
Adenosine Triphosphate: The Primordial Molecule That Controls Protein Homeostasis and Shapes the Genome–Proteome Interface
Abstract
:
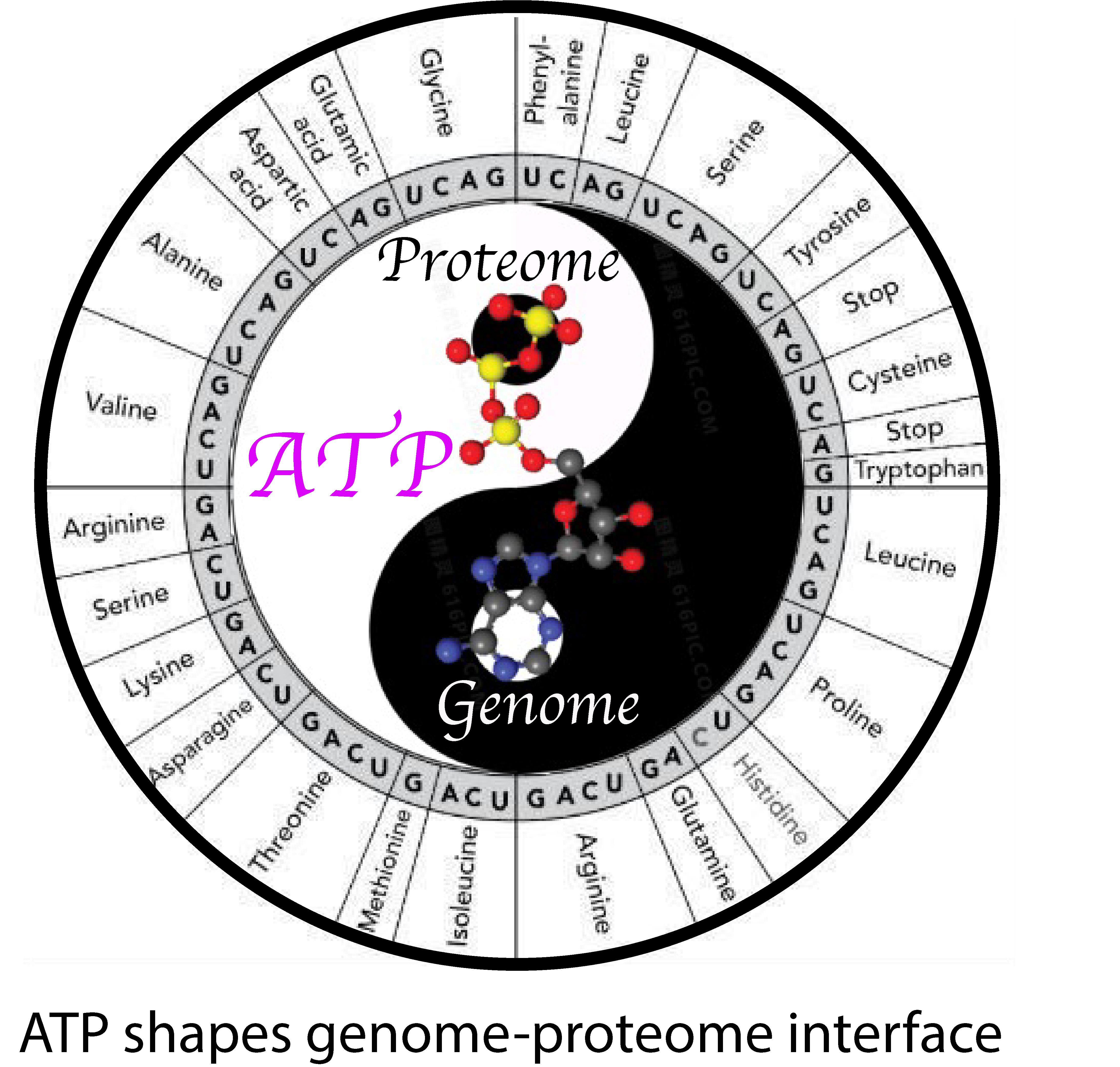{kind=link}
{kind=link}
{kind=link}
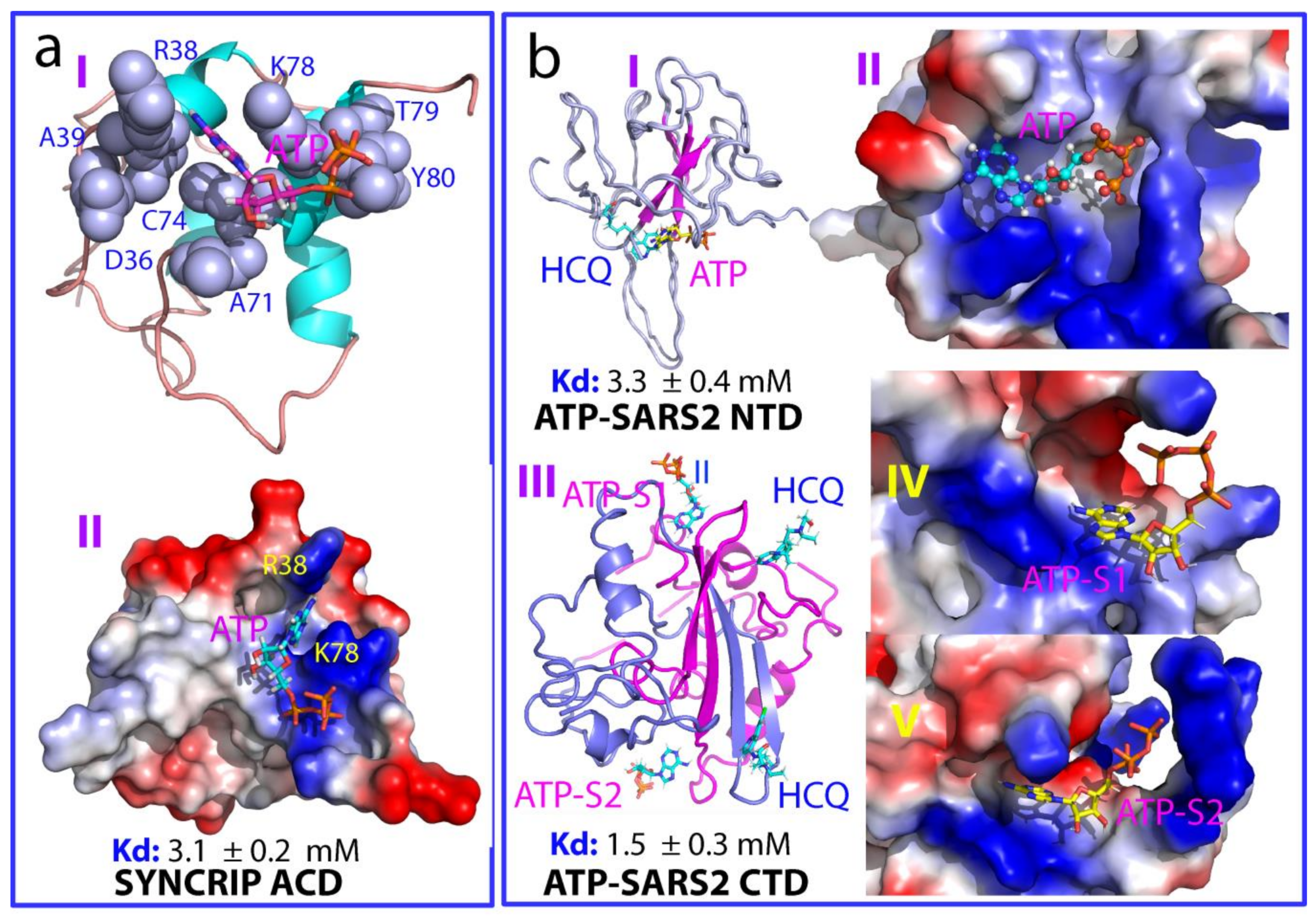{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. ATP Binds Folded Nucleic-Acid-Binding Domains
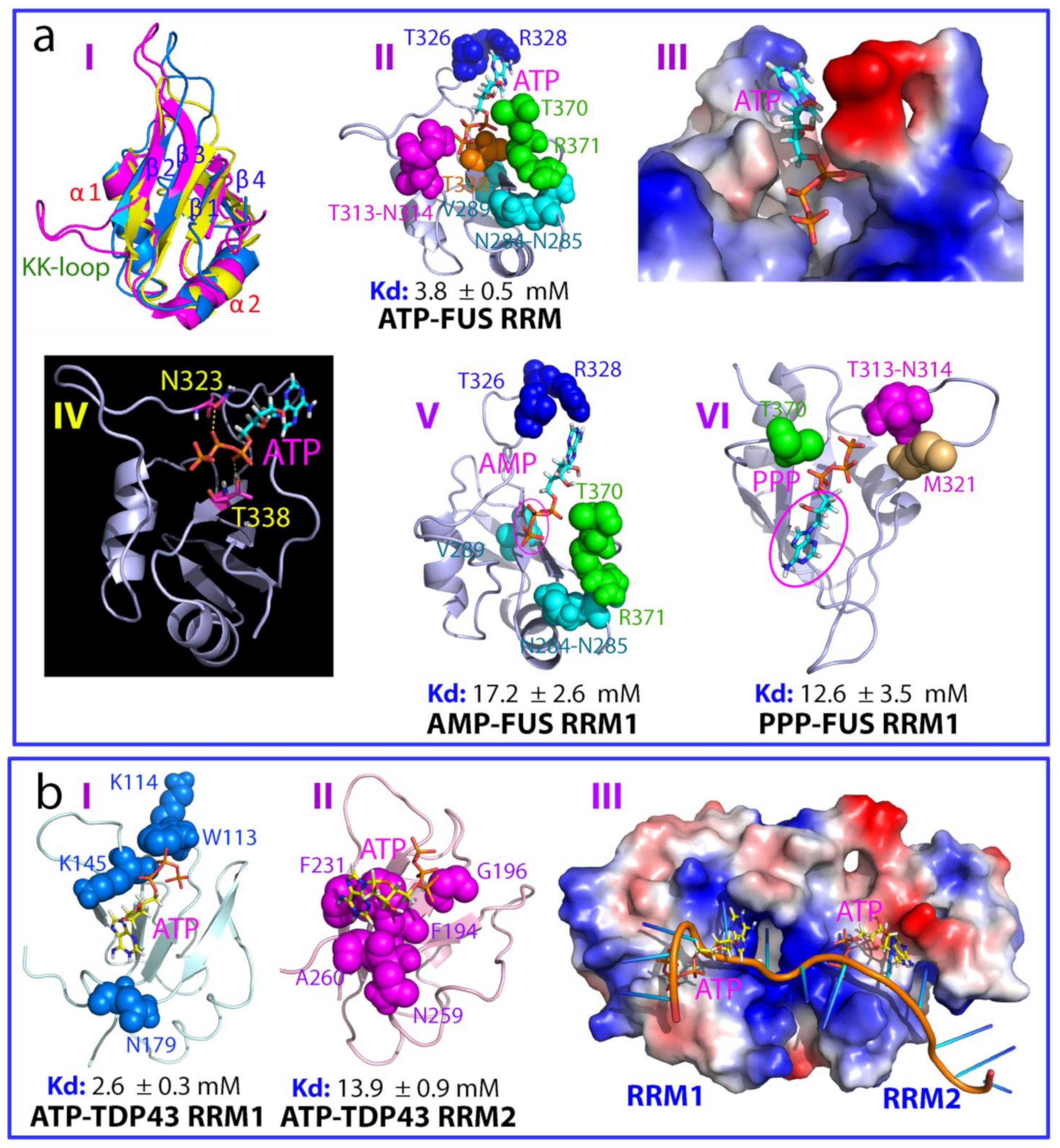
2.1. ATP Binds the Nucleic-Acid-Binding Pocket of the FUS RRM Domain
2.2. ATP Binds the Nucleic-Acid-Binding Pocket of TDP-43 RRM1 and RRM2
2.3. ATP Binds NTD and CTD of SARS-CoV-2 Nucleocapsid Protein
3. ATP and Nucleic Acids Interplay in Modulating LLPS
3.1. ATP and Nucleic Acids Interplay in Modulating the LLPS of FUS IDRs
3.2. How ATP and Nucleic Acids Interplay to Modulate the LLPS of the TDP-43 PLD
3.3. How ATP and Nucleic Acids Interplay to Modulate the LLPS of the SARS-CoV-2 N Protein
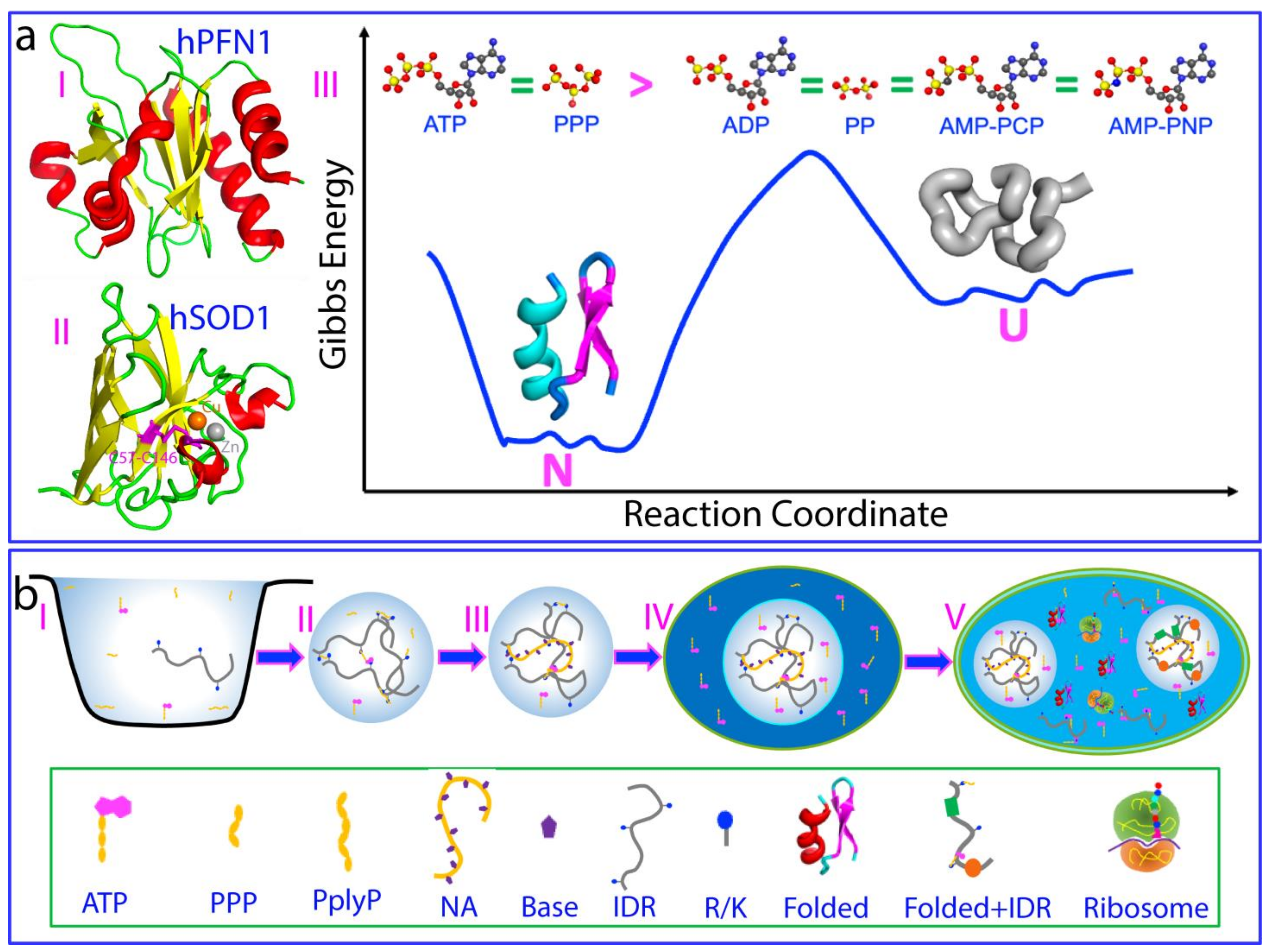
4. ATP Induces Protein Folding with the Highest Efficiency
5. ATP Action from Prebiotic Evolution to Modern Cells
Funding
Acknowledgments
Conflicts of Interest
References
- Todd, A.R. Where There’s Life There’s Phosphorus; Nakamura, K., Kageyama, M., Oshima, T., Eds.; Science and Society Press: Tokyo, Japan, 1981; p. 275. [Google Scholar]
- Westheimer, F.H. Why nature chose phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Bowler, M.W.; Cliff, M.; Walthoand, J.P.; Blackburn, G.M. Why did nature select phosphate for its dominant roles in biology? New J. Chem. 2010, 34, 784–794. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Sharma, P.K.; Prasad, R.B.; Warshel, A. Why nature really chose phosphate. Q. Rev. Biophys. 2013, 46, 1–132. [Google Scholar] [CrossRef] [PubMed]
- Leningher, A. Principles of Biochemistry; W. H. Freeman and Company: New York, NY, USA, 2005. [Google Scholar]
- Pinna, S.; Kunz, C.; Halpern, A.; Harrison, S.A.; Jordan, S.F.; Ward, J.; Werner, F.; Lane, N. A prebiotic basis for ATP as the universal energy currency. PLoS Biol. 2022, 20, e3001437. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline Pgranules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, 6357. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Handwerger, K.E.; Gall, J.G. Subnuclear organelles: New insights into form and function. Trends Cell Biol. 2006, 16, 19–26. [Google Scholar] [CrossRef]
- Overbeek, J.T.G.; Voorn, M.J. Phase separation in polyelectrolyte solutions. Theory of complex coacervation. J. Cell. Comp. Physiol. 1957, 49, 7–26. [Google Scholar] [CrossRef]
- Taratuta, V.G.; Holschbach, A.; Thurston, G.M.; Blankschtein, D.; Benedek, G.B. Liquid-liquid phase separation of aqueous lysozyme solutions: Effects of pH and salt identity. J. Phys. Chem. 1990, 94, 2140–2472. [Google Scholar] [CrossRef]
- Song, J. Molecular mechanisms of phase separation and amyloidosis of ALS/FTD-linked FUS and TDP-43. Aging Dis. 2023. [Google Scholar] [CrossRef] [PubMed]
