


Mutations in Glycosyltransferases and Glycosidases: Implications for Associated Diseases

Abstract
1. Introduction
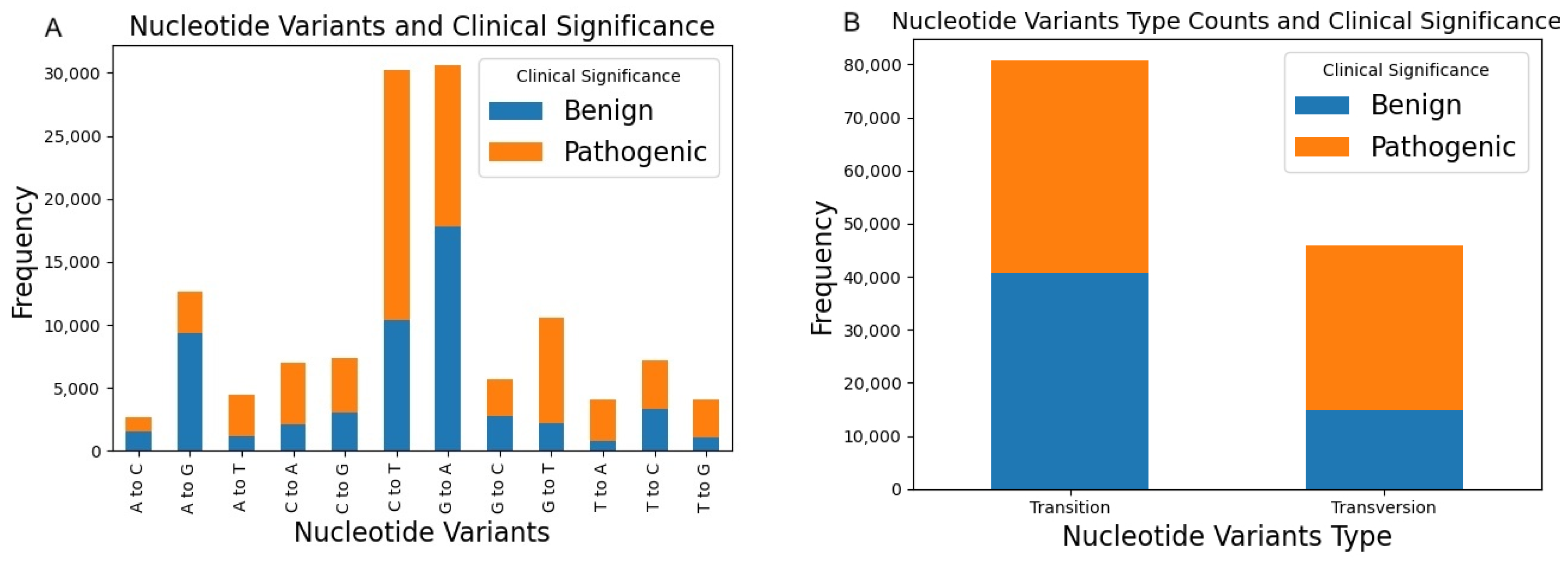
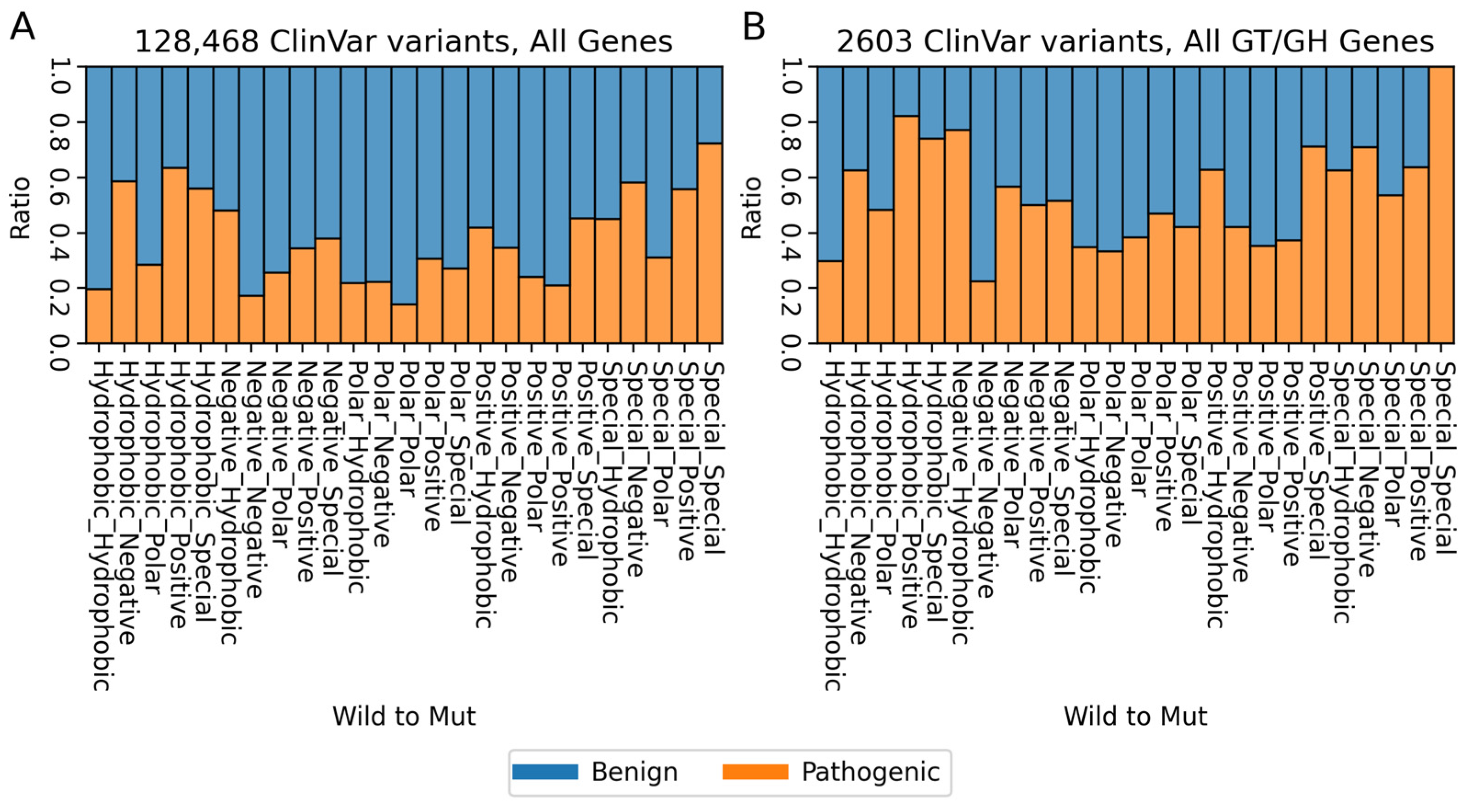
2. Variants of Human Glycosyltransferases and Glycoside Hydrolases in ClinVar
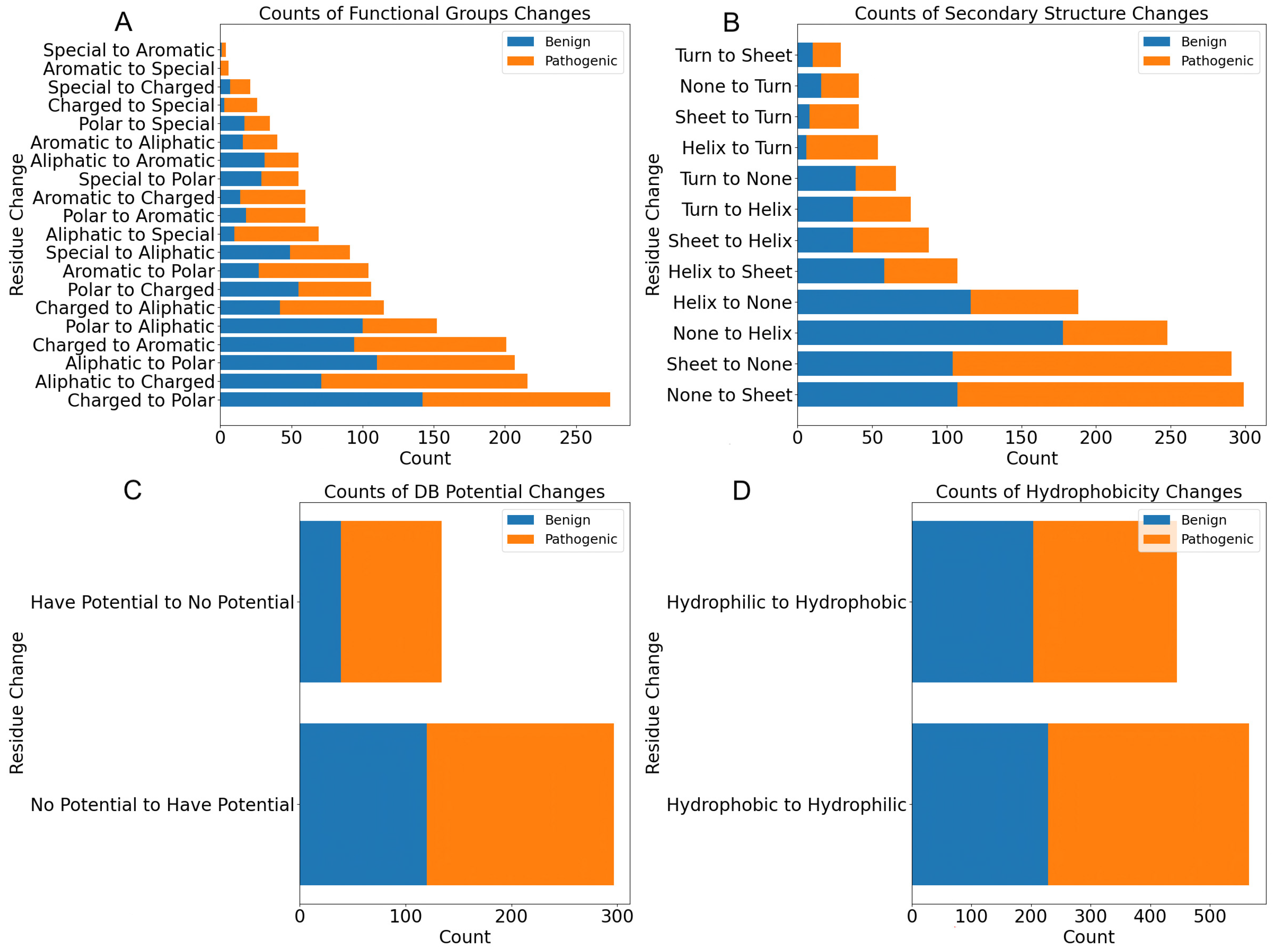
3. Comprehensive Structural Analysis of Glycosyltransferases and Glycoside Hydrolases (GT/GHs)
3.1. Alpha-Galactosidase A, GLA
3.2. Lysosomal Alpha-Glucosidase, GAA
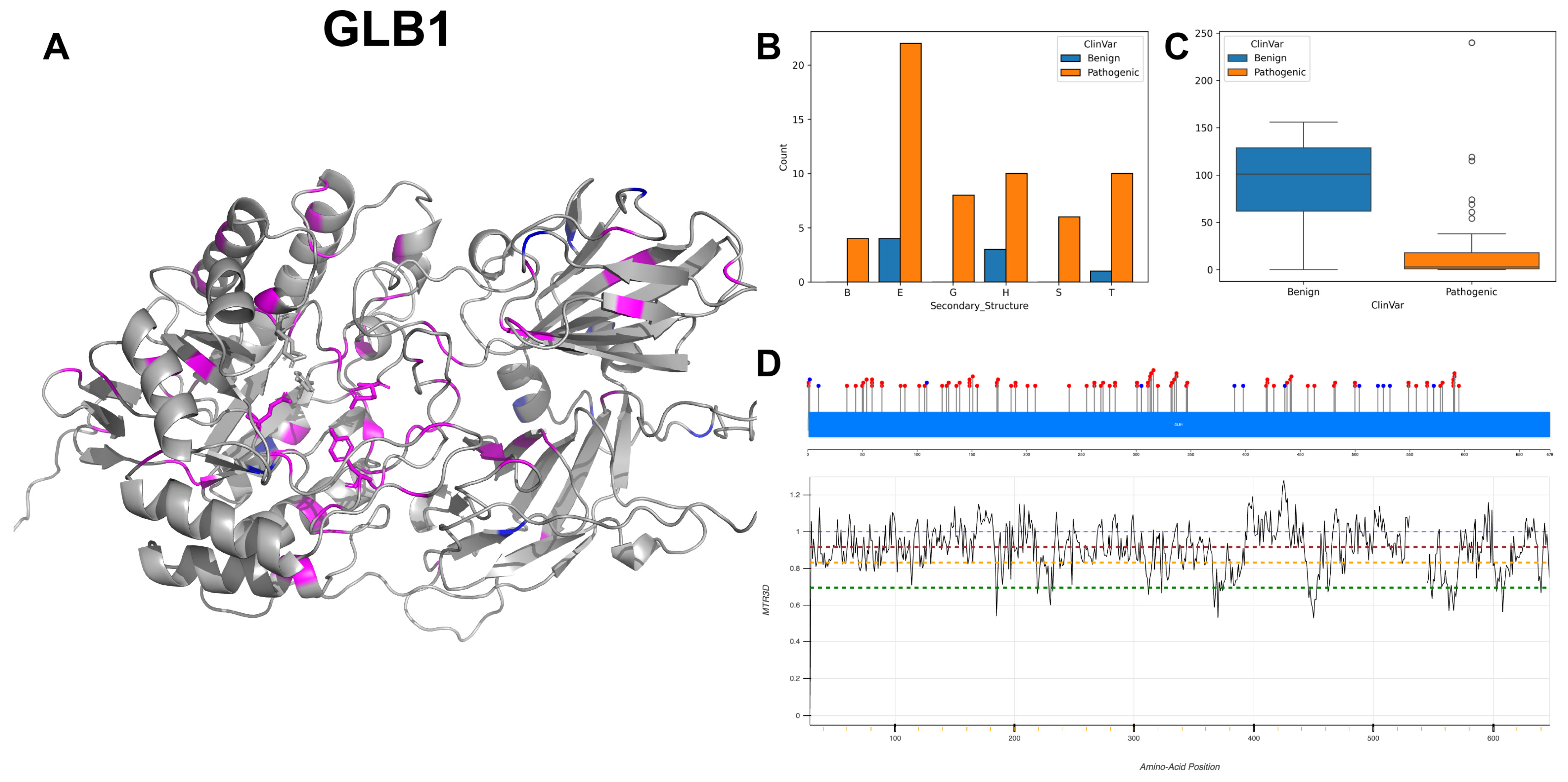
3.3. Beta-Galactosidase, GLB1
3.4. Galactocerebrosidase, GALC
3.5. Alpha-N-Acetylgucosaminidase, NAGLU
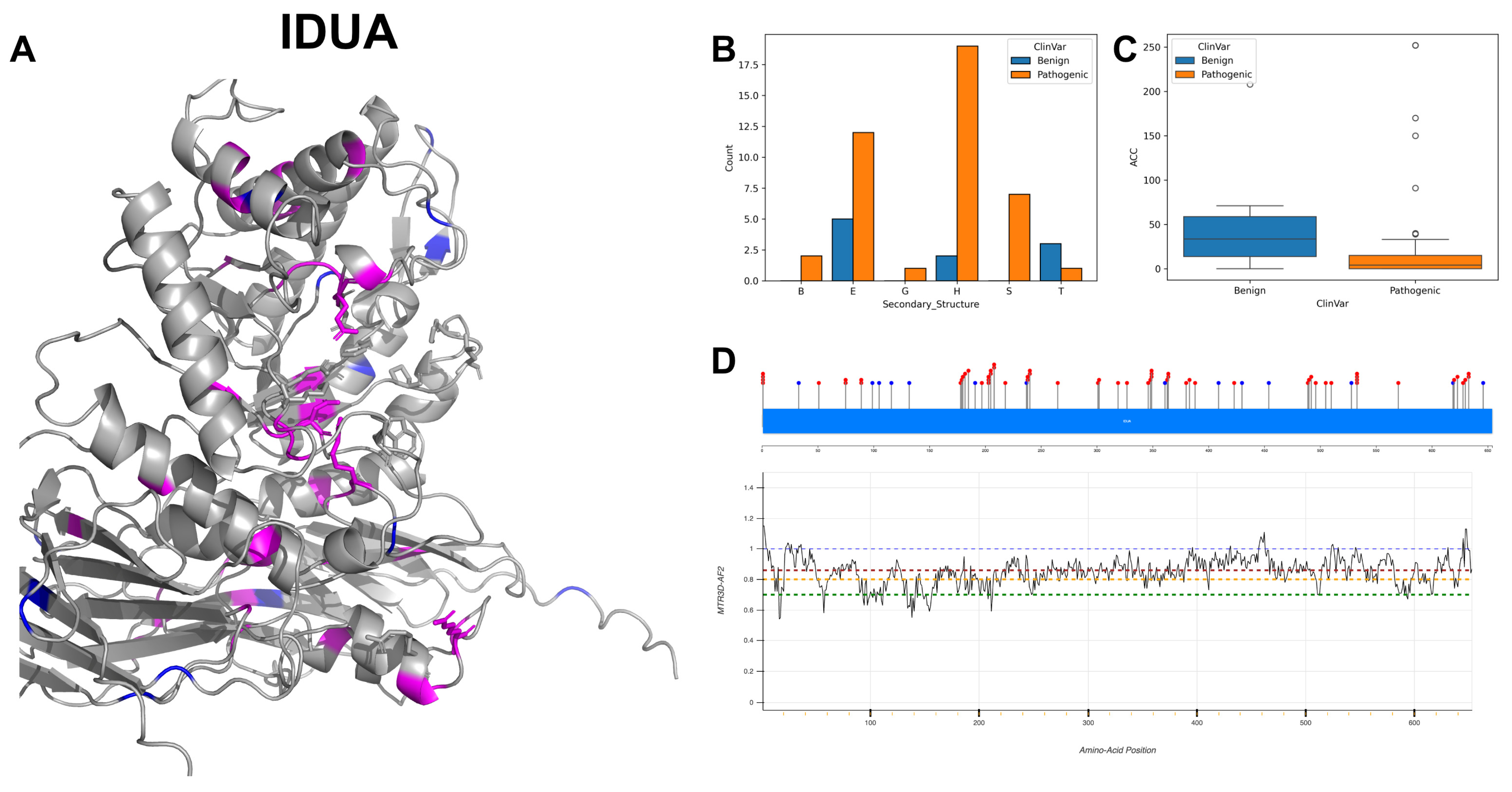
3.6. Alpha-L-Iduronidase, IDUA
3.7. Lysosomal Acid Glucosylceramidase, GBA1
3.8. Putative Bifunctional UDP-N-Acetylglucosamine Transferase and Deubiquitinase ALG13, ALG13
3.9. Exostosin-1, EXT1
3.10. Beta-Hexosaminidase Subunit Alpha, HEXA
3.11. Bifunctional UDP-N-Acetylglucosamine 2-Epimerase/N-Acetylmannosamine Kinase, GNE
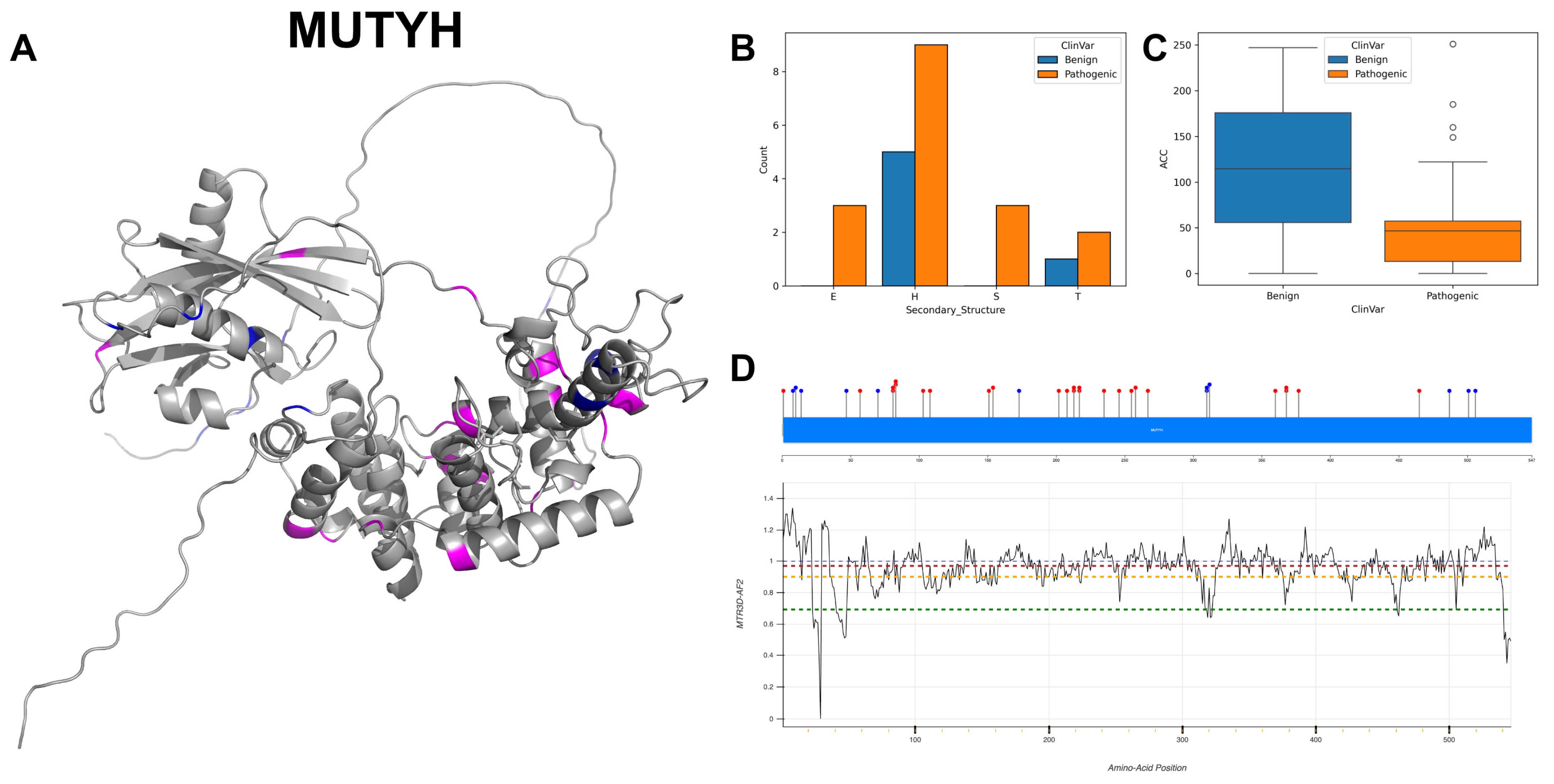
3.12. Adenine DNA Glycosylase, MUTYH
3.13. Lysosomal Alpha-Mannosidase, MAN2B1
3.14. Beta-Hexosaminidase Subunit Beta, HEXB
3.15. 1,4-Alpha-Glucan-Branching Enzyme, GBE1
3.16. Phosphatidylinositol N-Acetylglucosaminyltransferase Subunit A, PIGA
3.17. Glycogen Debranching Enzyme, AGL
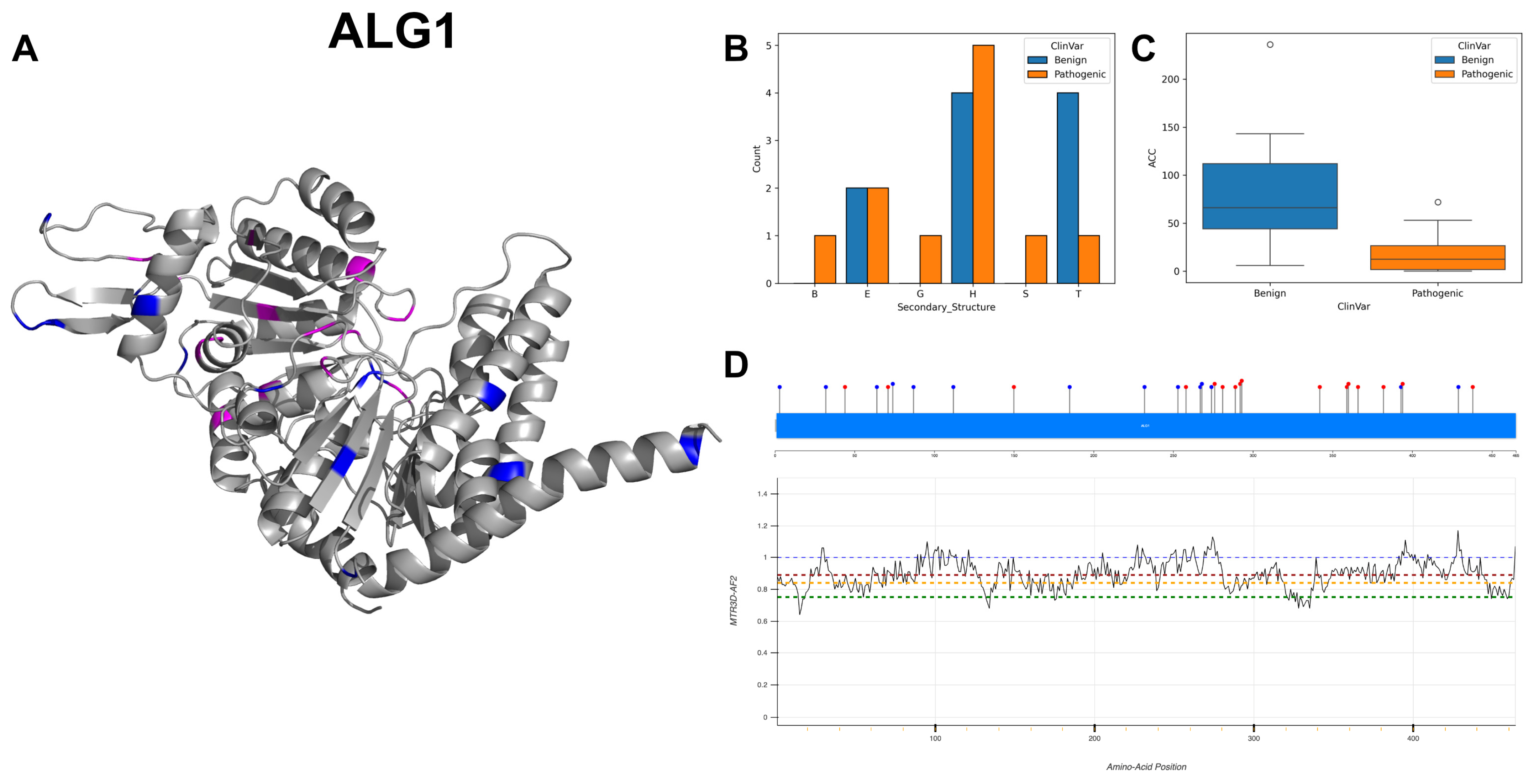
3.18. Chitobiosyldiphosphodolichol Beta-Mannosyltransferase, ALG1
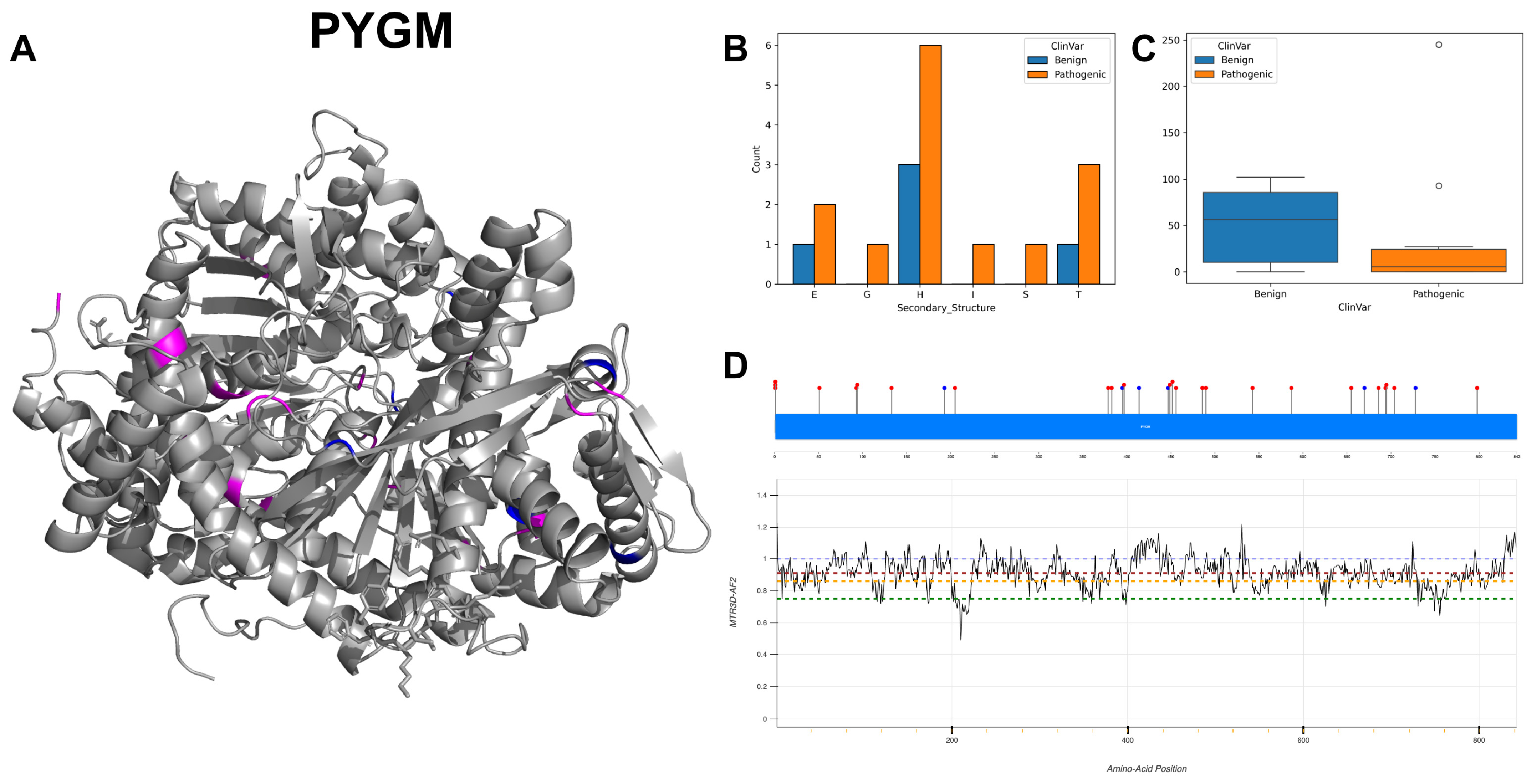
3.19. Glycogen Phosphorylase, Muscle Form, PYGM
3.20. Sucrase-Isomaltase, Intestinal, SI
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dods, R.; Dods, R. Posttranslational Modifications (PTMs): Alteration of the Three-Dimensional Structure of Proteins. Concepts Biosci. Eng. 2019, 151–179. [Google Scholar]
- Trincone, A.; Giordano, A. Glycosyl Hydrolases and Glycosyltransferases in the Synthesis of Oligosaccharides. Curr. Org. Chem. 2006, 10, 1163–1193. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Venkat, A.; Tehrani, D.; Taujale, R.; Yeung, W.; Gravel, N.; Moremen, K.W.; Kannan, N. Modularity of the hydrophobic core and evolution of functional diversity in fold A glycosyltransferases. J. Biol. Chem. 2022, 298, 102212. [Google Scholar] [CrossRef] [PubMed]
- Joshi, H.J.; Hansen, L.; Narimatsu, Y.; Freeze, H.H.; Henrissat, B.; Bennett, E.; Wandall, H.H.; Clausen, H.; Schjoldager, K.T. Glycosyltransferase genes that cause monogenic congenital disorders of glycosylation are distinct from glycosyltransferase genes associated with complex diseases. Glycobiology 2018, 28, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Moll, T.; Ramesh, T.; Castelli, L.; Beer, A.; Robins, H.; Fox, I.; Niedermoser, I.; Van Damme, P.; Moisse, M.; et al. Mutations in the Glycosyltransferase Domain of GLT8D1 Are Associated with Familial Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 26, 2298–2306.e5. [Google Scholar] [CrossRef] [PubMed]
