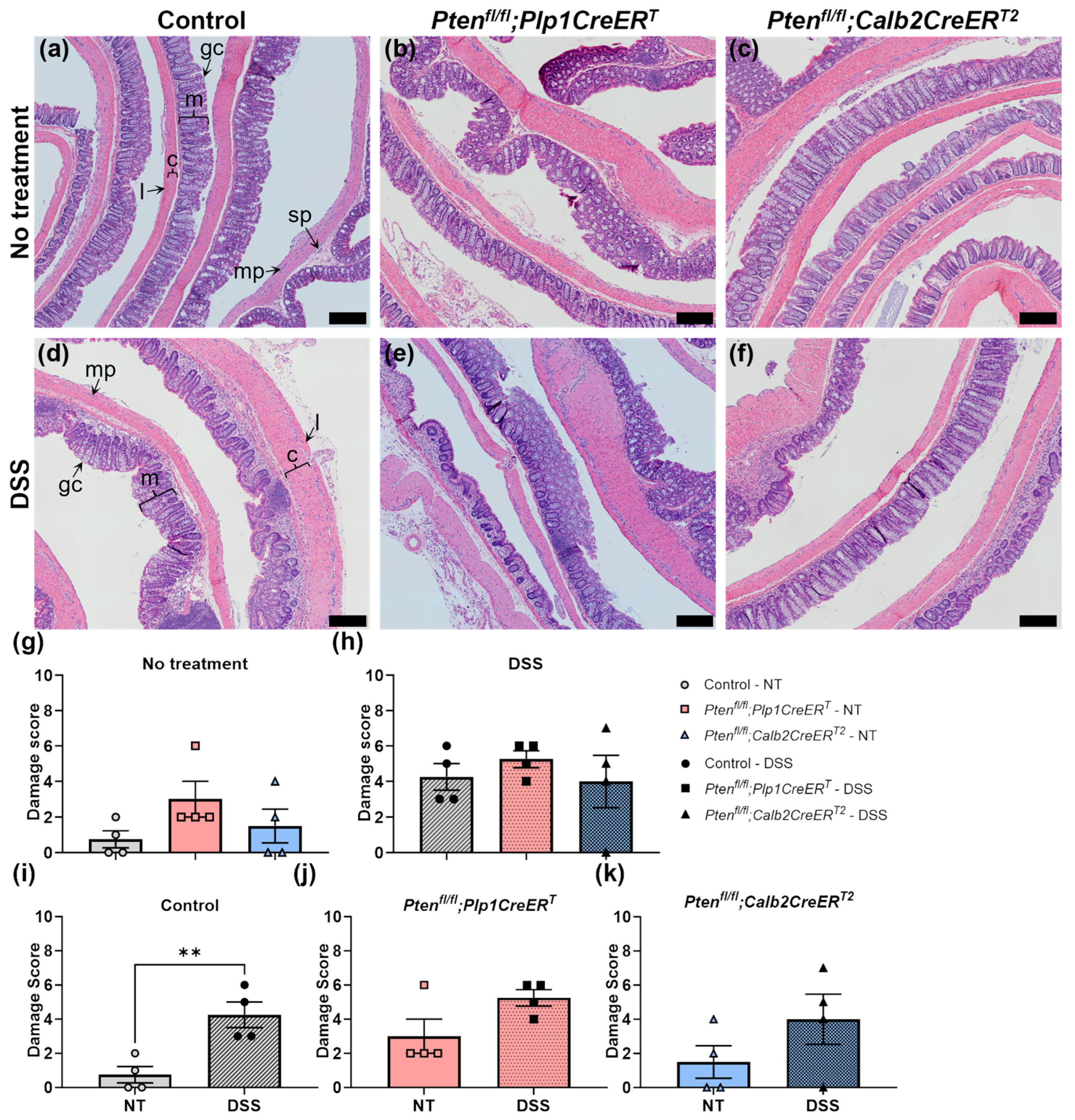
2.4.1. Damage Scores
2.4.2. Immunofluorescence
2.5. Wholemount Preparation
2.5.1. Immunofluorescence
2.5.2. Image Analysis
2.6. Imaging
2.7. Statistical Analysis
3. Results
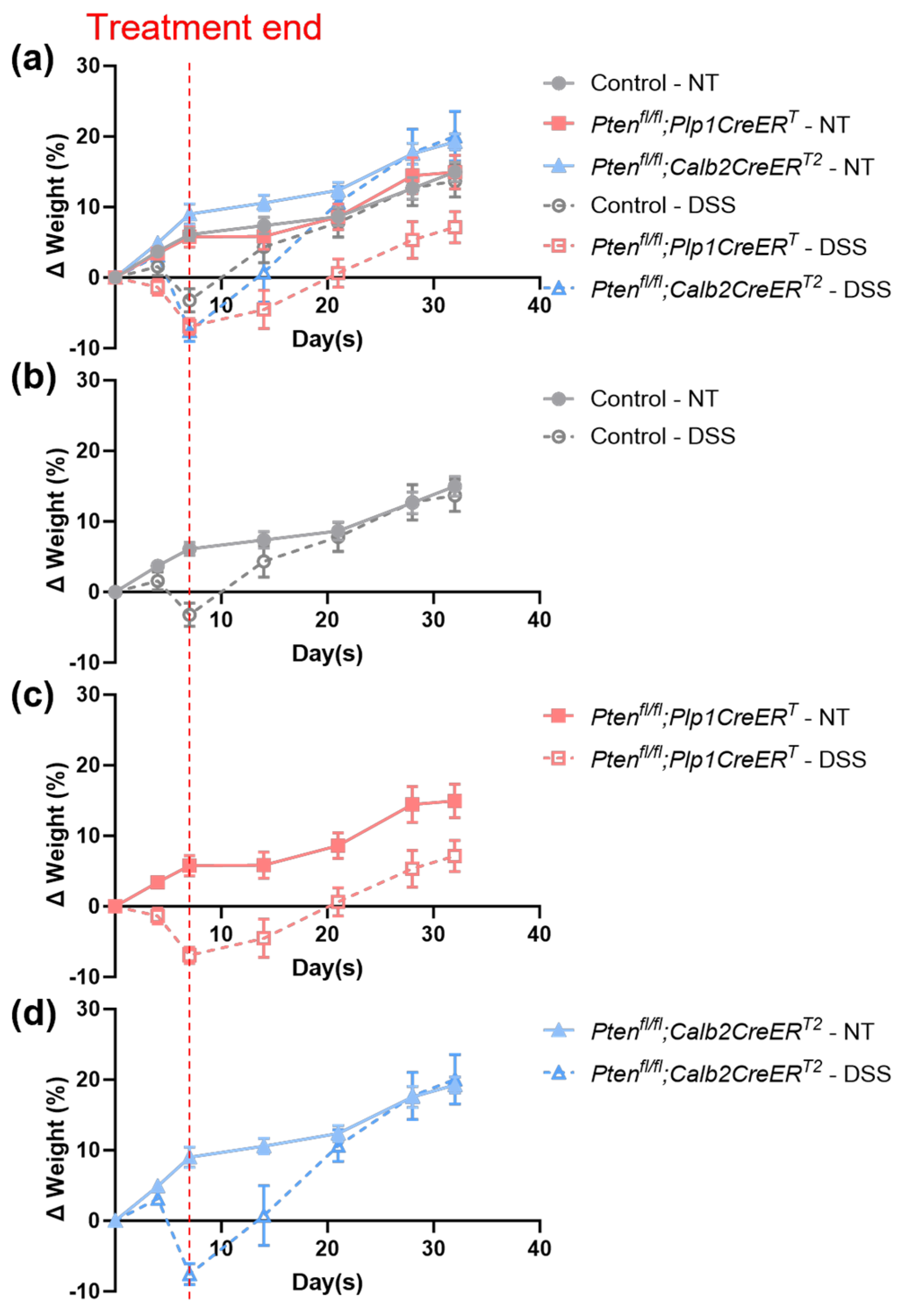
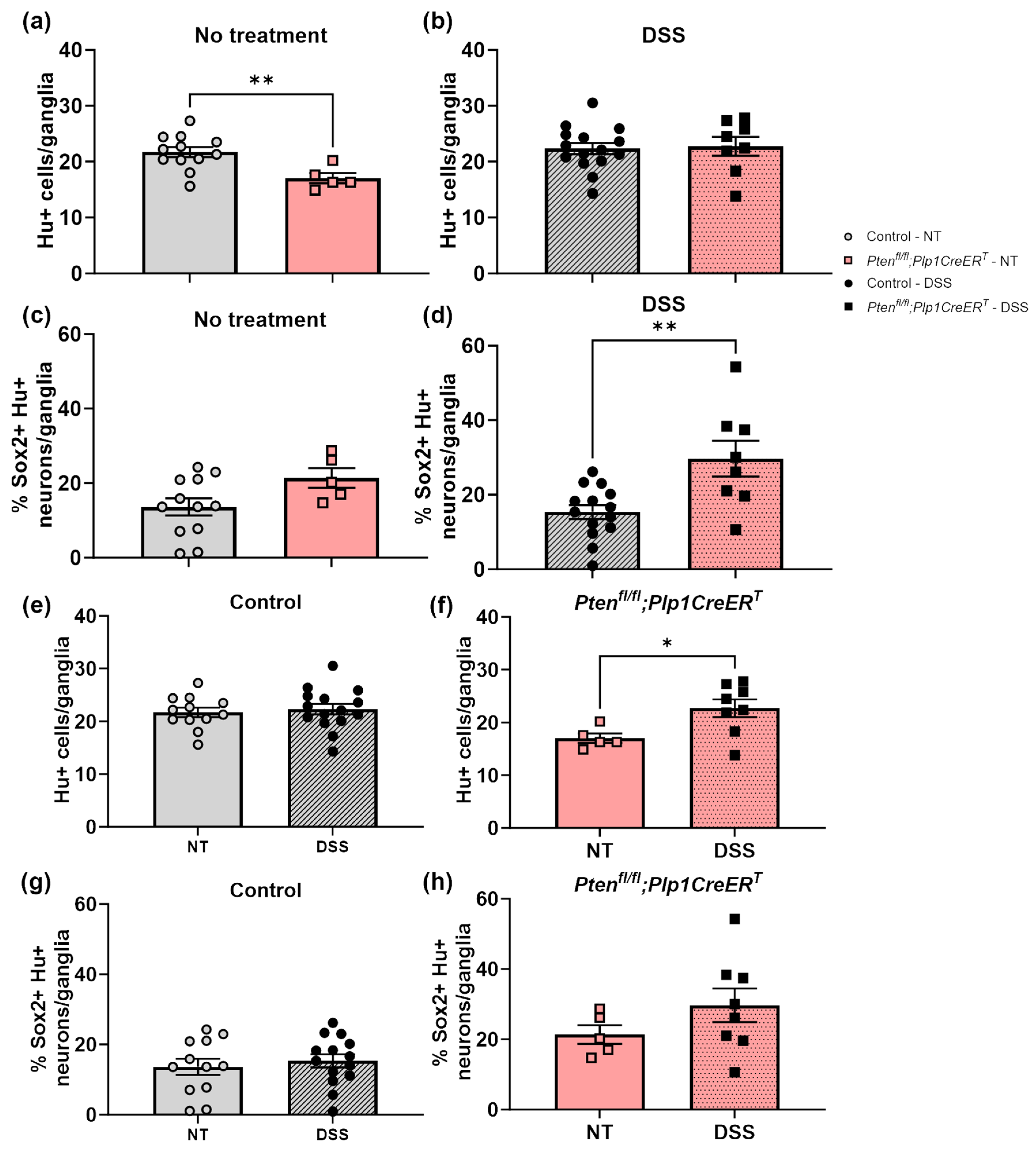
3.1. Mice Lacking Pten in Plp1-Expressing Cells Were Not Able to Recover Weight Lost Due to DSS Treatment during the Recovery Period
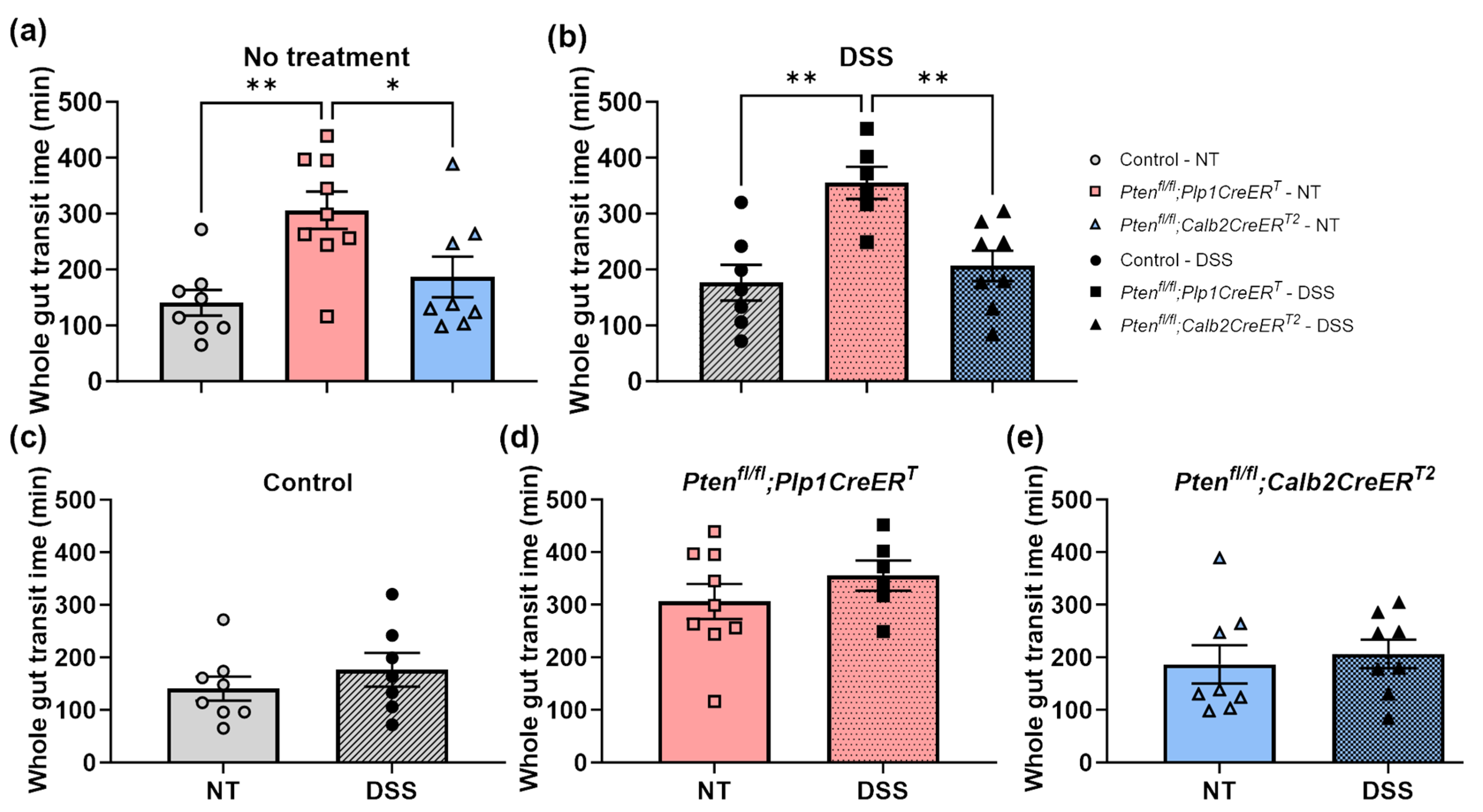
3.2. In Mice Lacking Pten in Plp1-Expressing Cells, the Rate of Colonic Migrating Motor Complexes Was Slightly Elevated during the Recovery Period after DSS Treatment and Intestinal Transit Times Were Slowed When Compared to DSS-Treated Control and Ptenfl/fl;Calb2CreERT2 Mice
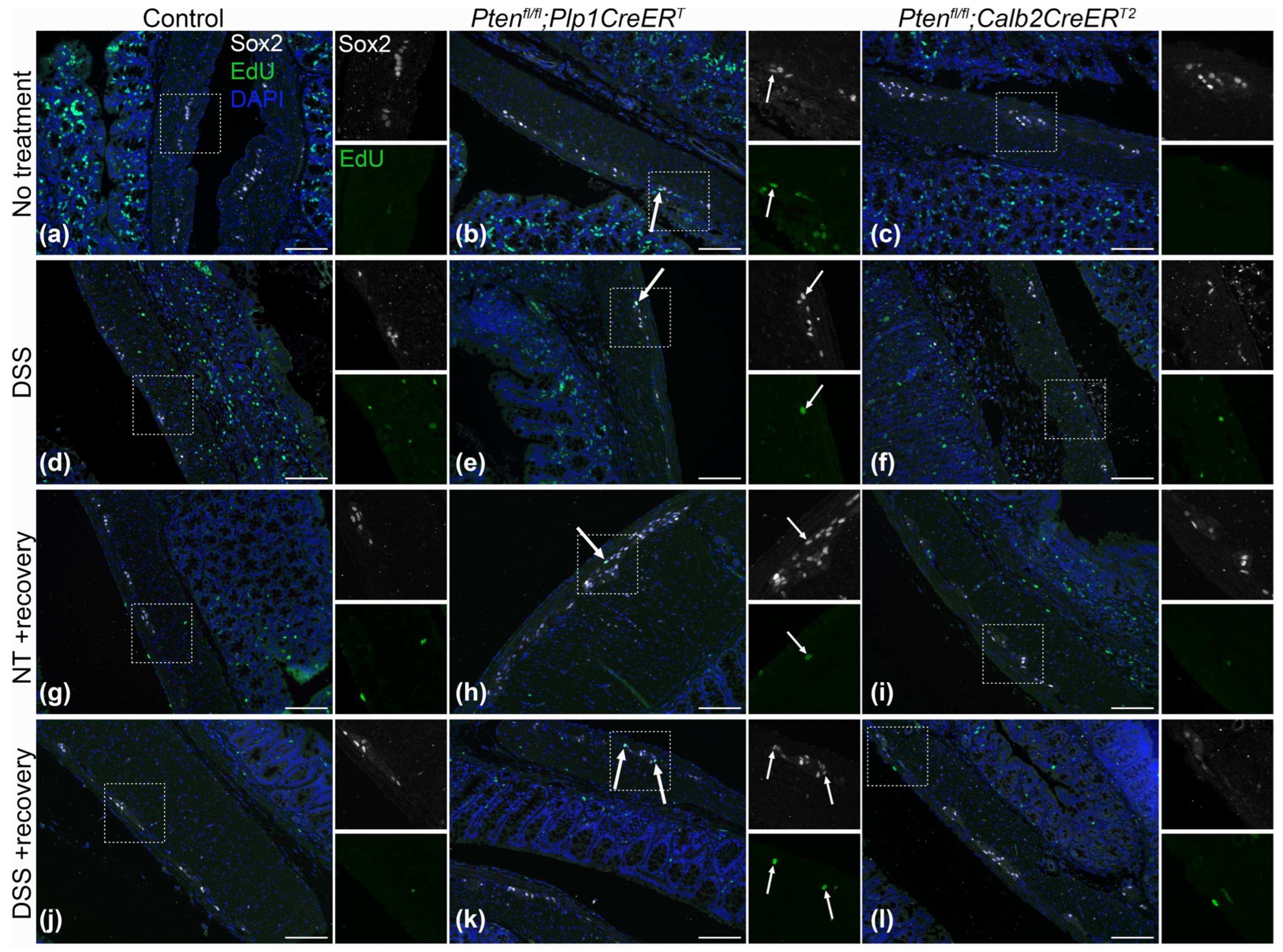
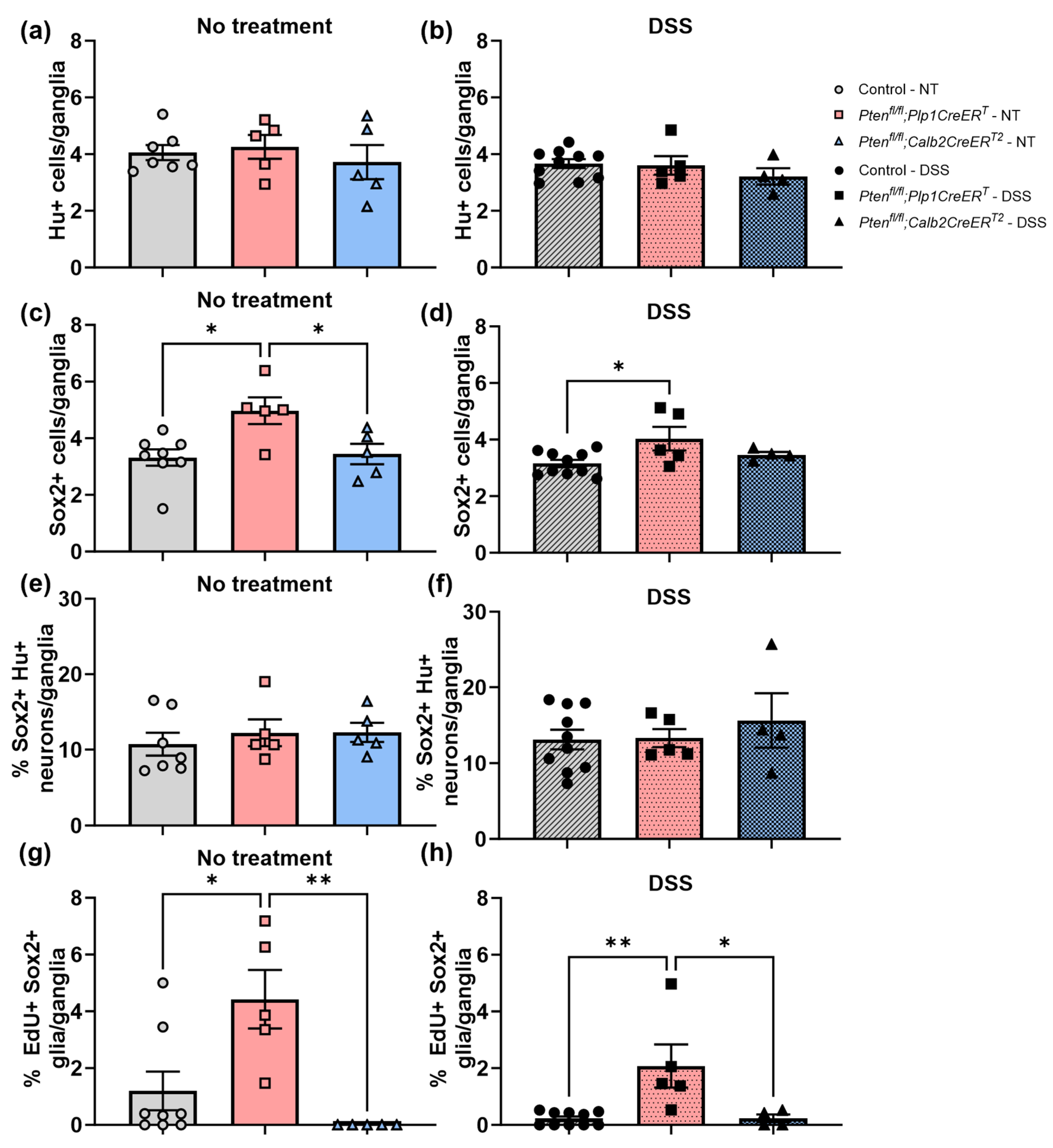
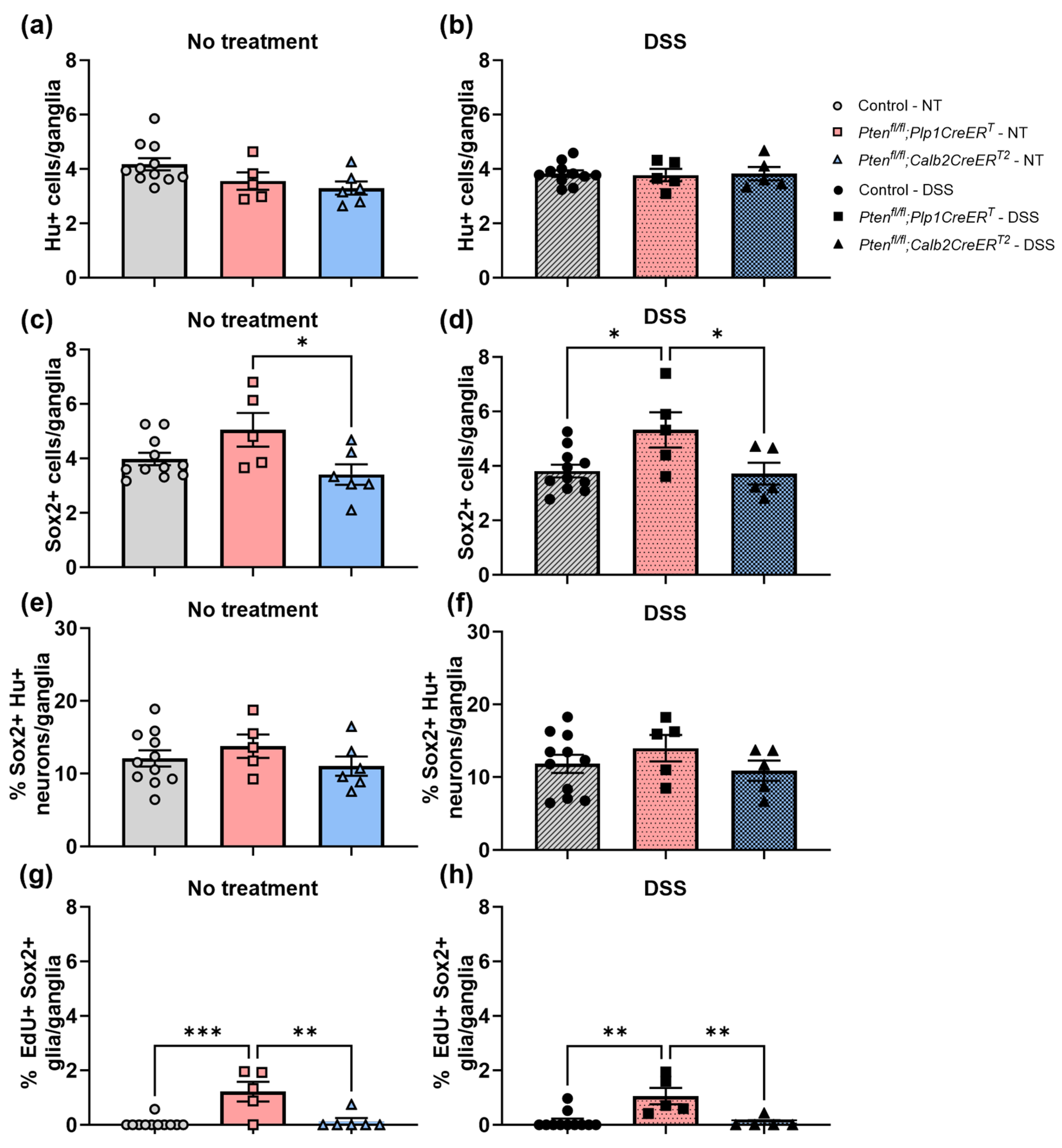
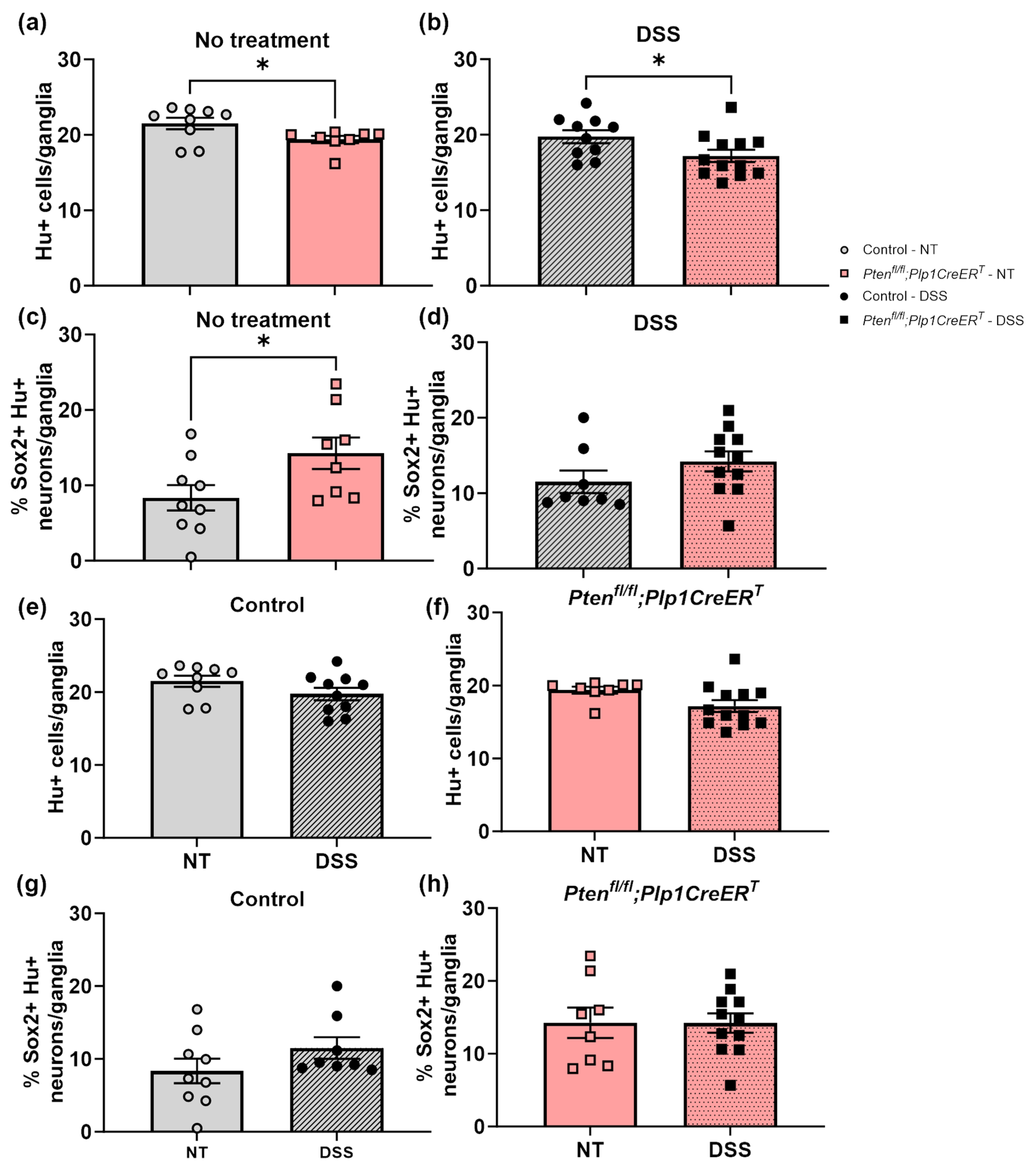
3.3. Immunofluorescent Assessment of the ENS Revealed That Pten Inhibition in Plp1+ Glial Cells Induced Gliogenesis in the Colon at Baseline and after DSS Treatment, and Neurogenesis in the Proximal Colon 3–4 Weeks Post-DSS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 286–294. [Google Scholar] [CrossRef]