- Toretsky, J.A.; Wright, P.E. Assemblages: Functional units formed by cellular phase separation. J. Cell Biol. 2014, 206, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. Molecular grammar underlying the driving forces for phase separation of prion-like RNA binding proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Song, J. A review of the effects of ATP and hydroxychloroquine on the phase separation of the SARS-CoV-2 nucleocapsid protein. Biophys. Rev. 2022, 14, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.P.; Farber, P.J.; Sekhar, A.; Lin, Y.-H.; Huang, R.; Bah, A.; Nott, T.J.; Chan, H.S.; Baldwin, A.J.; Forman-Kay, J.D.; et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. USA 2017, 114, E8194–E8203. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef]
- Portz, B.; Lee, B.L.; Shorter, J. FUS and TDP-43 Phases in Health and Disease. Trends Biochem. Sci. 2021, 46, 550–563. [Google Scholar] [CrossRef]
- Keating, C.D. Aqueous phase separation as a possible route to compartmentalization of biological molecules. Acc. Chem. Res. 2012, 45, 2114–2124. [Google Scholar] [CrossRef] [PubMed]
- Maharana, S.; Wang, J.; Papadopoulos, D.K.; Richter, D.; Pozniakovsky, A.; Poser, I.; Bickle, M.; Rizk, S.; Guillén-Boixet, J.; Franzmann, T.M.; et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 2018, 360, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Milin, A.N.; Deniz, A.A. Reentrant phase transitions and non- equilibrium dynamics in membraneless organelles. Biochemistry 2018, 57, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Ukmar-Godec, T.; Hutten, S.; Grieshop, M.P.; Rezaei-Ghaleh, N.; Cima-Omori, M.-S.; Biernat, J.; Mandelkow, E.; Söding, J.; Dormann, D.; Zweckstetter, M. Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat. Commun. 2019, 10, 2909. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lim, L.; Lu, Y.; Song, J. A unified mechanism for LLPS of ALS/FTLD-causing FUS as well as its modulation by ATP and oligonucleic acids. PLoS Biol. 2019, 17, e3000327. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lim, L.; Song, J. ATP enhances at low concentrations but dissolves at high concentrations liquid-liquid phase separation (LLPS) of ALS/FTD-causing FUS. Biochem. Biophys. Res. Commun. 2018, 504, 545–551. [Google Scholar] [CrossRef]
- Wang, L.; Kang, J.; Lim, L.; Wei, Y.; Song, J. TDP-43 NTD can be induced while CTD is significantly enhanced by ssDNA to undergo liquid-liquid phase separation. Biochem. Biophys. Res. Commun. 2018, 499, 189–195. [Google Scholar] [CrossRef]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a biological hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef]
- Rice, A.M.; Rosen, M.K. ATP controls the crowd. Science 2017, 356, 701–702. [Google Scholar] [CrossRef]
- Mehringer, J.; Do, T.-M.; Touraud, D.; Hohenschutz, M.; Khoshsima, A.; Horinek, D.; Kunz, W. Hofmeister versus Neuberg: Is ATP really a biological hydrotrope? Cell Rep. Phys. Sci. 2021, 2, 100343. [Google Scholar] [CrossRef]