- Takata, R.; Tokita, K.; Mori, S.; Shimoda, R.; Harada, N.; Ichinose, H.; Kaneko, S.; Igarashi, K.; Samejima, M.; Tsumuraya, Y.; et al. Degradation of carbohydrate moieties of arabinogalactan-proteins by glycoside hydrolases from Neurospora crassa. Carbohydr. Res. 2010, 345, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.-C.; Oh, D.-K. Classification of glycosidases that hydrolyze the specific positions and types of sugar moieties in ginsenosides. Crit. Rev. Biotechnol. 2016, 36, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, M.; Kitamura, M.; Hondoh, H.; Kang, M.-S.; Mori, H.; Kimura, A.; Tanaka, I.; Yao, M. Catalytic Mechanism of Retaining α-Galactosidase Belonging to Glycoside Hydrolase Family 97. J. Mol. Biol. 2009, 392, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.M.; Oster, M.E.; Hebert, D.N. Protein Quality Control in the Endoplasmic Reticulum. Protein J. 2019, 38, 317–329. [Google Scholar] [CrossRef]
- Anstee, D.J. The relationship between blood groups and disease. Blood 2010, 115, 4635–4643. [Google Scholar] [CrossRef]
- Harrus, D.; Kellokumpu, S.; Glumoff, T. Crystal structures of eukaryote glycosyltransferases reveal biologically relevant enzyme homooligomers. Cell. Mol. Life Sci. 2018, 75, 833–848. [Google Scholar] [CrossRef]
- Mittler, M.; Bechthold, A.; Schulz, G.E. Structure and action of the C-C bond-forming glycosyltransferase biosynthesis of the UrdGT2 involved in the antibiotic urdamycin. J. Mol. Biol. 2007, 372, 67–76. [Google Scholar] [CrossRef]
- Ondruskova, N.; Cechova, A.; Hansikova, H.; Honzik, T.; Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129751. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef]
- Diekmann, L.; Pfeiffer, K.; Naim, H.Y. Congenital lactose intolerance is triggered by severe mutations on both alleles of the lactase gene. BMC Gastroenterol. 2015, 15, 36. [Google Scholar] [CrossRef]
- Nordstrom, L.J.; Clark, C.A.; Andersen, B.; Champlin, S.M.; Schwinefus, J.J. Effect of Ethylene Glycol, Urea, and N-Methylated Glycines on DNA Thermal Stability: The Role of DNA Base Pair Composition and Hydration. Biochemistry 2006, 45, 9604–9614. [Google Scholar] [CrossRef]
- Stoltzfus, A.; Norris, R.W. On the Causes of Evolutionary Transition:Transversion Bias. Mol. Biol. Evol. 2016, 33, 595–602. [Google Scholar] [CrossRef]
- Vitkup, D.; Sander, C.; Church, G.M. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003, 4, R72. [Google Scholar] [CrossRef]
- Silk, M.; Pires, D.; Rodrigues, C.M.; D’souza, E.N.; Olshansky, M.; Thorne, N.; Ascher, D. MTR3D: Identifying regions within protein tertiary structures under purifying selection. Nucleic Acids Res. 2021, 49, W438–W445. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Alexandrov, V.; Lehnert, U.; Echols, N.; Milburn, D.; Engelman, D.; Gerstein, M. Normal modes for predicting protein motions: A comprehensive database assessment and associated Web tool. Protein Sci. 2005, 14, 633–643. [Google Scholar] [CrossRef]
- Mayrose, I.; Graur, D.; Ben-Tal, N.; Pupko, T. Comparison of Site-Specific Rate-Inference Methods for Protein Sequences: Empirical Bayesian Methods Are Superior. Mol. Biol. Evol. 2004, 21, 1781–1791. [Google Scholar] [CrossRef]
- Garman, S.C.; Garboczi, D.N. The Molecular Defect Leading to Fabry Disease: Structure of Human α-Galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef]