- Ramalho, F.S.; Santos, G.C.; Ramalho, L.N.; Kajiwara, J.K.; Zucoloto, S. Myenteric neuron number after acute and chronic denervation of the proximal jejunum induced by benzalkonium chloride. Neurosci. Lett. 1993, 163, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Hanani, M.; Ledder, O.; Yutkin, V.; Abu-Dalu, R.; Huang, T.Y.; Hartig, W.; Vannucchi, M.G.; Faussone-Pellegrini, M.S. Regeneration of myenteric plexus in the mouse colon after experimental denervation with benzalkonium chloride. J. Comp. Neurol. 2003, 462, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Laranjeira, C.; Sandgren, K.; Kessaris, N.; Richardson, W.; Potocnik, A.; Vanden Berghe, P.; Pachnis, V. Glial cells in the mouse enteric nervous system can undergo neurogenesis in response to injury. J. Clin. Investig. 2011, 121, 3412–3424. [Google Scholar] [CrossRef] [PubMed]
- Belkind-Gerson, J.; Hotta, R.; Nagy, N.; Thomas, A.R.; Graham, H.; Cheng, L.; Solorzano, J.; Nguyen, D.; Kamionek, M.; Dietrich, J.; et al. Colitis induces enteric neurogenesis through a 5-HT4-dependent mechanism. Inflamm. Bowel Dis. 2015, 21, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Hotta, R.; Cheng, L.; Graham, H.K.; Nagy, N.; Belkind-Gerson, J.; Mattheolabakis, G.; Amiji, M.M.; Goldstein, A.M. Delivery of enteric neural progenitors with 5-HT4 agonist-loaded nanoparticles and thermosensitive hydrogel enhances cell proliferation and differentiation following transplantation in vivo. Biomaterials 2016, 88, 1–11. [Google Scholar] [CrossRef]
