1. Introduction
Polyamines (PAs) comprise a collection of aliphatic compounds, characterized by the presence of multiple amino groups [
1]. PAs, found universally in all living organisms, are vital for sustaining fundamental physiological functions [
1,
2,
3]. Among them, spermidine (Spd) is considered indispensable for biological growth and development, exhibiting significant benefits in promoting health, combating aging, and extending lifespan [
4,
5,
6]. Manipulating the duration of inhibitor exposure in vivo emerges as a superior strategy to elucidate the physiological significance of PAs [
7]. Previous reports demonstrated that cyclohexylamine (CHA) effectively hinders the synthesis of Spd in cells [
7,
8,
9]. Whether in prokaryotes or eukaryotes, the introduction of CHA results in a notable reduction in intracellular Spd content, thereby significantly impeding the growth of organisms and compromising their resilience in the face of adversity, as observed in
Cryptococcus neoformans (yeast) [
7],
Escherichia coli (bacteria) [
8], and
Strombidium parasulcatum (ciliate) [
9]. These negative physiological effects caused by external addition of CHA can be alleviated through exogenous supplementation of Spd, providing further evidence that Spd is a pivotal biomolecule influencing biological growth [
7,
8,
9].
PAs serve as a significant contributor to the dissolved organic matter (DOM) in the marine environment [
10,
11]. Dynamic changes in the environmental concentration of PAs are highly related to planktons, with particular emphasis on diatoms and bacterioplankton communities [
10,
11,
12,
13]. Diatoms, noteworthy microalgae in the marine environment, exhibit ecological success due to their exceptional efficiency in carbon sequestration and crucial role in driving primary productivity, making them indispensable organisms within the marine ecosystem [
14,
15]. Many reports indicate that diatoms possess the capability to produce and release PAs, and algal blooms, which are predominantly composed of diatoms, can influence the PA composition in the environment [
10,
13]. Furthermore, the population densities of certain bacterioplanktons, such as Roseobacter, have been reported to elevate in response to increasing concentrations of PAs in seawater [
12]. Recent research reveals that secondary metabolites produced by diatoms play a role in stimulating the growth of specific bacteria, including Spd [
16,
17,
18]. Additionally, bacterioplankton has been demonstrated to release plant hormones and nitrogen sources that support the growth of diatoms, contributing to the establishment of mutualism [
19]. These reports suggest that the secretion of PAs through diatoms contributes to the promotion of ocean carbon cycling and the regulation of marine ecosystems.
Spd synthesis is primarily dominated by the spermidine synthase (SDS)-based synthetic pathway in the majority of organisms [
1,
2,
3]. Originating from arginine, it undergoes conversion into ornithine via arginase, followed by the removal of one CO
2 through ornithine decarboxylase (ODC) to initiate the synthesis of the foundational PA—putrescine (Put) [
20]. The substrate for the PA extension reactions, decarboxylated S-adenosylmethionine (dcSAM), is also a derivative of amino acid [
21]. It is synthesized from methionine through the enzymatic actions of methionine adenosyltransferase (MAT) and S-adenosylmethionine decarboxylase (SAMDC) [
1,
2,
3]. The elongation reaction of Put is facilitated by SDS, an aminopropyl transferase (APT) that transfers the aminopropyl group from dcSAM to Put, resulting in the formation of Spd [
22]. Several genome databases of diatoms are accessible, and the PA synthetic pathway has been predicted [
20,
23]. Nevertheless, only one diatom SDS in
Thalassiosira pseudonana (XP_002294888) has been verified for in vitro activity, despite the presence of an additional candidate SDS gene (XP_002287929) in its genome [
24]. Indeed, the in vivo investigation of the diatom Spd synthetic pathway has not been thorough to date [
20]. Utilizing
Phaeodactylum tricornutum as a model species, this study investigates enzyme activities and evaluates physiological functions associated with its two SDS candidate genes (
Scheme 1). This research contributes to a more comprehensive understanding of the role of SDS in the physiological processes of diatoms.
2. Materials and Methods
2.1. General Chemical Reagents
All experimental materials, including nutrient salts, vitamins, antibiotics and polyamine standards with reagent grade purity, were purchased from Sigma-Aldrich (St. Louis, MO, USA). dcSAM was obtained from Toronto Research Chemicals (Ontario, Canada). Perchloric acid (HClO4), toluene, and HPLC grade acetonitrile (ACN) were purchased from J.T. Baker Company (Phillipsburg, NJ, USA). Restriction enzymes and Gibson Assembly® Master Mix were purchased from New England Biolabs (Ipswich, MA, USA). The PCR-related reagents used in the experiments were obtained from BiOptic (Taipei, Taiwan).
2.2. Algal Cultivation and Enumeration
P. tricornutum (strain CCMP 632), Chaetoceros muelleri (strain CCMP 1316), Isochrysis galbana (strain CCMP 1323), and T. pseudonana (strain CCMP 1335) were purchased from Provasoli-Guillard National Center for Marine Algae and Microbiota (NCMA). All cultured microalgae were cultivated in L1 medium prepared in sterilized seawater and placed in a full-light incubator with a constant temperature of 20°C and an irradiance level of 115 photons m−2 s−1.