- Hogarth, V.; Dhoat, S.; Mehta, A.B.; Orteu, C.H. Late-onset Fabry disease associated with angiokeratoma of Fordyce and multiple cherry angiomas. Clin. Exp. Dermatol. 2011, 36, 506–508. [Google Scholar] [CrossRef]
- Lukas, J.; Scalia, S.; Eichler, S.; Pockrandt, A.-M.; Dehn, N.; Cozma, C.; Giese, A.; Rolfs, A. Functional and Clinical Consequences of Novel α-Galactosidase A Mutations in Fabry Disease. Hum. Mutat. 2016, 37, 43–51. [Google Scholar] [CrossRef]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase—A guide for the treatment of Pompe disease. Nat. Commun. 2017, 8, 1111. [Google Scholar] [CrossRef]
- Schröder, B.; Wrocklage, C.; Pan, C.; Jäger, R.; Kösters, B.; Schäfer, H.; Elsässer, H.; Mann, M.; Hasilik, A. Integral and Associated Lysosomal Membrane Proteins. Traffic 2007, 8, 1676–1686. [Google Scholar] [CrossRef]
- Montalvo, A.; Bembi, B.; Donnarumma, M.; Filocamo, M.; Parenti, G.; Rossi, M.; Merlini, L.; Buratti, E.; De Filippi, P.; Dardis, A.; et al. Mutation profile of theGAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum. Mutat. 2006, 27, 999–1006. [Google Scholar] [CrossRef]
- Jia, X.; Shao, L.; Liu, C.; Chen, T.; Peng, L.; Cao, Y.; Zhang, C.; Yang, X.; Zhang, G.; Gao, J.; et al. GAA compound heterozygous mutations associated with autophagic impairment cause cerebral infarction in Pompe disease. Aging 2020, 12, 4268–4282. [Google Scholar] [CrossRef]
- Ohto, U.; Usui, K.; Ochi, T.; Yuki, K.; Satow, Y.; Shimizu, T. Crystal structure of human beta-galactosidase: Structural basis of Gm1 gangliosidosis and morquio B diseases. J. Biol. Chem. 2012, 287, 1801–1812. [Google Scholar] [CrossRef]
- Suzuki, H.; Ohto, U.; Higaki, K.; Mena-Barragán, T.; Aguilar-Moncayo, M.; Mellet, C.O.; Nanba, E.; Fernandez, J.M.G.; Suzuki, Y.; Shimizu, T. Structural basis of pharmacological chaperoning for human beta-galactosidase. J. Biol. Chem. 2014, 289, 14560–14568. [Google Scholar] [CrossRef]
- Willemsen, R.; Hoogeveen, A.T.; Sips, H.J.; Van Dongen, J.M.; Galjaard, H. Immunoelectron microscopical localization of lysosomal beta-galactosidase and its precursor forms in normal and mutant human fibroblasts. Eur. J. Cell Biol. 1986, 40, 9–15. [Google Scholar]
- Morreau, H.; Galjart, N.J.; Gillemans, N.; Willemsen, R.; Van Der Horst, G.T.; D’Azzo, A. Alternative splicing of beta-galactosidase mRNA generates the classic lysosomal enzyme and a beta-galactosidase-related protein. J. Biol. Chem. 1989, 264, 20655–20663. [Google Scholar] [CrossRef]
- Tappino, B.; Biancheri, R.; Mort, M.; Regis, S.; Corsolini, F.; Rossi, A.; Stroppiano, M.; Lualdi, S.; Fiumara, A.; Bembi, B.; et al. Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum. Mutat. 2010, 31, E1894–E1914. [Google Scholar] [CrossRef]
- Furuya, H.; Kukita, Y.-J.; Nagano, S.; Sakai, Y.; Yamashita, Y.; Fukuyama, H.; Inatomi, Y.; Saito, Y.; Koike, R.; Tsuji, S.; et al. Adult onset globoid cell leukodystrophy (Krabbe disease): Analysis of galactosylceramidase cDNA from four Japanese patients. Hum. Genet. 1997, 100, 450–456. [Google Scholar] [CrossRef]