- Belkind-Gerson, J.; Graham, H.K.; Reynolds, J.; Hotta, R.; Nagy, N.; Cheng, L.; Kamionek, M.; Shi, H.N.; Aherne, C.M.; Goldstein, A.M. Colitis promotes neuronal differentiation of Sox2+ and PLP1+ enteric cells. Sci. Rep. 2017, 7, 2525. [Google Scholar] [CrossRef]
- Vicentini, F.A.; Keenan, C.M.; Wallace, L.E.; Woods, C.; Cavin, J.B.; Flockton, A.R.; Macklin, W.B.; Belkind-Gerson, J.; Hirota, S.A.; Sharkey, K.A. Intestinal microbiota shapes gut physiology and regulates enteric neurons and glia. Microbiome 2021, 9, 210. [Google Scholar] [CrossRef]
- Sulis, M.L.; Parsons, R. PTEN: From pathology to biology. Trends Cell Biol. 2003, 13, 478–483. [Google Scholar] [CrossRef]
- Stiles, B.; Groszer, M.; Wang, S.; Jiao, J.; Wu, H. PTENless means more. Dev. Biol. 2004, 273, 175–184. [Google Scholar] [CrossRef]
- Fraser, M.M.; Zhu, X.; Kwon, C.H.; Uhlmann, E.J.; Gutmann, D.H.; Baker, S.J. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004, 64, 7773–7779. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, Y.X.; Zeng, Y.Q.; Zhang, S.F.; Teng, Z.Q.; Gao, J.; Saijilafu; Liu, C.M. miR-26a promotes axon regeneration in the mammalian central nervous system by suppressing PTEN expression. Acta Biochim. Biophys. Sin. 2021, 53, 758–765. [Google Scholar] [CrossRef]
- Puig, I.; Champeval, D.; De Santa Barbara, P.; Jaubert, F.; Lyonnet, S.; Larue, L. Deletion of Pten in the mouse enteric nervous system induces ganglioneuromatosis and mimics intestinal pseudoobstruction. J. Clin. Investig. 2009, 119, 3586–3596. [Google Scholar] [CrossRef]