- Hautke, A.; Ebbinghaus, S. The emerging role of ATP as a cosolute for biomolecular processes. Biol. Chem. 2023, 404, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed. Engl. 2011, 50, 4808–4842. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.-L.; Shan, Y.; Zhang, P.; Ding, H.-M.; Ma, Y.-Q. Uncovering the molecular mechanism for dual effect of ATP on phase separation in FUS solution. Sci. Adv. 2022, 8, eabo7885. [Google Scholar] [CrossRef] [PubMed]
- Song, J. ATP energy-independently controls protein homeostasis with unique structure and diverse mechanisms. Protein Sci. 2021, 30, 1277–1293. [Google Scholar] [CrossRef] [PubMed]
- Mogami, G.; Wazawa, T.; Morimoto, N.; Kodama, T.; Suzuki, M. Hydration properties of adenosine phosphate series as studied by microwave dielectric spectroscopy. Biophys. Chem. 2011, 154, 1–7. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kang, J.; Song, J. ATP antagonizes the crowding-induced destabilization of the human eye-lens protein γS-crystallin. Biochem. Biophys. Res. Commun. 2020, 526, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lim, L.; Song, J. ATP induces folding of ALS-causing C71G-hPFN1 and nascent hSOD1. Commun. Chem. 2023, 2, 223. [Google Scholar] [CrossRef] [PubMed]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2021, 22, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.L.J.; Riback, J.A.; Bascetin, R.; Brangwynne, C.P. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2021, 22, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Honson, D.; Guttman, M. Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat. Rev. Mol. Cell Biol. 2021, 22, 653–670. [Google Scholar] [CrossRef]
- Emmanouilidis, L.; Esteban-Hofer, L.; Jeschke, G.; Allain, F.H.T. Structural biology of RNA-binding proteins in the context of phase separation: What NMR and EPR can bring? Curr. Opin. Struct. Biol. 2021, 70, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Cech, T.R.; Parker, R.R. Biochemical properties and biological functions of FET proteins. Annu. Rev. Biochem. 2015, 84, 355–379. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Cléry, A.; Blatter, M.; Allain, F.H. RNA recognition motifs: Boring? Not quite. Curr. Opin. Struct. Biol. 2008, 18, 290–298. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Masters, P.S. Coronavirus genomic RNA packaging. Virology 2019, 537, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’Kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020, 94, e01925-19. [Google Scholar] [CrossRef] [PubMed]
- Iserman, C.; Roden, C.A.; Boerneke, M.A.; Sealfon, R.S.; McLaughlin, G.A.; Jungreis, I.; Fritch, E.J.; Hou, Y.J.; Ekena, J.; Weidmann, C.A.; et al. Genomic RNA Elements Drive Phase Separation of the SARS-CoV-2 Nucleocapsid. Mol. Cell 2020, 80, 1078–1091. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R., 3rd; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef]