To monitor the proliferation of cells, the microalgae were cultured in advance to the exponential growth phase and then inoculated to ensure the consistency of the health status of the algae in each experiment. Next, 2 × 107 cell-cultured algae were cultured in 200 mL of fresh medium (starting density 105 cell/mL). The algal samples were collected from the culture flasks daily for counting the cell density during the cultivation process. At the same time, the algae samples were also collected on the 4th day (log phase) to analyze the levels of the PAs.
In order to determine the cell density, 1 mL of the algal sample was stained with Lugol’s reagent and placed in a Sedgewick-Rafter counting chamber (Wildlife Supply Company, Yulee, FL, USA) for 5 min. The cell number was determined with an optical microscope (Optiphot-2, Nikon, Tokyo, Japan) at 100× magnification.
2.3. Extraction, Modification, and Analysis of PAs from Microalgae
The algal PA extraction method was referenced from our previous study with slight modification [
9]. PA standards, including diaminopropane (Dap), Put, cadaverine (Cad), nor-spermidine (NSpd), Spd, nor-spermine (NSpm), and thermospermine (TSpm), were prepared in serial solutions at concentrations of 0.4, 0.8, 1.6, 3.2, and 6.4 μM (
Figure S1, Supplementary Materials). A cultured medium containing 2 × 10
8 algae cells was sampled through centrifugation (3000×
g, 10 min, 4 °C), followed by removal of the suspension. The pellet was reconstituted in 450 μL of 5% perchloric acid with 5 μM DAH (internal control). Subsequently, algal cells were disrupted using 0.01 mm diameter glass beads (BioSpec, Bartlesville, MA, USA) with a volume of approximately 100 μL in a ball mill (MM 301, Retsch, Germany) for 10 min, operating at a frequency of 30 Hz. Following separation from cellular debris via high-speed centrifugation (16,000×
g, 5 min, 4 °C), the transfer of 200 μL from the suspension to a new Eppendorf tube constituted the collection of algal PA extracts.
Subsequently, 275 μL of the derivatization reagent was added (with a final concentration of K2CO3 at 250 mM and dansyl chloride at 10 mM) and kept for continuous shaking at 100 rpm overnight in the dark. Then, 200 μL of 2 M proline was introduced and vortexed at 100 rpm for 1 h in darkness to facilitate the adsorption of free dansyl chloride. The derivatized PAs were further separated using 150 μL of toluene. Following the removal of the liquid-phase solvent using a vacuum dryer, the residue was subsequently re-dissolved in 1 mL of ACN.
Derivatized PA samples were analyzed via high-performance liquid chromatography (HPLC; Prominence-i LC-2030C Plus, Shimadzu, Kyoto, Japan), employing a C
18 column (ZORBAX Eclipse XDB-C18, 5 µm, 4.6 × 150 mm) and fluorescence detector (RF-20Axs, Shimadzu; excitation/emission, 340/515). In accordance with a previous study [
9], the mobile phase was employed at a flow rate of 1 mL/min, and a gradient process was established between H
2O and ACN for signal separation in detection. The entire gradient spanned 50 min. From 0–2.5 min, 65% ACN was used. Between 2.5 and 3.0 min, there was a linear increase in ACN to 75% and it was maintained from 3.0 to 15.0 min. At 15.0 min, there was a linear increase to 85% ACN and it was maintained as such from 16.0 to 22.0 min. From 22.0 to 37.0 min, ACN linearly increased to 100% and it was consistently maintained between 37.0 and 47.0 min. At 47.0 min, ACN linearly decreased to 65%. Between 47.1 and 50.0 min, the mobile phase returned to 65% ACN (
Table S1, Supplementary Materials).
2.4. Alignment Analysis
The amino acid sequences of spermidine synthase (SDS) and spermine synthase (SMS) used in this study were obtained from the National Center for Biotechnology Information (NCBI) from encompassed organisms such as Homo sapiens (HsSDS, NP_003123; HsSMS, NP_001245352), Arabidopsis thaliana (AtSDS1 and AtSDS2, NP_973900 and NP_177188; AtSMS, NP_001190527), and P. tricornutum (PtSDS1 and PtSDS2, XP_002185179 and XP_002185737). The amino acid sequences were aligned using the ClustalW method, and similarity analysis was conducted with DNASTAR Lasergene software.
2.5. 3D Structure Similarity Analysis
The DALI server was utilized for the similarity analysis of the N-terminal domains of PtSDS1 and PtSDS2 against the Dail-PDB25 subset Protein Data Bank. Both predicted models of PtSDS1 and PtSDS2 were downloaded from AlphaFold Protein Structure database (AFDB;
Supplementary Files 1 and 2) [
25,
26,
27]. To generate the query structure, the N-terminal domains of PtSDS1 (residues 14–74;
Supplementary File 3) and PtSDS2 (residues 21–102;
Supplementary File 4) were extracted.