- Fu, M.; Sato, Y.; Lyons-Warren, A.; Zhang, B.; Kane, M.A.; Napoli, J.L.; Heuckeroth, R.O. Vitamin A facilitates enteric nervous system precursor migration by reducing Pten accumulation. Development 2010, 137, 631–640. [Google Scholar] [CrossRef]
- O’Donnell, A.M.; Puri, P. A role for Pten in paediatric intestinal dysmotility disorders. Pediatr. Surg. Int. 2011, 27, 491–493. [Google Scholar] [CrossRef]
- Becker, L.; Peterson, J.; Kulkarni, S.; Pasricha, P.J. Ex vivo neurogenesis within enteric ganglia occurs in a PTEN dependent manner. PLoS ONE 2013, 8, e59452. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Micci, M.A.; Leser, J.; Shin, C.; Tang, S.C.; Fu, Y.Y.; Liu, L.; Li, Q.; Saha, M.; Li, C.; et al. Adult enteric nervous system in health is maintained by a dynamic balance between neuronal apoptosis and neurogenesis. Proc. Natl. Acad. Sci. USA 2017, 114, E3709–E3718. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Nelms, B.D.; Dong, L.; Salinas-Rios, V.; Rutlin, M.; Gershon, M.D.; Corfas, G. Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 2015, 63, 2040–2057. [Google Scholar] [CrossRef]
- Feng, J.; Hibberd, T.J.; Luo, J.; Yang, P.; Xie, Z.; Travis, L.; Spencer, N.J.; Hu, H. Modification of Neurogenic Colonic Motor Behaviours by Chemogenetic Ablation of Calretinin Neurons. Front. Cell Neurosci. 2022, 16, 799717. [Google Scholar] [CrossRef]
