
Recent Findings on Therapeutic Cancer Vaccines: An Updated Review

Abstract
1. Introduction
2. Tumor Microenvironment and Cancer Vaccine Mechanisms
3. Cancer Immune Cycle
4. Escaping from the Cancer Immune Cycle
5. Tumor Antigen Classifications
6. Different Cancer Vaccine Platforms
6.1. Peptide-Based Vaccines
6.2. Recombinant (Pathogen) Vaccines: Viral and Bacterial-Based Vaccines
6.3. Cell-Based Vaccines: Dendritic Cells (DCs), Stem Cells, and Chimeric Antigen Receptor (CAR) T Cell Therapy
6.4. DC Subsets and Their Roles in Priming and Activating T Cells
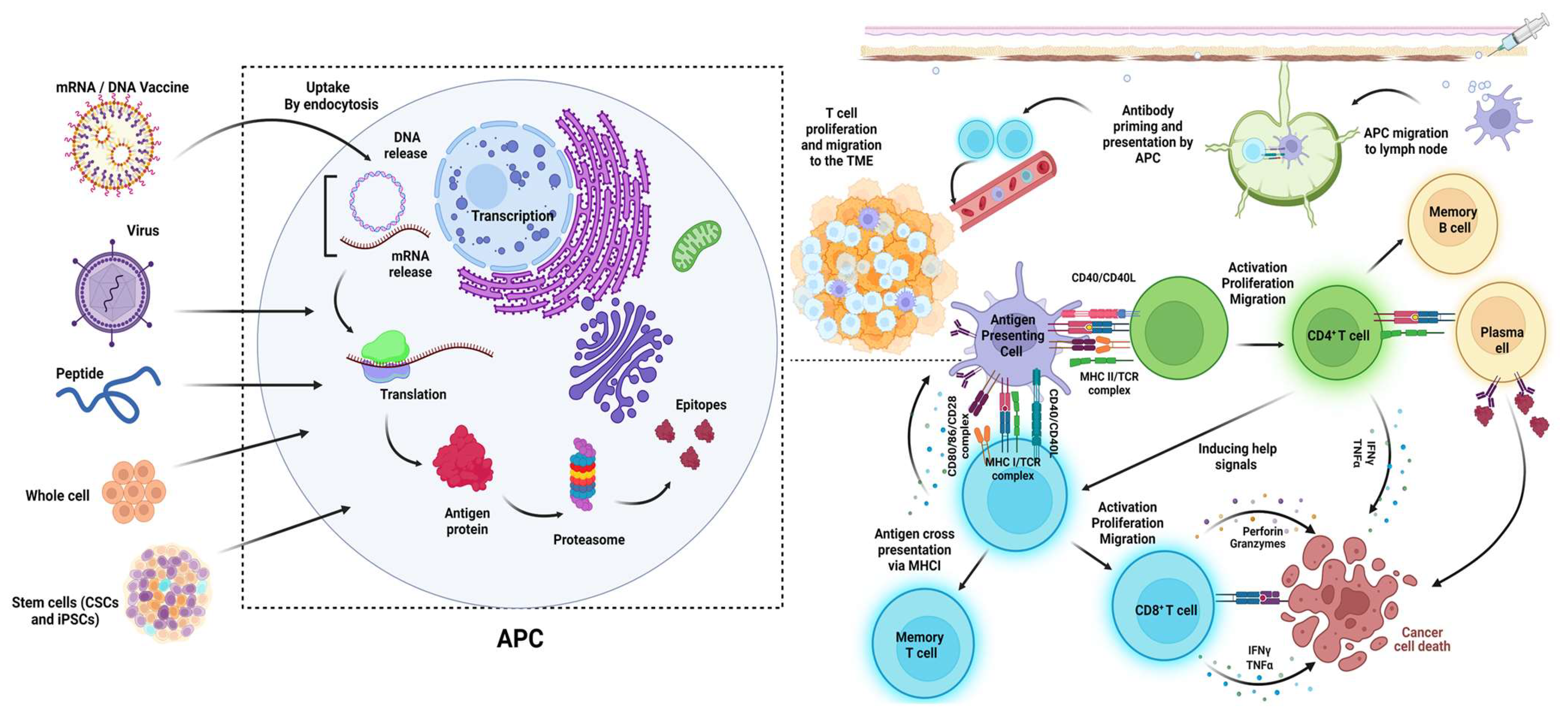
6.5. Nucleic Acid-Based Vaccines: DNA and mRNA Vaccines
6.5.1. DNA-Based Cancer Vaccines
6.5.2. mRNA-Based Vaccines
6.6. Personalized Cancer Vaccines
7. Combining Artificial Intelligence and Cold Plasma Technology as Novel Modality Tools to Develop Cancer Vaccines
8. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moore, Z.S.; Seward, J.F.; Lane, J.M. Smallpox. Lancet 2006, 367, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.S.; Stern, P.L. Opportunities and challenges for human papillomavirus vaccination in cancer. Nat. Rev. Cancer 2018, 18, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Pol, S. Hepatitis: HBV vaccine—The first vaccine to prevent cancer. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Chodon, T.; Koya, R.C.; Odunsi, K. Active Immunotherapy of Cancer. Immunol. Investig. 2015, 44, 817–836. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Lu, C.; Liu, Y.; Ali, N.M.; Zhang, B.; Cui, X. The role of innate immune cells in the tumor microenvironment and research progress in anti-tumor therapy. Front. Immunol. 2023, 13, 1039260. [Google Scholar] [CrossRef]
- Sun, B.; Hyun, H.; Li, L.T.; Wang, A.Z. Harnessing nanomedicine to overcome the immunosuppressive tumor microenvironment. Acta Pharmacol. Sin. 2020, 41, 970–985. [Google Scholar] [CrossRef]
- Kleponis, J.; Skelton, R.; Zheng, L. Fueling the engine and releasing the break: Combinational therapy of cancer vaccines and immune checkpoint inhibitors. Cancer Biol. Med. 2015, 12, 201–208. [Google Scholar] [PubMed]
- Kang, W.; Feng, Z.; Luo, J.; He, Z.; Liu, J.; Wu, J.; Rong, P. Tertiary Lymphoid Structures in Cancer: The Double-Edged Sword Role in Antitumor Immunity and Potential Therapeutic Induction Strategies. Front. Immunol. 2021, 12, 689270. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class i and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Gaudino, S.J.; Kumar, P. Cross-talk between antigen presenting cells and T cells impacts intestinal homeostasis, bacterial infections, and tumorigenesis. Front. Immunol. 2019, 10, 422031. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Kanai, M.; Choi, W.; Li, X.; Sakaue, S.; Yamamoto, K.; Ogawa, K.; Gutierrez-Arcelus, M.; Gregersen, P.K.; Stuart, P.E.; et al. A high-resolution HLA reference panel capturing global population diversity enables multi-ancestry fine-mapping in HIV host response. Nat. Genet. 2021, 53, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Velleuer, E. Molecular Immunology: How Science Works; Springer Nature: Berlin/Heidelberg, Germany, 2022; ISBN 9783031040252. [Google Scholar]