- Ouesleti, S.; Coutinho, M.F.; Ribeiro, I.; Miled, A.; Mosbahi, D.S.; Alves, S. Update of the spectrum of mucopolysaccharidoses type III in Tunisia: Identification of three novel mutations and in silico structural analysis of the missense mutations. World J. Pediatr. 2017, 13, 374–380. [Google Scholar] [CrossRef]
- Beesley, C.E.; Jackson, M.; Young, E.P.; Vellodi, A.; Winchester, B.G. Molecular defects in Sanfilippo syndrome type B (mucopolysaccharidosis IIIB). J. Inherit. Metab. Dis. 2005, 28, 759–767. [Google Scholar] [CrossRef]
- Bie, H.; Yin, J.; He, X.; Kermode, A.R.; Goddard-Borger, E.D.; Withers, S.G.; James, M.N. Insights into mucopolysaccharidosis I from the structure and action of alpha-L-iduronidase. Nat. Chem. Biol. 2013, 9, 739–745. [Google Scholar] [CrossRef]
- Maita, N.; Tsukimura, T.; Taniguchi, T.; Saito, S.; Ohno, K.; Taniguchi, H.; Sakuraba, H. Human α-l-iduronidase uses its own N-glycan as a substrate-binding and catalytic module. Proc. Natl. Acad. Sci. USA 2013, 110, 14628–14633. [Google Scholar] [CrossRef]
- Bertola, F.; Filocamo, M.; Casati, G.; Mort, M.; Rosano, C.; Tylki-Szymanska, A.; Tüysüz, B.; Gabrielli, O.; Grossi, S.; Scarpa, M.; et al. IDUA mutational profiling of a cohort of 102 European patients with mucopolysaccharidosis type I: Identification and characterization of 35 novel α-L-iduronidase (IDUA) alleles. Hum. Mutat. 2011, 32, E2189–E2210. [Google Scholar] [CrossRef]
- Premkumar, L.; Sawkar, A.R.; Boldin-Adamsky, S.; Toker, L.; Silman, I.; Kelly, J.W.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-beta-glucosidase covalently bound to conduritol-B-epoxide. Implications for Gaucher disease. J. Biol. Chem. 2005, 280, 23815–23819. [Google Scholar] [CrossRef]
- Yap, T.L.; Velayati, A.; Sidransky, E.; Lee, J.C. Membrane-bound α-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol. Genet. Metab. 2013, 108, 56–64. [Google Scholar] [CrossRef]
- Jian, J.; Tian, Q.Y.; Hettinghouse, A.; Zhao, S.; Liu, H.; Wei, J.; Grunig, G.; Zhang, W.; Setchell, K.D.R.; Sun, Y.; et al. Progranulin Recruits HSP70 to beta-Glucocerebrosidase and Is Therapeutic Against Gaucher Disease. EBioMedicine 2016, 13, 212–224. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef]
- Miočić, S.; Filocamo, M.; Dominissini, S.; Montalvo, A.L.E.; Vlahoviček, K.; Deganuto, M.; Mazzotti, R.; Cariati, R.; Bembi, B.; Pittis, M.G. Identification and functional characterization of five novel mutant alleles in 58 Italian patients with Gaucher disease type 1. Hum. Mutat. 2005, 25, 100. [Google Scholar] [CrossRef]
- Liou, B.; Kazimierczuk, A.; Zhang, M.; Scott, C.R.; Hegde, R.S.; Grabowski, G.A. Analyses of variant acid beta-glucosidases: Effects of Gaucher disease mutations. J. Biol. Chem. 2006, 281, 4242–4253. [Google Scholar] [CrossRef]
- Latham, T.E.; Theophilus, B.D.M.; Grabowski, G.A.; Smith, F.I. Heterogeneity of mutations in the acid beta-glucosidase gene of Gaucher disease patients. DNA Cell Biol. 1991, 10, 15–21. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, R.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. DNAJC6 Mutations Associated with Early-Onset Parkinson’s Disease. Ann. Neurol. 2016, 79, 244–256. [Google Scholar]