- Dang, M.; Li, Y.; Song, J. ATP biphasically modulates LLPS of SARS-CoV-2 nucleocapsid protein and specifically binds its RNA-binding domain. Biochem Biophys. Res. Commun. 2021, 19, 50–55. [Google Scholar]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. PLoS Pathog. 2020, 16, e1009100. [Google Scholar] [CrossRef]
- Peng, Y.; Du, N.; Lei, Y.; Dorje, S.; Qi, J.; Luo, T.; Gao, G.F.; Song, H. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J. 2020, 39, e105938. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Song, J. Structural basis of anti-SARS-CoV-2 activity of HCQ: Specific binding to N protein to disrupt its interaction with nucleic acids and LLPS. QRB Discov. 2021, 2, e13. [Google Scholar] [CrossRef] [PubMed]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid–liquid phase separation by intrinsically disordered protein regions of viruses: Roles in viral life cycle and control of virus–host interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef] [PubMed]
- Guseva, S.; Milles, S.; Jensen, M.R.; Schoehn, G.; Ruigrok, R.W.; Blackledge, M. Structure, dynamics and phase separation of measles virus RNA replication machinery. Curr. Opin. Virol. 2020, 41, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Scoca, V.; Di Nunzio, F. Membraneless organelles restructured and built by pandemic viruses: HIV-1 and SARS-CoV-2. J. Mol. Cell Biol. 2021, 13, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Monette, A.; Niu, M.; Chen, L.; Rao, S.; Gorelick, R.J.; Mouland, A.J. Pan-retroviral nucleocapsid-mediated phase separation regulates genomic RNA positioning and trafficking. Cell Rep. 2020, 31, 107520. [Google Scholar] [CrossRef] [PubMed]
- Salladini, E.; Gondelaud, F.; Nilsson, J.F.; Pesce, G.; Bignon, C.; Murrali, M.G.; Fabre, R.; Pierattelli, R.; Kajava, A.V.; Horvat, B.; et al. Identification of a region in the common amino-terminal domain of Hendra Virus P, V, andWproteins responsible for phase transition and amyloid formation. Biomolecules 2021, 11, 1324. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.A.; Whelan, S.P. Phase transitions drive the formation of vesicular stomatitis virus replication compartments. mBio 2018, 9, e02290-17. [Google Scholar] [CrossRef] [PubMed]
- Hirai, Y.; Tomonaga, K.; Horie, M. Borna disease virus phosphoprotein triggers the organization of viral inclusion bodies by liquid-liquid phase separation. Int. J. Biol. Macromol. 2021, 192, 55–63. [Google Scholar] [CrossRef]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudrière-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef]
- Bianchi, G.; Brocca, S.; Longhi, S.; Uversky, V. Liaisons dangereuses: Intrinsic Disorder in Cellular Proteins Recruited to Viral Infection-Related Biocondensates. Int. J. Mol. Sci. 2023, 24, 2151. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Niu, C.; Ren, J.; Zhang, J.; Xie, X.; Zhu, H.; Feng, W.; Gong, W. The RRM domain of human fused in sarcoma protein reveals a non-canonical nucleic acid binding site. Biochim. Biophys. Acta 2013, 1832, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Loughlin, F.E.; Lukavsky, P.J.; Kazeeva, T.; Reber, S.; Hock, E.