- Hilligan, K.L.; Ronchese, F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell. Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Eiz-Vesper, B.; Schmetzer, H.M. Antigen-presenting cells: Potential of proven und new players in immune therapies. Transfus. Med. Hemotherapy 2020, 47, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Saxena, V.; Li, L.; Paluskievicz, C.; Kasinath, V.; Bean, A.; Abdi, R.; Jewell, C.M.; Bromberg, J.S. Role of lymph node stroma and microenvironment in T cell tolerance. Immunol. Rev. 2019, 292, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Riven, I.; Feigelson, S.W.; Kartvelishvily, E.; Tohya, K.; Miyasaka, M.; Alon, R.; Haran, G. Three-dimensional localization of T-cell receptors in relation to microvilli using a combination of superresolution microscopies. Proc. Natl. Acad. Sci. USA 2016, 113, E5916–E5924. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, J.; Abdi, R.; Jewell, C.M.; Bromberg, J.S. Lymph node fibroblastic reticular cells steer immune responses. Trends Immunol. 2021, 42, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.; Harrington, K.J.; Zocca, M.B.; Ehrnrooth, E.; Cohen, E.E.W. The changing landscape of therapeutic cancer vaccines-novel platforms and neoantigen identification. Clin. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Miao, L.; Sui, J.; Hao, Y.; Huang, G. Nanoparticle cancer vaccines: Design considerations and recent advances. Asian J. Pharm. Sci. 2020, 15, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2021, 11, 615240. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, P.J.; Bilusic, M. Cancer Vaccines. Hematol. Oncol. Clin. N. Am. 2019, 33, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell responses to TP53 “Hotspot” Mutations and unique neoantigens expressed by human ovarian cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef]
- Barnett, L.G.; Simkins, H.M.A.; Barnett, B.E.; Korn, L.L.; Johnson, A.L.; Wherry, E.J.; Wu, G.F.; Laufer, T.M. B Cell Antigen Presentation in the Initiation of Follicular Helper T Cell and Germinal Center Differentiation. J. Immunol. 2014, 192, 3607–3617. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Campillo, M.T.; Molina-Hernández, V.; Bautista, M.J.; Pacheco, I.L.; Zafra, R.; Buffoni, L.; Martínez-Moreno, F.J.; Martínez-Moreno, A.; Pérez, J. Characterization of dendritic cells and follicular dendritic cells in the hepatic lymph nodes and liver of sheep experimentally infected with Fasciola hepatica. Vet. Res. 2020, 51, 33. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Silina, K.; van den Broek, M.; Hirahara, K.; Yanagita, M. The roles of tertiary lymphoid structures in chronic diseases. Nat. Rev. Nephrol. 2023, 9, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Warnock, R.A.; Askari, S.; Butcher, E.C.; Von Andrian, U.H. Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J. Exp. Med. 1998, 187, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Hess, E.; Duheron, V.; Decossas, M.; Lézot, F.; Berdal, A.; Chea, S.; Golub, R.; Bosisio, M.R.; Bridal, S.L.; Choi, Y.; et al. RANKL Induces Organized Lymph Node Growth by Stromal Cell Proliferation. J. Immunol. 2012, 188, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Luther, S.A.; Ansel, K.M.; Cyster, J.G. Overlapping roles of CXCL13, interleukin 7 receptor α, and CCR7 ligands in lymph node development. J. Exp. Med. 2003, 197, 1191–1198. [Google Scholar] [CrossRef]
- Yoshida, H.; Naito, A.; Inoue, J.I.; Satoh, M.; Santee-Cooper, S.M.; Ware, C.F.; Togawa, A.; Nishikawa, S.; Nishikawa, S.I. Different cytokines induce surface lymphotoxin-αβ on IL-7 receptor-α cells that differentially engender lymph nodes and Peyer’s patches. Immunity 2002, 17, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Cepok, S.; Sellner, J.; Grummel, V.; Weber, M.S.; Korn, T.; Berthele, A.; Hemmer, B. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J. Neuroinflammation 2012, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, S. Tertiary lymphoid structures are critical for cancer prognosis and therapeutic response. Front. Immunol. 2023, 13, 1063711. [Google Scholar] [CrossRef] [PubMed]
- Meier, D.; Bornmann, C.; Chappaz, S.; Schmutz, S.; Otten, L.A.; Ceredig, R.; Acha-Orbea, H.; Finke, D. Ectopic Lymphoid-Organ Development Occurs through Interleukin 7-Mediated Enhanced Survival of Lymphoid-Tissue-Inducer Cells. Immunity 2007, 26, 643–654. [Google Scholar] [CrossRef]
- Jacquelot, N.; Tellier, J.; Sl, N.; Gt, B. Tertiary lymphoid structures and B lymphocytes in cancer prognosis and response to immunotherapies. Oncoimmunology 2021, 10, 1900508. [Google Scholar] [CrossRef]
- He, M.; He, Q.; Cai, X.; Liu, J.; Deng, H.; Li, F.; Zhong, R.; Lu, Y.; Peng, H.; Wu, X.; et al. Intratumoral tertiary lymphoid structure (TLS) maturation is influenced by draining lymph nodes of lung cancer. J. Immunother. Cancer 2023, 11, e005539. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M. Antigen presentation by MHC-dressed cells. Front. Immunol. 2014, 5, 120340. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Canonical and Non-Canonical Functions of the Autophagy Machinery in MHC Restricted Antigen Presentation. Front. Immunol. 2022, 13, 868888. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Sijts, E.J.A.M.; Kloetzel, P.M. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell. Mol. Life Sci. 2011, 68, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, N.; Van den Eynde, B.J. Proteasome subtypes and regulators in the processing of antigenic peptides presented by class I molecules of the major histocompatibility complex. Biomolecules 2014, 4, 994–1025. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Garbi, N.; Reinheckel, T.; Moldenhauer, G.; Hämmerling, G.J.; Momburg, F. A Transporter Associated with Antigen-Processing Independent Vacuolar Pathway for the MHC Class I-Mediated Presentation of Endogenous Transmembrane Proteins. J. Immunol. 2007, 178, 7932–7942. [Google Scholar] [CrossRef]