- Chantret, I.; Dancourt, J.; Barbat, A.; Moore, S.E. Two proteins homologous to the N- and C-terminal domains of the bacterial glycosyltransferase Murg are required for the second step of dolichyl-linked oligosaccharide synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 9236–9242. [Google Scholar] [CrossRef]
- Bickel, T.; Lehle, L.; Schwarz, M.; Aebi, M.; Jakob, C.A. Biosynthesis of lipid-linked oligosaccharides in Saccharomyces cerevisiae: Alg13p and Alg14p form a complex required for the formation of GlcNAc(2)-PP-dolichol. J. Biol. Chem. 2005, 280, 34500–34506. [Google Scholar] [CrossRef]
- Gao, X.-D.; Moriyama, S.; Miura, N.; Dean, N.; Nishimura, S.-I. Interaction between the C Termini of Alg13 and Alg14 Mediates Formation of the Active UDP-N-acetylglucosamine Transferase Complex. J. Biol. Chem. 2008, 283, 32534–32541. [Google Scholar] [CrossRef]
- Timal, S.; Hoischen, A.; Lehle, L.; Adamowicz, M.; Huijben, K.; Sykut-Cegielska, J.; Paprocka, J.; Jamroz, E.; van Spronsen, F.J.; Körner, C.; et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet. 2012, 21, 4151–4161. [Google Scholar] [CrossRef] [PubMed]
- Epi4K Consortium; Epilepsy Phenome/Genome Project. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef]
- Alsharhan, H.; He, M.; Edmondson, A.C.; Daniel, E.J.; Chen, J.; Donald, T.; Bakhtiari, S.; Amor, D.J.; Jones, E.A.; Vassallo, G.; et al. ALG13 X-linked intellectual disability: New variants, glycosylation analysis, and expanded phenotypes. J. Inherit. Metab. Dis. 2021, 44, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-D.; Tachikawa, H.; Sato, T.; Jigami, Y.; Dean, N. Alg14 Recruits Alg13 to the Cytoplasmic Face of the Endoplasmic Reticulum to Form a Novel Bipartite UDP-N-acetylglucosamine Transferase Required for the Second Step of N-Linked Glycosylation. J. Biol. Chem. 2005, 280, 36254–36262. [Google Scholar] [CrossRef]
- Wang, C.D.; Xu, S.; Chen, S.; Chen, Z.H.; Dean, N.; Wang, N.; Gao, X.D. An in vitro assay for enzymatic studies on human ALG13/14 heterodimeric UDP-N-acetylglucosamine transferase. Front. Cell Dev. Biol. 2022, 10, 1008078. [Google Scholar] [CrossRef]
- Esposito, T.; De Stefano, G.; Reccia, M.G.; Di Lorenzo, I.; Napolitano, F.; Scalabrì, F.; Lombardi, A.; Saleem, M.A.; Griffiths, L.R.; Gianfrancesco, F. Dysregulation of the Expression of Asparagine-Linked Glycosylation 13 Short Isoform 2 Affects Nephrin Function by Altering Its N-Linked Glycosylation. Nephron 2017, 136, 143–150. [Google Scholar] [CrossRef]
- Missaghian, P.; Dierker, T.; Khosrowabadi, E.; Axling, F.; Eriksson, I.; Ghanem, A.; Kusche-Gullberg, M.; Kellokumpu, S.; Kjellén, L. A dominant negative splice variant of the heparan sulfate biosynthesis enzyme NDST1 reduces heparan sulfate sulfation. Glycobiology 2022, 32, 518–528. [Google Scholar] [CrossRef]
- Senay, C.; Lind, T.; Muguruma, K.; Tone, Y.; Kitagawa, H.; Sugahara, K.; Lidholt, K.; Lindahl, U.; Kusche-Gullberg, M. The EXT1/EXT2 tumor suppressors: Catalytic activities and role in heparan sulfate biosynthesis. EMBO Rep. 2000, 1, 282–286. [Google Scholar] [CrossRef]