-M.; Colombo, M.; Von Schroetter, C.; Pauli, P.; Cléry, A.; Mühlemann, O.; et al. The Solution Structure of FUS Bound to RNA Reveals a Bipartite Mode of RNA Recognition with Both Sequence and Shape Specificity. Mol. Cell 2019, 73, 490–504.e6. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, D.; Campagne, S.; Schmidt, R.; Reber, S.; Mechtersheimer, J.; Gypas, F.; Schweingruber, C.; Colombo, M.; von Schroetter, C.; Loughlin, F.E.; et al. Aberrant interaction of FUS with the U1 snRNA provides a molecular mechanism of FUS induced amyotrophic lateral sclerosis. Nat. Commun. 2020, 11, 6341. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lim, L.; Song, J. RRM domain of ALS/FTD-causing FUS characteristic of irreversible unfolding spontaneously self-assembles into amyloid fibrils. Sci. Rep. 2017, 7, 1043. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lim, L.Z.; Song, J. ATP binds and inhibits the neurodegeneration-associated fibrillization of the FUS RRM domain. Commun. Biol. 2019, 2, 223. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Wu, F.; Harrich, D.; García-Martínez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Lim, L.; Wei, Y.; Song, J. TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. USA 2014, 111, 18619–18624. [Google Scholar] [CrossRef]
- Afroz, T.; Hock, E.-M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef]
- Kuo, P.H.; Doudeva, L.G.; Wang, Y.T.; Shen, C.K.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.H.; Chiang, C.H.; Wang, Y.T.; Doudeva, L.G.; Yuan, H.S. The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res. 2014, 42, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Kang, J.; Lim, L.; Song, J. ATP is a cryptic binder of TDP-43 RRM domains to enhance stability and inhibit ALS/AD-associated fibrillation. Biochem. Biophys. Res. Commun. 2020, 522, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Song, J. ALS-causing D169G mutation disrupts the ATP-binding capacity of TDP-43 RRM1 domain. Biochem. Biophys. Res. Commun. 2020, 524, 459–464. [Google Scholar] [CrossRef]
- Dang, M.; Li, Y.; Song, J. Tethering-induced destabilization and ATP-binding for tandem RRM domains of ALS-causing TDP-43 and hnRNPA1. Sci. Rep. 2021, 11, 1034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Usluer, S.; Zhang, F.; Lenard, A.J.; Bourgeois, B.M.; Madl, T. ATP regulates RNA-driven cold inducible RNA binding protein phase separation. Protein Sci. 2021, 30, 1438–1453. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kang, J.; Lim, L.; Song, J. ATP binds nucleic-acid-binding domains beyond RRM fold. Biochem. Biophys. Res. Commun. 2020, 522, 826–831. [Google Scholar] [CrossRef]
- Beuck, C.; Williamson, J.R.; Wüthrich, K.; Serrano, P. The acidic domain is a unique structural feature of the splicing factor SYNCRIP. Protein Sci. 2016, 25, 1545–1550. [Google Scholar] [CrossRef]