2.6. Plasmid Construction
The nine plasmids used in this study included pGEX-PtSDS1-His, pGEX-PtSDS2-His, pGEX-PtSDS1 (Y79F)-His, pGEX-PtSDS2 (F79Y)-His, pET-myc-PtSDS1-His, pNR-PtSDS1-EGFP, pNR-PtSDS2-EGFP, pNR-PtSAMDC-EGFP, and pNR-PtSAMDC-His. All plasmids were assembled by ligating amplified PCR products with the Gibson Assembly
® Cloning Kit (New England Biolabs). The primer sequences used in the PCR amplification reaction are described in
Supplementary Table S2. PtSDS1 and PtSDS2 were amplified using the specific primers of PtSDS1 or PtSDS2 genes, and
P. tricornutum gDNA as a template, and then introduced into the
NdeI/
XhoI,
EcoRI/
XhoI, and
EcoRI/
AgeI restriction enzyme site intervals of pET-21a (Millipore, Bedford, MA, USA), pGEX-His (GE Healthcare, Chicago, IL, USA), and pNR-EGFP [
28] to generate pGEX-PtSDS1-His, pGEX-PtSDS2-His, pGEX-PtSDS1 (Y79F)-His, pGEX-PtSDS2 (F79Y)-His, pET-myc-PtSDS1-His, pNR-PtSDS1-EGFP, and pNR-PtSDS2-EGFP, a total of six plasmids. As for pET-myc-PtSDS1-His, myc tag and PtSDS1 PCR products were imported into the
NdeI/
XhoI interval of pET-21a. In addition, the amplicon generated by the PCR reaction using PtSAMDC-specific primers against
P. tricornutum gDNA was inserted into the
EcoRI/
AgeI restriction site of pNR-EGFP to yield pNR-PtSAMDC-EGFP. Alternatively, the PtSAMDC amplicon was introduced into the
EcoRI/
HindIII restriction site along with the His tag gene to construct the pNR-PtSAMDC-His plasmid.
2.7. Recombinant Protein Expression and Purification
The pGEX-PtSDS1-His, pGEX-PtSDS2-His, pGEX-PtSDS1 (Y79F)-His, pGEX-PtSDS2 (F79Y)-His, and pET-myc-PtSDS1-His plasmids were transferred to the competent cells of ArcticExpress (DE3) RIL strain (Agilent Technologies Inc., Santa Clara, CA, USA). Following an overnight incubation at 37 °C, these transgenic bacterial strains were subsequently diluted tenfold with LB medium (a total volume of 250 mL) and cultivated at 30 °C until reaching the logarithmic growth phase (OD600 value ranging between 0.4 and 0.6). Isopropyl β-D-1-thiogalactopyranoside (IPTG) was then introduced to attain a final concentration of 200 μM, followed by induction at 14 °C while agitating at 200 rpm for a duration of 16–18 h.
After the induced cultivation, the LB medium was replaced with 1/10 volume of binding buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8) by centrifugation (5000× g, 4 °C, 10 min). The bacterial samples were treated with sonication (S4000, Misonix, Farmingdale, NY, USA) to disrupt the cells and then separated through high-speed centrifugation, with the resulting fraction of the suspension solution containing crude protein. Then, 1 mL of Ni-NTA agarose resin (high density nickel resin, BCL-QHNi-100, Qiagen, Hilden, Germany) was used for purifying the target proteins (i.e., GST-PtSDS1-His, GST-PtSDS2-His, myc-PtSDS1-His, GST-PtSDS1 (F79Y)-His, or GST-PtSDS2 (Y79F)-His) as per the purification protocol provided by the original manufacturer. In brief, after resin equilibration with the binding buffer, the crude extract was passed through the column, and the target recombinant protein was subsequently eluted using a wash buffer (binding buffer containing 25 mM imidazole). After verifying the purity of the eluate through sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), the recombinant proteins were concentrated using an ultra-centrifugal filter (10 kDa cutoff, Millipore) and exchanged with a Tris-HCl buffer (100 mM, pH 7.4). The protein concentration in the recombinant protein samples was determined using a bicinchoninic aid (BCA) assay. Subsequently, the recombinant protein samples were mixed with an equal volume of glycerol and stored at −20 °C.
2.8. Aminopropyl Transferase Activity Assay
The in vitro activity assay for spermidine synthase entailed combining 2 µg of recombinant protein with 10 µM Put and 40 µM decarboxylated S-adenosylmethionine (dcSAM) in a total volume of 50 µL. The mixture was then incubated at 37 °C for 30 min, and the reaction was halted by adding 10 µL of 30% perchloric acid. Subsequently, the PA derivatization modification was carried out, followed by HPLC quantification to assess the progress of the catalytic reaction.