- Bernard, M.A.; Hall, C.E.; Hogue, D.A.; Cole, W.G.; Scott, A.; Snuggs, M.B.; Clines, G.A.; Lovett, M.; Van Winkle, W.B.; Hecht, J.T.; et al. Diminished levels of the putative tumor suppressor proteins EXT1 and EXT2 in exostosis chondrocytes. Cell Motil. Cytoskelet. 2001, 48, 149–162. [Google Scholar] [CrossRef]
- Lemieux, M.J.; Mark, B.L.; Cherney, M.M.; Withers, S.G.; Mahuran, D.J.; James, M.N. Crystallographic structure of human beta-hexosaminidase A: Interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis. J. Mol. Biol. 2006, 359, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Tse, R.; Mahuran, D.J. Direct determination of the substrate specificity of the alpha-active site in heterodimeric beta-hexosaminidase A. Biochemistry 1996, 35, 3963–3969. [Google Scholar] [CrossRef]
- Dersh, D.; Iwamoto, Y.; Argon, Y. Tay–Sachs disease mutations in HEXA target the α chain of hexosaminidase A to endoplasmic reticulum–associated degradation. Mol. Biol. Cell 2016, 27, 3813–3827. [Google Scholar] [CrossRef]
- Martinez, J.; Nguyen, L.D.; Hinderlich, S.; Zimmer, R.; Tauberger, E.; Reutter, W.; Saenger, W.; Fan, H.; Moniot, S. Crystal Structures of N-Acetylmannosamine Kinase Provide Insights into Enzyme Activity and Inhibition. J. Biol. Chem. 2012, 287, 13656–13665. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Baron, P.; Prelle, A.; Serafini, M.; Moggio, M.; Fonzo, A.D.; Castagni, M.; Bresolin, N.; Comi, G.P. Novel missense mutation and large deletion of GNE gene in autosomal-recessive inclusion-body myopathy. Muscle Nerve 2003, 28, 113–117. [Google Scholar] [CrossRef]
- Leroy, J.G.; Seppala, R.; Huizing, M.; Dacremont, G.; De Simpel, H.; Van Coster, R.N.; Orvisky, E.; Krasnewich, D.M.; Gahl, W.A. Dominant Inheritance of Sialuria, an Inborn Error of Feedback Inhibition. Am. J. Hum. Genet. 2001, 68, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Komine, K.; Shimodaira, H.; Takao, M.; Soeda, H.; Zhang, X.; Takahashi, M.; Ishioka, C. Functional Complementation Assay for 47 MUTYH Variants in a MutY-Disrupted Escherichia coli Strain. Hum. Mutat. 2015, 36, 704–711. [Google Scholar] [CrossRef]
- D’agostino, V.G.; Minoprio, A.; Torreri, P.; Marinoni, I.; Bossa, C.; Petrucci, T.C.; Albertini, A.M.; Ranzani, G.N.; Bignami, M.; Mazzei, F. Functional analysis of MUTYH mutated proteins associated with familial adenomatous polyposis. DNA Repair 2010, 9, 700–707. [Google Scholar] [CrossRef]
- Dallosso, A.R.; Dolwani, S.; Jones, N.; Jones, S.; Colley, J.; Maynard, J.; Idziaszczyk, S.; Humphreys, V.; Arnold, J.; Donaldson, A.; et al. Inherited predisposition to colorectal adenomas caused by multiple rare alleles of MUTYH but not OGG1, NUDT1, NTH1 or NEIL 1, 2 or 3. Gut 2008, 57, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Malm, D.; Nilssen, O. Alpha-mannosidosis. Orphanet J. Rare Dis. 2008, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Stensland, H.M.F.R.; Klenow, H.B.; Van Nguyen, L.; Hansen, G.M.; Malm, D.; Nilssen, Ø. Identification of 83 novel alpha-mannosidosis-associated sequence variants: Functional analysis of MAN2B1 missense mutations. Hum. Mutat. 2012, 33, 511–520. [Google Scholar] [CrossRef]