- Hobor, F.F.; Dallmann, A.; Ball, N.J.; Cicchini, C.; Battistelli, C. A cryptic RNAbinding domain mediates Syncrip recognition and exosomal partitioning of miRNA targets. Nat. Commun. 2018, 9, 831. [Google Scholar] [CrossRef]
- Dang, M.; Song, J. CTD of SARS-CoV-2 N protein is a cryptic domain for binding ATP and nucleic acid that interplay in modulating phase separation. Protein Sci. 2021, 31, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Walinda, E.; Morimoto, D.; Kohn, B.; Scheler, U.; Shirakawa, M.; Sugase, K. Effects of weak nonspecific interactions with ATP on proteins. J. Am. Chem. Soc. 2021, 143, 11982–11993. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Ou, X.; Li, J. Mechanistic Insight on General Protein-Binding Ability of ATP and the Impacts of Arginine Residues. J. Phys. Chem. B 2022, 126, 4647–4658. [Google Scholar] [CrossRef] [PubMed]
- Scheeff, E.D.; Bourne, P.E. Structural evolution of the protein kinase-like superfamily. PLoS Comput. Biol. 2005, 1, e49. [Google Scholar] [CrossRef] [PubMed]
- Kanev, G.K.; de Graaf, C.; de Esch, I.J.; Leurs, R.; Würdinger, T.; Westerman, B.A.; Kooistra, A.J. The landscape of atypical and eukaryotic protein kinases. Trends Pharmacol. Sci. 2019, 40, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Michelitsch, M.D.; Weissman, J.S. A census of glutamine/asparagine-rich regions: Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA 2000, 97, 11910–11915. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low Complexity Domains. Cell 2017, 171, 615–627.e16. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; McKnight, S.L. The low-complexity domain of the FUS RNA binding protein self-assembles via the mutually exclusive use of two distinct cross-β cores. Proc. Natl. Acad. Sci. USA 2021, 118, e2114412118. [Google Scholar] [CrossRef]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell. 2015, 60, 231–241. [Google Scholar] [CrossRef]
- Murthy, A.C.; Tang, W.S.; Jovic, N.; Janke, A.M.; Seo, D.H.; Perdikari, T.M.; Mittal, J.; Fawzi, N.L. Molecular interactions contributing to FUS SYGQ LC-RGG phase separation and co-partitioning with RNA polymerase II heptads. Nat. Struct. Mol. Biol. 2021, 28, 923–935. [Google Scholar] [CrossRef]
- Song, J.; Ni, F. NMR for the design of functional mimetics of protein-protein interactions: One key is in the building of bridges. Biochem. Cell Biol. 1998, 76, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS mutations disrupt phase separation mediated by a-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhou, X.; Kato, M.; Liu, D.; Ghaemmaghami, S.; Tu, B.P.; McKnight, S.L. Redox mediated regulation of an evolutionarily conserved cross-β structure formed by the TDP43 low complexity domain. Proc. Natl. Acad. Sci. USA 2020, 117, 28727–28734. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Zhou, X.; Sutherland, L.; Kato, M.; Jaczynska, K.; Rizo, J.; McKnight, S.L. Oxidative regulation of TDP-43 self-association by a β-to-α conformational switch. Proc. Natl. Acad. Sci. USA 2023, 120, e2311416120. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.; Wei, Y.; Lu, Y.; Song, J. ALS-causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol. 2016, 14, e1002338. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Lim, L.; Kang, J.; Song, J. ATP biphasically modulates LLPS of TDP-43 PLD by specifically binding arginine residues. Commun. Biol. 2021, 4, 714. [Google Scholar] [CrossRef]
- Dang, M.; Li, T.; Zhou, S.; Song, J. Arg/Lys-containing IDRs are cryptic binding domains for ATP and nucleic acids that interplay to modulate LLPS. Commun. Biol. 2022, 5, 1315. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.B.; Barreau, A.; Rohatgi, R. Phase separation-deficient TDP43 remains functional in splicing. Nat. Commun. 2019, 10, 4890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.R.; Chiang, W.C.; Chou, P.C.; Wang, W.J.; Huang, J.R. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 2018, 293, 6090–6098. [Google Scholar] [CrossRef]
- Dang, M.; Li, T.; Song, J. ATP and nucleic acids competitively modulate LLPS of the SARS-CoV2 nucleocapsid protein. Commun. Biol. 2023, 6, 80. [Google Scholar] [CrossRef]
- Kim, T.H.; Payliss, B.J.; Nosella, M.L.; Lee, I.T.; Toyama, Y.; Forman-Kay, J.D.; Kay, L.E. Interaction hot spots for phase separation revealed by NMR studies of a CAPRIN1 condensed phase. Proc. Natl. Acad. Sci. USA 2021, 118, e2104897118. [Google Scholar] [CrossRef] [PubMed]
- Kota, D.; Prasad, R.; Zhou, H.X. Adenosine Triphosphate Mediates Phase Separation of Disordered Basic Proteins by Bridging Intermolecular Interaction Networks. J. Am. Chem. Soc. 2024, 146, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L. New directions in the study of Peptide h-bonds and Peptide solvation. Adv. Protein Chem. 2005, 72, ix–xi. [Google Scholar]
- Rose, G.D.; Fleming, P.J.; Banavar, J.R.; Maritan, A. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. USA 2006, 103, 16623–16633. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Song, J. Why do proteins aggregate? “Intrinsically insoluble proteins” and “dark mediators” revealed by studies on “insoluble proteins” solubilized in pure water. F1000Research 2013, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Kurzawa, N.; Werner, T.; Günthner, I.; Helm, D.; Huber, W.; Bantscheff, M.; Savitski, M.M. Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat. Commun. 2019, 10, 1155. [Google Scholar] [CrossRef]
- Hayes, M.H.; Peuchen, E.H.; Dovichi, N.J.; Weeks, D.L. Dual roles for ATP in the regulation of phase separated protein aggregates in Xenopus oocyte nucleoli. eLife 2018, 7, e35224. [Google Scholar] [CrossRef]