- Lesche, R.; Groszer, M.; Gao, J.; Wang, Y.; Messing, A.; Sun, H.; Liu, X.; Wu, H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 2002, 32, 148–149. [Google Scholar] [CrossRef]
- Doerflinger, N.H.; Macklin, W.B.; Popko, B. Inducible site-specific recombination in myelinating cells. Genesis 2003, 35, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; He, M.; Wu, P.; Kim, S.; Paik, R.; Sugino, K.; Kvitsiani, D.; Fu, Y.; Lu, J.; Lin, Y.; et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 2011, 71, 995–1013. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, M.; Hill-Yardin, E.; Ellis, M.; Zygorodimos, M.; Johnston, L.A.; Gwynne, R.M.; Bornstein, J.C. Video Imaging and Spatiotemporal Maps to Analyze Gastrointestinal Motility in Mice. J. Vis. Exp. 2016, 108, e53828. [Google Scholar] [CrossRef]
- Woods, C.; Flockton, A.R.; Wallace, L.E.; Keenan, C.M.; Macklin, W.B.; Sharkey, K.A.; Belkind-Gerson, J. Proteolipid protein 1 is involved in the regulation of intestinal motility and barrier function in the mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 324, G115–G130. [Google Scholar] [CrossRef]
- Hoffman, J.M.; Brooks, E.M.; Mawe, G.M. Gastrointestinal Motility Monitor (GIMM). J. Vis. Exp. 2010, 46, e2435. [Google Scholar] [CrossRef]
- Woods, C.; Kapur, R.P.; Bischoff, A.; Lovell, M.; Arnold, M.; Pena, A.; Flockton, A.; Sharkey, K.A.; Belkind-Gerson, J. Neurons populating the rectal extrinsic nerves in humans express neuronal and Schwann cell markers. Neurogastroenterol. Motil. 2021, 33, e14074. [Google Scholar] [CrossRef] [PubMed]