2.9. Pull-Down Assay of PtSDS1 and PtSDS2
Equimolar quantities of GST-PtSDS2-His recombinant protein were combined with myc-PtSDS1-His or GST-PtSDS1-His recombinant proteins in a 500 mL PBS buffer at 4°C for 18 h. Subsequently, Glutathione-Sepharose 4B beads (GE Healthcare) were introduced, and the mixture was gently agitated at room temperature for 30 min. After that, the beads were washed with PBS to eliminate nonspecifically bound proteins. Finally, the protein samples were washed with Laemmli sample buffer (GE Healthcare) and further analyzed using Western blotting.
Protein samples were resolved by 12% SDS-PAGE and then transferred to a polyvinylidene fluoride (PVDF) membrane, which was further incubated with an anti-Myc antibody (1:5000 in 5% skim milk) and an anti-mice immunoglobulin G (IgG) antibody (1:10,000 in 5% skim milk) as the primary and secondary antibodies, respectively. The signals were visualized using an ECL Plus chemiluminescence reagent (GE Healthcare) under an imaging system (UVP Biospectrum, Upland, CA, USA).
2.10. Gene Expression Pattern Analysis under Dark Synchronization
P. tricornutum in the exponential growth phase was inoculated at a density of 106 cells/mL into 1 L of fresh culture medium. The culture was then incubated for 24 h of dark synchronization, using an aluminum foil to block out the light source. Following the restoration of lighting conditions, 25 mL samples were collected every 3 h.
Before RNA extraction, the collected diatom samples were initially centrifuged (3000× g for 10 min at 4 °C) and processed in a ball mill (30 Hz for 10 min; MM 301, Retsch) with glass beads (0.01 mm diameter; BioSpec). Total RNA was extracted utilizing the RNeasy Plant Mini Kit (Qiagen) and RNase-free DNase I set (Qiagen), as per the instructions provided by manufacturer. RNA, quantified using nanodrop, was mixed with an equal volume of nucleic acid denaturant (2 × RNA loading dye, New England Biolabs), and heated for 10 min. Subsequently, the RNA quality was analyzed by agarose gel electrophoresis (2%).
The total RNA (500 ng) was reverse transcribed into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific Inc., Wilmington, DE, USA). The reverse transcription process was carried out at 42 °C for 60 min, followed by 70 °C incubation for 5 min. The procedures and conditions for quantitative PCR (QPCR) analysis were in accordance with the methods detailed in our prior research [
28]. In each analysis, a 20 μL reaction mixture consisted of the cDNA sample, specific primer pairs (300 nM), and 1 × SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). The specific primer pairs designed to detect the mRNA expression levels of PtSDS1, PtSDS2, and PtCycB1 were generated using Primer Express Software (Applied Biosystems, Thermo Fisher Scientific Inc., Foster City, CA, USA), whereas a reference gene, ribosomal protein small subunit 30S (RPS), was obtained from a prior study [
29]. All specific primers required for QPCR are listed in
Supplementary Table S3. The reactions were conducted using an Mx3000P QPCR system (Agilent Technologies, Santa Clara, CA, USA) with the following thermal profile: an initial cycle at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 sec and annealing/extension at 60 °C for 1 min. To confirm the specificity of the QPCR products, a melting temperature analysis was conducted on the same system, ranging from 55 °C to 95 °C.
The mRNA expression level was quantified by calculating the RNA molar ratio between a target gene (X) and a reference gene (R) using the following equation.
The methodology was elaborated in detail previously [
25]. In this equation, X
0 and R
0 correspond to the cDNA levels of the target and reference genes, respectively. M
X and M
R denote the molecular weights of X and R, while C
T,X and C
T,R represent the cycle threshold numbers for target and reference amplicons, respectively. Additionally, b
X and b
R indicate the slopes of standard curves for target and reference amplicons, respectively. Each standard curve was generated using serially diluted cDNA, which was reverse transcribed from 500 ng of total RNA (
Figure S2, Supplementary Information). The amplification efficiency (E) was calculated as [10
(−1/slope) − 1] × 100%.
2.11. The Multi-Pulse Electroporation Transformation for P. tricornutum
All operational procedures were based on a previous report with minor adjustments [
30].
P. tricornutum was pre-cultivated in EASW medium (
http://www3.botany.ubc.ca/cccm/NEPCC/esaw.html) until it reached the logarithmic growth phase. Diatom cells were harvested, followed by two washes with a 375 mM sorbitol solution. Subsequently, 2 × 10
8 cells were combined with 5 µg of linearized transgenic vector and 40 µg of salmon sperm DNA. After a 30-min incubation on ice, the sample was transferred to a 2-mm electroporation cuvette. Diatom electroporation was performed using the electrotransplantation system (Model PA-4000 Advanced PulseAgile, Cyto Pulse Sciences lnc., Glen Burnie, MA, USA), which generated multiple series of square wave pulses, including 8 poring pulses and 5 transferring pulses (
Table S4, Supplementary Materials). The transformed diatoms were promptly transferred to EASW medium and cultured for 24 h under relatively low light conditions. Subsequently, 5 × 10
7 diatom cells were smeared on 5 EASW agar plates (1.5%) containing 100 μg/mL Zeocin. About 20–100 brownish colonies appeared in 2–3 weeks under incubation at 20 °C with continuous illumination, which were individually transferred into 2 mL EASW medium containing 100 μg/mL Zeocin. After the 7-day screening period, PCR and green fluorescence measurement (excitation/emission 488/509 nm; SynergyMx, Biotek, Winooski, VT, USA) were used to detect surviving cells to verify the presence and expression of the reporter gene (EGFP). Additionally, a fluorescent microscope (Optiphot-2, Nikon, Tokyo, Japan) was used to observe the transgenic diatoms under 400× magnification.