- Sengupta, D.; Graham, M.; Liu, X.; Cresswell, P. Proteasomal degradation within endocytic organelles mediates antigen cross-presentation. EMBO J. 2019, 38, e99266. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front. Immunol. 2017, 8, 248429. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Hahn, M.; Michel, J.; Sankowski, R.; Kilian, M.; Kehl, N.; Günter, M.; Bunse, T.; Pusch, S.; von Deimling, A.; et al. Dysfunctional dendritic cells limit antigen-specific T cell response in glioma. Neuro Oncol. 2023, 25, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.M.S.; D’Aulerio, R.; Yong, T.; He, M.; Baptista, M.A.P.; Nylén, S.; Westerberg, L.S. Increased cross-presentation by dendritic cells and enhanced anti-tumour therapy using the Arp2/3 inhibitor CK666. Br. J. Cancer 2023, 128, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.; Joyner, D.; Mege, N.J.; Cusimano, G.M.; Bell, M.R.; Marcy, J.; Taramangalam, B.; Kim, K.M.; Lin, P.J.C.; Tam, Y.K.; et al. Lipid nanoparticles (LNP) induce activation and maturation of antigen presenting cells in young and aged individuals. Commun. Biol. 2023, 6, 188. [Google Scholar] [CrossRef] [PubMed]
- Haen, S.P.; Löffler, M.W.; Rammensee, H.G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Leal, Y.; Grubaugh, D.; Nogueira, C.V.; Lopetegui-González, I.; Del Valle, A.; Escalona, F.; Laborde, R.J.; Alvarez, C.; Fernández, L.E.; Starnbach, M.N.; et al. Antigen cross-presentation induced by the pore-forming protein sticholysin II encapsulated into liposomes. Front. Immunol. 2018, 9, 2473. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Usatorre, A.; De Palma, M. Dendritic cell cross-dressing and tumor immunity. EMBO Mol. Med. 2022, 14, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Pishesha, N.; Harmand, T.J.; Ploegh, H.L. A guide to antigen processing and presentation. Nat. Rev. Immunol. 2022, 22, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Schriek, P.; Villadangos, J.A. Trogocytosis and cross-dressing in antigen presentation. Curr. Opin. Immunol. 2023, 83, 102331. [Google Scholar] [CrossRef]
- Campana, S.; De Pasquale, C.; Carrega, P.; Ferlazzo, G.; Bonaccorsi, I. Cross-dressing: An alternative mechanism for antigen presentation. Immunol. Lett. 2015, 168, 349–354. [Google Scholar] [CrossRef]
- Merlotti, A.; Sadacca, B.; Arribas, Y.A.; Ngoma, M.; Burbage, M.; Goudot, C.; Houy, A.; Rocañín-Arjó, A.; Lalanne, A.; Seguin-Givelet, A.; et al. Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer. Sci. Immunol. 2023, 8, eabm6359. [Google Scholar] [CrossRef]
- Smyth, L.A.; Harker, N.; Turnbull, W.; El-Doueik, H.; Klavinskis, L.; Kioussis, D.; Lombardi, G.; Lechler, R. The Relative Efficiency of Acquisition of MHC:Peptide Complexes and Cross-Presentation Depends on Dendritic Cell Type. J. Immunol. 2008, 181, 3212–3220. [Google Scholar] [CrossRef]
- Dolan, B.P.; Gibbs, K.D.; Ostrand-Rosenberg, S. Dendritic Cells Cross-Dressed with Peptide MHC Class I Complexes Prime CD8+ T Cells. J. Immunol. 2006, 177, 6018–6024. [Google Scholar] [CrossRef] [PubMed]