- Heanue, T.A.; Pachnis, V. Prospective identification and isolation of enteric nervous system progenitors using Sox2. Stem Cells 2011, 29, 128–140. [Google Scholar] [CrossRef]
- Hamnett, R.; Dershowitz, L.B.; Sampathkumar, V.; Wang, Z.; Gomez-Frittelli, J.; De Andrade, V.; Kasthuri, N.; Druckmann, S.; Kaltschmidt, J.A. Regional cytoarchitecture of the adult and developing mouse enteric nervous system. Curr. Biol. 2022, 32, 4483–4492.e4485. [Google Scholar] [CrossRef]
- Kruger, G.M.; Mosher, J.T.; Bixby, S.; Joseph, N.; Iwashita, T.; Morrison, S.J. Neural crest stem cells persist in the adult gut but undergo changes in self-renewal, neuronal subtype potential, and factor responsiveness. Neuron 2002, 35, 657–669. [Google Scholar] [CrossRef]
- Bondurand, N.; Natarajan, D.; Thapar, N.; Atkins, C.; Pachnis, V. Neuron and glia generating progenitors of the mammalian enteric nervous system isolated from foetal and postnatal gut cultures. Development 2003, 130, 6387–6400. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rodriguez, R.; Belkind-Gerson, J. Cultured nestin-positive cells from postnatal mouse small bowel differentiate ex vivo into neurons, glia, and smooth muscle. Stem Cells 2004, 22, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; He, S.; Quintana, E.; Kim, Y.G.; Nunez, G.; Morrison, S.J. Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J. Clin. Investig. 2011, 121, 3398–3411. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, H.; Garton, D.R.; Andressoo, J.O. Myenteric Neurons Do Not Replicate in Small Intestine Under Normal Physiological Conditions in Adult Mouse. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 27–34. [Google Scholar] [CrossRef]
- Domanskyi, A.; Geissler, C.; Vinnikov, I.A.; Alter, H.; Schober, A.; Vogt, M.A.; Gass, P.; Parlato, R.; Schutz, G. Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. FASEB J. 2011, 25, 2898–2910. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Jia, J.; Zhou, H.; Zhang, C.; Liu, L.; Liu, J.; Lu, L.; Li, X.; Kang, Y.; Lou, Y.; et al. PTEN modulates neurites outgrowth and neuron apoptosis involving the PI3K/Akt/mTOR signaling pathway. Mol. Med. Rep. 2019, 20, 4059–4066. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Name | Strain | Jax Stock No. | RRID * No. |
|---|---|---|---|
| Ptenflox | C;129S4-Ptentm1Hwu/J | 006440 | IMSR_JAX:006440 |
| Plp1CreERT | B6.Cg-Tg(Plp1-cre/ERT)3Pop/J | 005975 | IMSR_JAX:005975 |
| Calb2CreERT2 | B6(Cg)-Calb2tm2.1(cre/ERT2)Zjh/J | 013730 | IMSR_JAX:013730 |
| Antibody | Host | Dilution | Source | Cat. No. | RRID * No. |
|---|---|---|---|---|---|
| ANNA-1 (Hu) | Human | 1:10,000 | Mayo Clinic, Rochester, MN, USA | n/a | AB_2314657 |
| Sox2 | Goat | 1:100 | R&D Systems, San Diego, CA, USA | AF2018 | AB_355110 |