2.12. PCR Verification of Transgenic Genes
Diatom genome was extracted using EasyPrep HY Genomic DNA Extraction Kit (Biotools, Taipei, Taiwan) as per the manufacturer’s instructions. Each PCR reaction contained 1 ng of genomic DNA, 500 nM specific primer pairs, and 1 × Gran Turismo PreMix (Ten Giga Bio, Keelung, Taiwan). The primers used to detect EGFP and the reference gene ribosomal protein small subunit 30S (RPS) are listed in
Table S3. The reaction was carried out in a thermal cycler (GeneAtlas G02, ASTEC, Fukuoka, Japan) and the PCR conditions were as follows: 10 min preheating at 95 °C; an amplification process with 32 cycles of 95 °C for 20 sec, 60 °C for 20 sec, and 72 °C for 30 sec; a final cycle of 72 °C for 5 min; then holding at 14 °C. The PCR products were analyzed by gel electrophoresis using an agarose gel (1%).
2.13. Diatom Protein Extraction and Analysis
A total of 5 × 108 diatom cells were harvested and centrifuged (3000× g, 10 min, 4 °C) to isolate the pellet. The pellet was resuspended in 200 μL of lysis buffer (10 mM 2-mercaptoethanol, 50 mM phosphate buffer, 0.1% Triton X-100, pH 7.3) with added glass beads (0.01 mm diameter; BioSpec) and mixed using a ball mill (30 Hz, 10 min; Retsch). Subsequent high-speed centrifugation (10,000× g, 10 min, 4 °C) yielded the crude protein suspension. The protein concentrations were assessed using the BCA reagent (Thermo Fisher) following the manufacturer’s instructions.
The expression level of EGFP, EGFP-fusion protein, or PtSAMDC-His was determined using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. A total of 25 μg of crude protein was separated by 12% SDS-PAGE, followed by either Coomassie Brilliant Blue staining or transfer onto a PVDF membrane. After treatment with a blocking buffer (5% skim milk), the PVDF membrane underwent sequential incubation with primary antibodies (anti-EGFP/anti-His; 1:5000 in 5% skim milk) and secondary antibodies (anti-rabbit/anti-mouse immunoglobulin antibodies; 1:5000 in 5% skim milk). The ECL signals on PVDF membrane were detected using an ECL reagent (Millipore) with an imaging system.
2.14. Statistical Analysis
All experiments and analyses were validated in three biological replicates. The results were represented as the standard deviation (SD) of the mean (mean ± SD). The Student’s t-test was used to determine comparative studies of means. Values of p < 0.05 indicated statistically significant differences.