- Lim, L.; Kang, J.; Song, J. Extreme diversity of 12 cations in folding ALS-linked hSOD1 unveils novel hSOD1-dependent mechanisms for Fe2+/Cu2+-induced cytotoxicity. Sci. Rep. 2023, 13, 19868. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Wholey, W.-Y.; Wagner, N.O.; Cremers, C.M.; Mueller-Schickert, A.; Hock, N.T.; Krieger, A.G.; Smith, E.M.; Bender, R.A.; Bardwell, J.C.; et al. Polyphosphate is a primordial chaperone. Mol. Cell 2014, 53, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.N.; Gómez-García, M.R.; Kornberg, A. Inorganic polyphosphate: Essential for growth and survival. Annu. Rev. Biochem. 2009, 78, 605–647. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, J.; Ran, X.; Fang, M.; Shi, J.; Qin, H.; Goh, J.M.; Song, J. Resurrecting abandoned proteins with pure water: CD and NMR studies of protein fragments solubilized in salt-free water. Biophys. J. 2006, 91, 4201–4209. [Google Scholar] [CrossRef] [PubMed]
- Thandapani, P.; O’Connor, T.R.; Bailey, T.L.; Richard, S. Defining the RGG/RG motif. Mol. Cell 2013, 50, 613–623. [Google Scholar] [CrossRef]
- Rajyaguru, P.; Parker, R. RGG motif proteins: Modulators of mRNA functional states. Cell Cycle 2012, 11, 2594–2599. [Google Scholar] [CrossRef]
- Hautke, A.; Voronin, A.; Idiris, F.; Riel, A.; Lindner, F.; Lelièvre, A.; Zhu, J.; Appel, B.; Fatti, E.; Weis, K.; et al. CAG-repeat RNA hairpin folding and recruitment into nuclear speckles with a pivotal role of ATP as a cosolute. J. Am. Chem. Soc. 2023, 145, 9571–9583. [Google Scholar] [CrossRef]
- Baldwin, R.L. Dynamic hydration shell restores Kauzmann’s 1959 explanation of how the hydrophobic factor drives protein folding. Proc. Natl. Acad. Sci. USA 2014, 111, 13052–13056. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Onuchic, J.N. Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 389–415. [Google Scholar] [CrossRef]
- Chong, S.H.; Ham, S. Distinct role of hydration water in protein misfolding and aggregation revealed by fluctuating thermodynamics analysis. Acc. Chem. Res. 2015, 48, 956–965. [Google Scholar] [CrossRef]
- Laage, D.; Elsaesser, T.; Hynes, J.T. Water dynamics in the hydration shells of biomolecules. Chem Rev. 2017, 117, 10694–10725. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.H. Complexity: A Very Short Introduction, Very Short Introductions; Oxford Academic: Oxford, UK, 2014. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J. Adenosine Triphosphate: The Primordial Molecule That Controls Protein Homeostasis and Shapes the Genome–Proteome Interface. Biomolecules 2024, 14, 500. https://doi.org/10.3390/biom14040500
Song J. Adenosine Triphosphate: The Primordial Molecule That Controls Protein Homeostasis and Shapes the Genome–Proteome Interface. Biomolecules. 2024; 14(4):500. https://doi.org/10.3390/biom14040500
Chicago/Turabian StyleSong, Jianxing. 2024. "Adenosine Triphosphate: The Primordial Molecule That Controls Protein Homeostasis and Shapes the Genome–Proteome Interface" Biomolecules 14, no. 4: 500. https://doi.org/10.3390/biom14040500
APA StyleSong, J. (2024). Adenosine Triphosphate: The Primordial Molecule That Controls Protein Homeostasis and Shapes the Genome–Proteome Interface. Biomolecules, 14(4), 500. https://doi.org/10.3390/biom14040500