- Harshyne, L.A.; Zimmer, M.I.; Watkins, S.C.; Barratt-Boyes, S.M. A Role for Class A Scavenger Receptor in Dendritic Cell Nibbling from Live Cells. J. Immunol. 2003, 170, 2302–2309. [Google Scholar] [CrossRef]
- Wakim, L.M.; Bevan, M.J. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature 2011, 471, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Rabella, M.; Garcia-Garijo, A.; Palomero, J.; Yuste-Estevanez, A.; Erhard, F.; Farriol-Duran, R.; Martín-Liberal, J.; Ochoa-de-Olza, M.; Matos, I.; Gartner, J.J.; et al. Exploring the Immunogenicity of Noncanonical HLA-I Tumor Ligands Identified through Proteogenomics. Clin. Cancer Res. 2023, 29, 2250–2265. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; He, J.; Liu, J.; Yan, X.; Fan, K. Nanozyme for tumor therapy: Surface modification matters. Exploration 2021, 1, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, S.; Malik, S.; Luu, R.; Loudig, O.; Mitchell, M.; Okafo, G.; Bhat, K.; Prideaux, B.; Eugenin, E.A. Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci. Rep. 2021, 11, 14556. [Google Scholar] [CrossRef] [PubMed]
- Pio, R.; Ajona, D.; Ortiz-Espinosa, S.; Mantovani, A.; Lambris, J.D. Complementing the cancer-immunity cycle. Front. Immunol. 2019, 10, 445513. [Google Scholar] [CrossRef] [PubMed]
- Weigelin, B.; Friedl, P. T cell-mediated additive cytotoxicity—Death by multiple bullets. Trends Cancer 2022, 8, 980–987. [Google Scholar] [CrossRef]
- Coënon, L.; Villalba, M. From CD16a Biology to Antibody-Dependent Cell-Mediated Cytotoxicity Improvement. Front. Immunol. 2022, 13, 913215. [Google Scholar] [CrossRef] [PubMed]
- Macor, P.; Capolla, S.; Tedesco, F. Complement as a biological tool to control tumor growth. Front. Immunol. 2018, 9, 2203. [Google Scholar] [CrossRef] [PubMed]
- Skowyra, A.; Śladowski, D.; Majszyk-Ionescu, J.; Grabska-Liberek, I. The role of the complement system in ocular diseases. Klin. Oczna 2017, 2017, 52–56. [Google Scholar]
- Wang, J.; Zhang, S.; Wang, Y.; Zhu, Y.; Xu, X.; Guo, J. Alternative Complement Pathway Signature Determines Immunosuppression and Resistance to Immunotherapy Plus Tyrosine Kinase Inhibitor Combinations in Renal Cell Carcinoma. Urol. Oncol. Semin. Orig. Investig. 2023, 41, e13–e51. [Google Scholar] [CrossRef] [PubMed]
- Lerner, E.; Woroniecka, K.; Anniballe, V.D.; Wilkinson, D.; Mohan, A.; Cook, S.; Tomaszewski, W.; Cui, X.; Khasraw, M.; Gunn, M.D.; et al. A Novel MHC-Independent Mechanism of Tumor Cell Killing by CD8+ T Cells. bioRxiv 2023. [Google Scholar] [CrossRef]
- Patel, S.A.; Minn, A.J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.A.; Baer, J.M.; Knolhoff, B.L.; Nywening, T.M.; Panni, R.Z.; Su, X.; Weilbaecher, K.N.; Hawkins, W.G.; Ma, C.; Fields, R.C.; et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat. Commun. 2018, 9, 1250. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S. Lifting the innate immune barriers to antitumor immunity. J. Immunother. Cancer 2020, 8, e000695. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Möller, A.; Smyth, M.J. Inflammation and immune surveillance in cancer. Semin. Cancer Biol. 2012, 22, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Damhofer, H.; Helin, K. EZH2i unlocks PDAC immune surveillance. Nat. Cancer 2023, 4, 781–783. [Google Scholar] [CrossRef]