4. Discussion
This work demonstrates the significant role of Spd produced through the SDS-based synthetic pathway in the growth of diatoms.
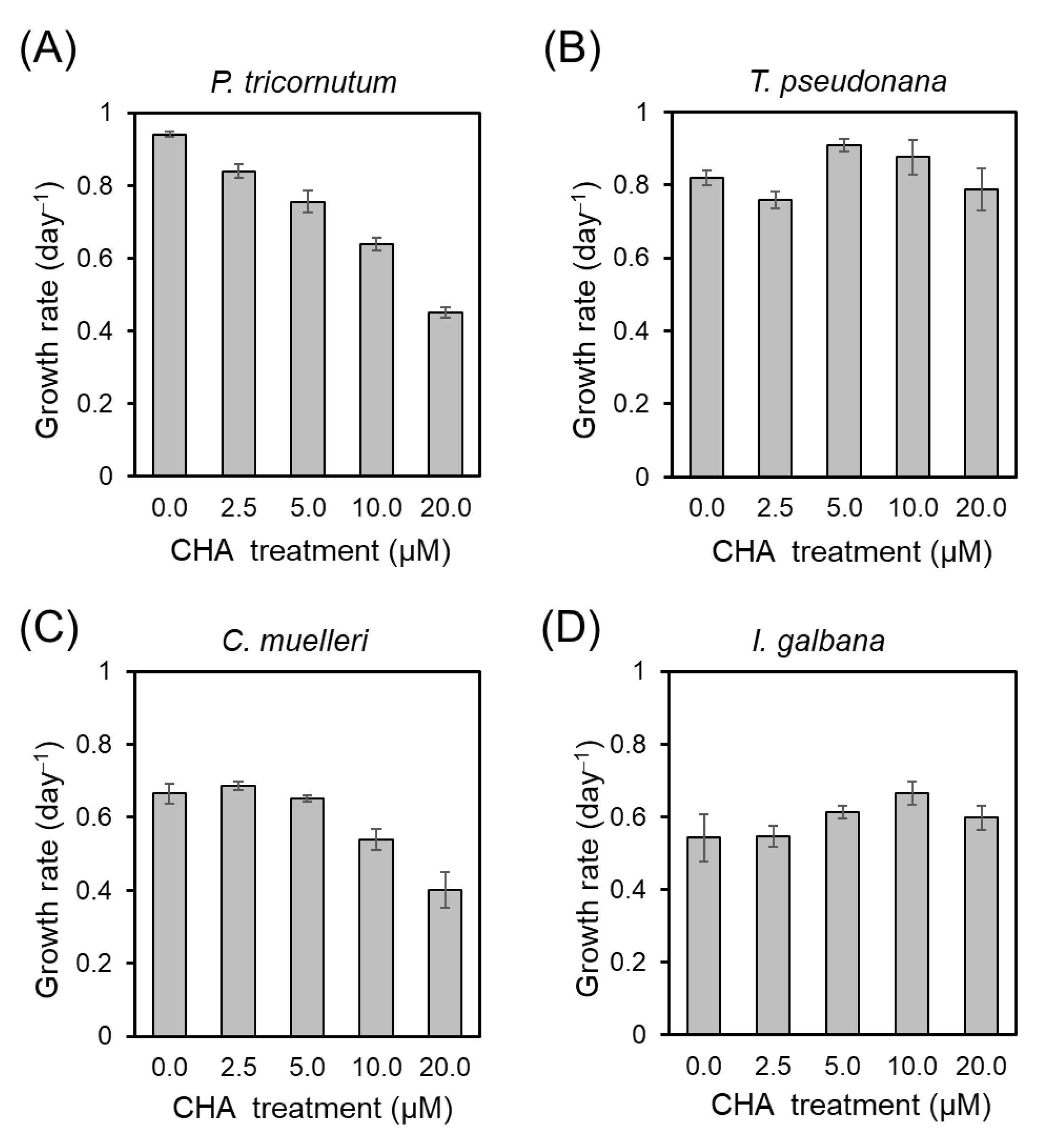
Figure 1 demonstrates varying impacts of CHA on the growth of four microalgae, including
P. tricornutum,
T. pseudonana,
C. muelleri, and
L. galbana. Notably, the growth of
P. tricornutum exhibited the most significant response to CHA, followed by
C. muelleri. In contrast, the growth rates of
T. pseudonana and
L. galbana remained relatively unchanged even under high concentrations of CHA (
Figure 1). The Spd levels relative to the total cellular PAs among the four microalgae, ranked from highest to lowest, were
P. tricornutum,
C. muelleri,
T. pseudonana, and
L. galbana (
Table 1). This pattern aligns with the trends observed in
Figure 1. These data suggest that Spd may only be highly relevant to the growth of some microalgae.
Previous research highlighted the presence of an uncommon PA, homospermidine (HSpd), in certain eukaryotic cells, serving as a homologous analog to Spd. Knocking out the SDS gene in yeast results in growth inhibition due to reduced Spd production, a suppression phenomenon that can be effectively reversed by adding HSpd [
32]. This study lends support to the potential for HSpd to physiologically substitute for Spd. In addition, eukaryotic initiation factor 5 A (eiF5a) is an important translation factor involved in eukaryotic protein synthesis, necessitating activation through modification by Spd [
33,
34]. Notably, in prokaryotic protein synthesis, the translation elongation factor P (EF-P), analogous to eiF5a, is employed, and its synthesis does not rely on Spd modification [
35]. Interestingly, microalgae often harbor prokaryotic genes utilizing horizontal gene transfer mechanisms to acquire bacterial genes [
36]. In fact, the presence of EF-P in the genome of
T. pseudonana is expected (PID 5581 in
T. pseudonana database of JGI). This observation leads us to speculate that EF-P may serve as a substitute for the function of eiF5a in certain microalgae. Consequently, the occurrence of Spd in some microalgae is notably scarce. For Spd-rich microalgae, such as
P. tricornutum (
Table 1), the regulation of PtSDS is essential for physiological processes.