| Human Alexa Fluor 488 | Donkey | 1:400 | Jackson ImmunoResearch, West Grove, PA, USA | 709-545-149 | AB_2340566 |
| Goat Alexa Fluor 647 | Donkey | 1:400 | Invitrogen, San Diego, CA, USA | A21447 | AB_2535864 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woods, C.; Flockton, A.R.; Belkind-Gerson, J. Phosphatase and Tensin Homolog Inhibition in Proteolipid Protein 1-Expressing Cells Stimulates Neurogenesis and Gliogenesis in the Postnatal Enteric Nervous System. Biomolecules 2024, 14, 346. https://doi.org/10.3390/biom14030346
Woods C, Flockton AR, Belkind-Gerson J. Phosphatase and Tensin Homolog Inhibition in Proteolipid Protein 1-Expressing Cells Stimulates Neurogenesis and Gliogenesis in the Postnatal Enteric Nervous System. Biomolecules. 2024; 14(3):346. https://doi.org/10.3390/biom14030346
Chicago/Turabian StyleWoods, Crystal, Amanda R. Flockton, and Jaime Belkind-Gerson. 2024. "Phosphatase and Tensin Homolog Inhibition in Proteolipid Protein 1-Expressing Cells Stimulates Neurogenesis and Gliogenesis in the Postnatal Enteric Nervous System" Biomolecules 14, no. 3: 346. https://doi.org/10.3390/biom14030346
APA StyleWoods, C., Flockton, A. R., & Belkind-Gerson, J. (2024). Phosphatase and Tensin Homolog Inhibition in Proteolipid Protein 1-Expressing Cells Stimulates Neurogenesis and Gliogenesis in the Postnatal Enteric Nervous System. Biomolecules, 14(3), 346. https://doi.org/10.3390/biom14030346