- Mark, B.L.; Mahuran, D.J.; Cherney, M.M.; Zhao, D.; Knapp, S.; James, M.N. Crystal structure of human beta-hexosaminidase B: Understanding the molecular basis of Sandhoff and Tay-Sachs disease. J. Mol. Biol. 2003, 327, 1093–1109. [Google Scholar] [CrossRef] [PubMed]
- Canibano-Fraile, R.; Harlaar, L.; dos Santos, C.A.; Hoogeveen-Westerveld, M.; Demmers, J.A.A.; Snijders, T.; Lijnzaad, P.; Verdijk, R.M.; van der Beek, N.A.M.E.; van Doorn, P.A.; et al. Lysosomal glycogen accumulation in Pompe disease results in disturbed cytoplasmic glycogen metabolism. J. Inherit. Metab. Dis. 2023, 46, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Magoulas, P.L.; El-Hattab, A.W.; Roy, A.; Bali, D.S.; Finegold, M.J.; Craigen, W.J. Diffuse reticuloendothelial system involvement in type IV glycogen storage disease with a novel GBE1 mutation: A case report and review. Hum. Pathol. 2012, 43, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Siripanyaphinyo, U.; Hong, Y.; Tashima, Y.; Maeda, Y.; Kinoshita, T. The Initial Enzyme for Glycosylphosphatidylinositol Biosynthesis Requires PIG-Y, a Seventh Component. Mol. Biol. Cell 2005, 16, 5236–5246. [Google Scholar] [CrossRef]
- Fauth, C.; Steindl, K.; Toutain, A.; Farrell, S.; Witsch-Baumgartner, M.; Karall, D.; Joset, P.; Böhm, S.; Baumer, A.; Maier, O.; et al. A recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2. Am. J. Med. Genet. A 2016, 170A, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Gregory, B.L.; Shelton, G.D.; Bali, D.S.; Chen, Y.T.; Fyfe, J.C. Glycogen storage disease type IIIa in curly-coated retrievers. J. Vet. Intern. Med. 2007, 21, 40–46. [Google Scholar]
- Goldstein, J.L.; Austin, S.L.; Boyette, K.; Kanaly, A.; Veerapandiyan, A.; Rehder, C.; Kishnani, P.S.; Bali, D.S. Molecular analysis of the AGL gene: Identification of 25 novel mutations and evidence of genetic heterogeneity in patients with Glycogen Storage Disease Type III. Anesthesia Analg. 2010, 12, 424–430. [Google Scholar] [CrossRef]
- Shen, J.J.; Chen, Y.T. Molecular Characterization of Glycogen Storage Disease Type III. Curr. Mol. Med. 2002, 2, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Shiryaev, S.A.; Rymen, D.; Eklund, E.A.; Raymond, K.; Kircher, M.; Abdenur, J.E.; Alehan, F.; Midro, A.T.; Bamshad, M.J.; et al. ALG1-CDG: Clinical and Molecular Characterization of 39 Unreported Patients. Hum. Mutat. 2016, 37, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Kranz, C.; Denecke, J.; Lehle, L.; Sohlbach, K.; Jeske, S.; Meinhardt, F.; Rossi, R.; Gudowius, S.; Marquardt, T. Congenital Disorder of Glycosylation Type Ik (CDG-Ik): A Defect of Mannosyltransferase I. Am. J. Hum. Genet. 2004, 74, 545–551. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andreu, A.L.; Nogales-Gadea, G.; Cassandrini, D.; Arenas, J.; Bruno, C. McArdle disease: Molecular genetic update. Acta Myol. 2007, 26, 53–57. [Google Scholar] [PubMed]
- Lukacs, C.M.; Oikonomakos, N.G.; Crowther, R.L.; Hong, L.-N.; Kammlott, R.U.; Levin, W.; Li, S.; Liu, C.-M.; Lucas-McGady, D.; Pietranico, S.; et al. The crystal structure of human muscle glycogen phosphorylase a with bound glucose and AMP: An intermediate conformation with T-state and R-state features. Proteins Struct. Funct. Bioinform. 2006, 63, 1123–1126. [Google Scholar] [CrossRef]