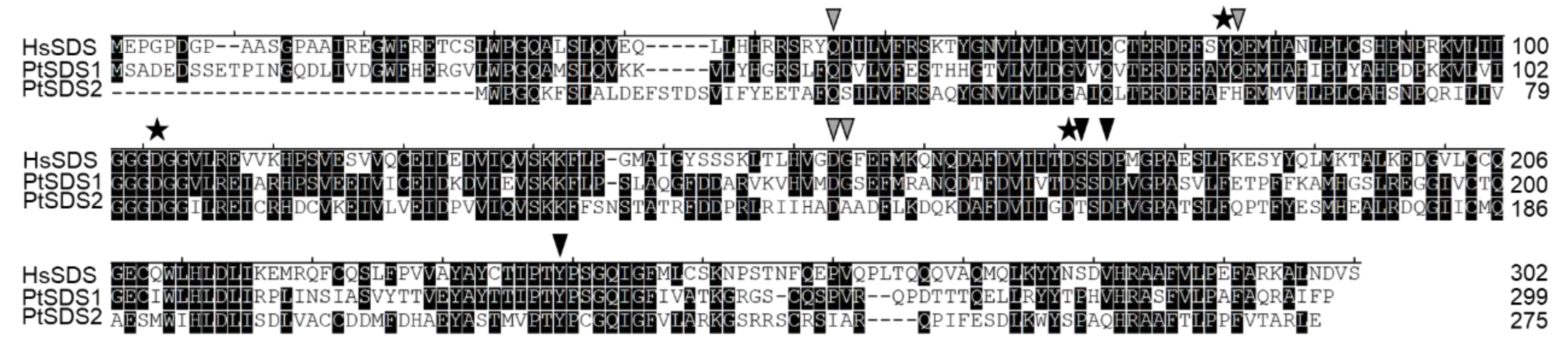
In
Figure 3, the sequence alignment highlights a distinct difference in the active site between PtSDS1 and PtSDS2, where Tyr79 is replaced by Phe79. This substitution may lead to reduced PtSDS2 activity due to the absence of the hydroxyl group in the side chain. Previously, the hydroxyl group of Tyr 79 is thought to augment the capability of Asp173 to deprotonate the N1 amine group in Put [
22]. This interaction plays a crucial role in enhancing the activity of spermidine synthase. In fact, the activity of PtSDS2 was 155-fold lower than the that of PtSDS1 (
Table 2). Further point mutation experiments demonstrated a similar trend. PtSDS1 exhibited approximately 10 times higher activity than PtSDS1-Y79F, while PtSDS2-F79Y displayed a catalytic activity around 10 times higher than PtSDS2. This underscores that the Tyr79 site is critical for the catalytic activity of PtSDS (
Table 2). Similar Y79F substitutions are also present in the SDS of other pennate diatoms, like one of the four SDS candidate genes in
Fragilariopsis cylindrus (OEU17362). Further exploration is warranted in the future to investigate whether similar distinctions exist in other related species. Nevertheless, the Y79F point mutation failed to induce a greater than 100-fold difference in activity between PtSDS1 and PtSDS2. This indicates that, apart from the active site, variations in other substrate binding sites, such as D80H, G156A, and S174T (
Figure 3), might also play a role in influencing protein activity to some extent.
PtSDS1 exhibits substrate selectivity by catalyzing Put and Cad, functioning as an atypical spermidine synthase (
Table 2). In vitro activity assays conducted on SDS from rats,
E. coli, and
Thermotoga maritima revealed similar characteristics [
22,
37,
38]. Notably, in all cases, the
Km values for Cad were significantly higher than those for Put. Moreover, employing CHA to inhibit SDS activity resulted in a substantial elevation of Put, with no impact on Cad (
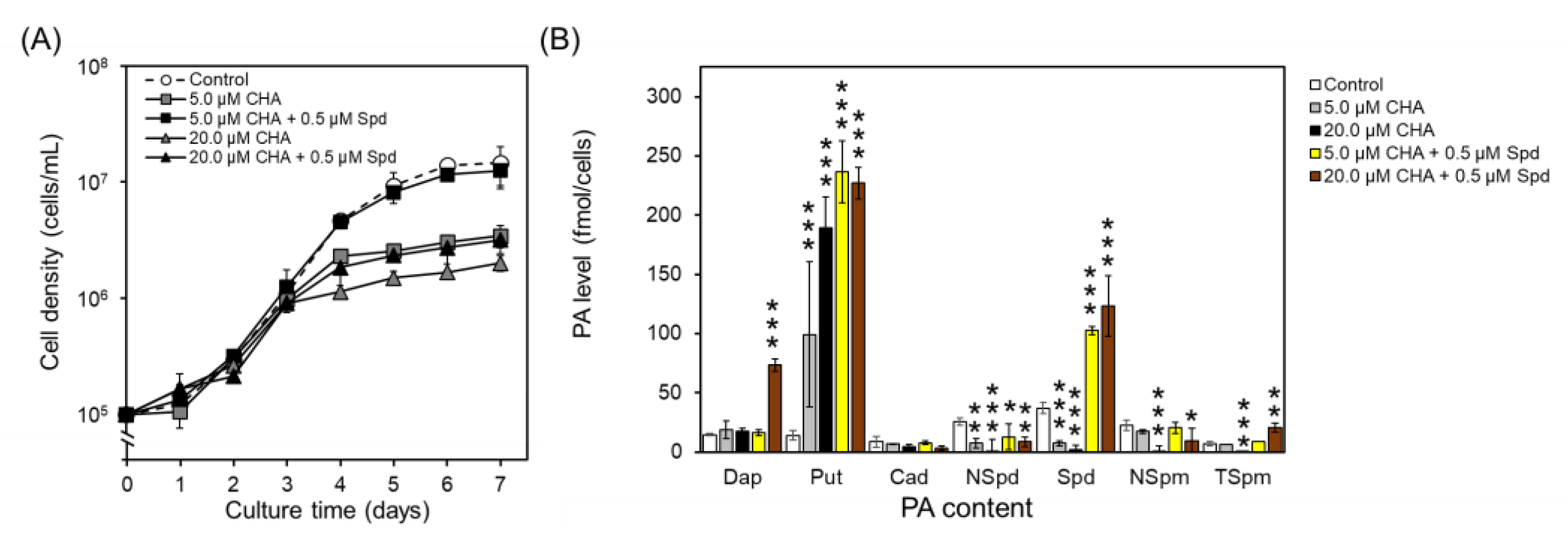
Figure 2). These results imply that under physiological conditions, Cad does not serve as the primary catalytic target for SDS, but rather, Put dominates. Following CHA treatment, Spd, as well as other higher PAs, including NSpd, NSpm, and TSpm, exhibited significant reductions (
Figure 2). Upon Spd supplementation, these PAs showed partial restoration, suggesting that NSpd, NSpm, and TSpm might be downstream metabolites influenced by Spd and regulated by other enzymes. Intriguingly, a parallel trend emerged in the PtSDS1/PtSAMDC co-overexpressing strain. In contrast to the wild type, this strain exhibited not only elevated levels of Spd but also a corresponding increase in NSpd and NSpm within the PA composition, providing additional support for the aforementioned argument (
Figure 6). These data underscore the significance of SDS in the PA synthetic pathway of diatoms.
Eukaryotes, along with certain viruses, depend on the SDS-based biosynthetic pathway to produce higher-order PAs, such as Spd [
1,
2,
3]. A recent report revealed that cyanobacteria possess an alternative pathway for Spd synthesis known as the carboxyaminopropylagmatine-based pathway, including carboxyaminopropylagmatine dehydrogenase (CAPADH), carboxyaminopropylagmatine decarboxylase (CAPADC), and aminopropylagmatine ureohydrolase (APAUH), to convert arginine into Spd [
39]. Homologs of these enzymes do not exist in diatoms, suggesting that they are more similar to eukaryotic regulatory mechanisms [
20,
39]. This study reveals that individual expressions of PtSDS1, PtSDS2, and PtSAMDC in
P. tricornutum do not alter PA composition or cell growth (
Figure 6). However, co-expression of PtSDS1 and SAMDC leads to changes in PA composition and an increased growth rate. This observation highlights that, in the SDS-based biosynthetic pathway, the cellular Spd content may be constrained by dcSAM. Similar regulation exists in other microalgae [
20]. For instance, the inhibition of SAMDC activity using methylglyoxal bis-guanylhydrazone (MGBG) in
Chattonella antiqua markedly constrained both cellular Spd content and cell growth [
40]. Notably, alongside PtSAMDC,
P. tricornutum also harbors three SAMDC-APT fusion genes [
23]. This suggests the potential existence of additional regulatory mechanisms in diatoms for the synthesis of distinct PAs.
PtSDS1 and PtSDS2, both found in the cytoplasm in
P. tricornutum, exhibit distinct regulatory influences on the cell cycle (
Figure 5). The expression of the
PtSDS1 gene remained consistently stable during cell cycle, whereas the
PtSDS2 gene exhibited an increase during the G
1 phase and a subsequent decrease during the S phase. In various organisms, Spd has been observed to fulfill a physiological function by stabilizing and safeguarding DNA [
1,
2]. This regulatory mechanism may aid diatom cells in preparing for DNA replication during the G
1 phase [
41]. The lack of a notable impact on activity when PtSDS1 and PtSDS2 were combined (
Figure S4B, Supplementary Materials) suggests that PtSDS2 may harbor additional physiological functions. This could involve the formation of metabolite channeling with other functionally correlated catalytic enzymes, a phenomenon referred to as metabolome [
42]. Forming a metabolome offers several advantages, including enhanced efficiency in utilizing substrates by enzymes, mitigation of harmful intermediate accumulation, minimized influence of surrounding molecules on metabolic flux, and prevention of substrate or intermediate competition by other metabolic pathways [
43]. The presence of a metabolome in the TCA cycle, Calvin cycle, and urea cycle facilitates the more efficient execution of catalytic reactions and resource allocation within the intricate intracellular environment [
44,
45,
46]. This, in turn, enhances an organism’s capacity to adapt to environmental changes [
43]. Considering the prediction that at least eight enzymes within the genome of
P. tricornutum are implicated in the synthesis of distinct PAs [
23], our hypothesis is that these enzymes are likely to establish a metabolome or other complex relationship. An essential focus for future research involves investigating whether these proteins can form a metabolon through protein-protein interactions (PPI), potentially contributing to the formation of biologically relevant PAs with more intricate structures, such as long-chain polyamines (LCPAs) [
20,
23,
47].



 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}