- Sim, L.; Willemsma, C.; Mohan, S.; Naim, H.Y.; Pinto, B.M.; Rose, D.R. Structural Basis for Substrate Selectivity in Human Maltase-Glucoamylase and Sucrase-Isomaltase N-terminal Domains. J. Biol. Chem. 2010, 285, 17763–17770. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | UniProt ID | Protein Name | Family * (Fold) | Variant Counts | Number of Benign | Number of Pathogenic |
|---|---|---|---|---|---|---|
| GLA | P06280 | Alpha-galactosidase A | GH27 (GH-D) | 207 | 5 | 202 |
| GAA | P10253 | Lysosomal Alpha-glucosidase | GH31 (GH-D) | 167 | 17 | 150 |
| GLB1 | P16278 | Beta-galactosidase | GH35 (GH-A) | 97 | 12 | 85 |
| GALC | P54803 | Galactocerebrosidase | GH59 (GH-A) | 90 | 10 | 80 |
| NAGLU | P54802 | Alpha-N-acetylglucosaminidase | GH89 | 83 | 11 | 72 |
| IDUA | P35475 | Alpha-L-iduronidase | GH39 (GH-A) | 75 | 14 | 61 |
| GBA1 | P04062 | Lysosomal acid glucosylceramidase | GH30 (GH-A) | 73 | 6 | 67 |
| ALG13 | Q9NP73 | Putative bifunctional UDP-N-acetylglucosamine transferase and deubiquitinase ALG13 | GT1 (GT-B) | 56 | 50 | 6 |
| EXT1 | Q16394 | Exostosin-1 | GT47; GT64 (GT-B; GT-A) | 49 | 15 | 34 |
| HEXA | P06865 | Beta-hexosaminidase subunit alpha | GH20 (GH-K) | 45 | 4 | 41 |
| GNE | Q9Y223 | Bifunctional UDP-N-Acetylglucosamine 2-Epimerase/N-Acetylmannosamine Kinase | 39 | 1 | 38 | |
| MUTYH | Q9UIF7 | Adenine DNA glycosylase | 38 | 12 | 26 | |
| MAN2B1 | O00754 | Lysosomal alpha-mannosidase | GH38 | 36 | 22 | 14 |
| HEXB | P07686 | Beta-hexosaminidase subunit beta | GH20 (GH-K) | 33 | 13 | 20 |
| GBE1 | Q04446 | 1,4-alpha-glucan-branching enzyme | CBM48; GH13; (GH-H) | 31 | 14 | 17 |
| PIGA | P37287 | Phosphatidylinositol N-acetylglucosaminyltransferase subunit A | GT4 (GT-B) | 31 | 16 | 15 |
| AGL | P35573 | Glycogen debranching enzyme | GH13; GH133; (GH-H) | 30 | 21 | 9 |
| ALG1 | Q9BT22 | Chitobiosyldiphosphodolichol beta-mannosyltransferase | GT33 (GT-B) | 30 | 14 | 16 |
| PYGM | P11217 | Glycogen phosphorylase, muscle form | GT35 (GT-B) | 30 | 6 | 24 |
| SI | P14410 | Sucrase-isomaltase, intestinal | GH31 (GH-D) | 30 | 28 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, X.; Kovacs, A.S.; Myung, Y.; Ascher, D.B. Mutations in Glycosyltransferases and Glycosidases: Implications for Associated Diseases. Biomolecules 2024, 14, 497. https://doi.org/10.3390/biom14040497
Gu X, Kovacs AS, Myung Y, Ascher DB. Mutations in Glycosyltransferases and Glycosidases: Implications for Associated Diseases. Biomolecules. 2024; 14(4):497. https://doi.org/10.3390/biom14040497
Chicago/Turabian StyleGu, Xiaotong, Aaron S. Kovacs, Yoochan Myung, and David B. Ascher. 2024. "Mutations in Glycosyltransferases and Glycosidases: Implications for Associated Diseases" Biomolecules 14, no. 4: 497. https://doi.org/10.3390/biom14040497
APA StyleGu, X., Kovacs, A. S., Myung, Y., & Ascher, D. B. (2024). Mutations in Glycosyltransferases and Glycosidases: Implications for Associated Diseases. Biomolecules, 14(4), 497. https://doi.org/10.3390/biom14040497