- Jongsma, M.L.M.; Neefjes, J.; Spaapen, R.M. Playing hide and seek: Tumor cells in control of MHC class I antigen presentation. Mol. Immunol. 2021, 136, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Ushijima, T. Accumulation of genetic and epigenetic alterations in normal cells and cancer risk. NPJ Precis. Oncol. 2019, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, R.; Yang, A.G.; Zheng, G. Diversity of immune checkpoints in cancer immunotherapy. Front. Immunol. 2023, 14, 1121285. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune checkpoint therapy—Current perspectives and future directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.N.; Rathmell, J.C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 2023, 41, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, O.S.; Spagnuolo, L.; Garner, H.; Voorwerk, L.; Isaeva, O.I.; van Dyk, E.; Bakker, N.; Chalabi, M.; Klaver, C.; Duijst, M.; et al. IL-5-producing CD4+ T cells and eosinophils cooperate to enhance response to immune checkpoint blockade in breast cancer. Cancer Cell 2023, 41, 106–123.e10. [Google Scholar] [CrossRef]
- Lin, W.; Chen, L.; Zhang, H.; Qiu, X.; Huang, Q.; Wan, F.; Le, Z.; Geng, S.; Zhang, A.; Qiu, S.; et al. Tumor-intrinsic YTHDF1 drives immune evasion and resistance to immune checkpoint inhibitors via promoting MHC-I degradation. Nat. Commun. 2023, 14, 265. [Google Scholar] [CrossRef]
- Doyle, H.A.; Mamula, M.J. Post-translational protein modifications in antigen recognition and autoimmunity. Trends Immunol. 2001, 22, 443–449. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Li, J.; Ivey, G.; Li, Y.F.; El-Gamil, M.; Lalani, A.; et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 2019, 9, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Coukos, G.; Bassani-Sternberg, M. Identification of tumor antigens with immunopeptidomics. Nat. Biotechnol. 2022, 40, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.S.; Ma, J.; Klatt, M.G.; Dündar, F.; Bandlamudi, C.; Razavi, P.; Wen, H.Y.; Weigelt, B.; Zumbo, P.; Fu, S.N.; et al. Immunogenicity and therapeutic targeting of a public neoantigen derived from mutated PIK3CA. Nat. Med. 2022, 28, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.H.M.; Vo, N.T.; Vo, P.H.; Nguyen, H.D. Antigen Discovery. In Fish Vaccines Health Management for Sustainable Aquaculture; CRC Press: Boca Raton, FL, USA, 2023; pp. 65–74. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, M.; Zhang, H.; Cao, S.; Li, Y.; Jiang, S.; Song, Y.; Liu, S. Detection of p53 mutation and serum monitoring alert caused by Marek’s disease virus in poultry. BMC Vet. Res. 2020, 16, 303. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. P53 Isoforms Can Regulate P53 Transcriptional Activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.P.; Brandao, P.; Chapado, M.J.; Hamid, S.; Narayanan, R. Open reading frames associated with cancer in the dark matter of the human genome. Cancer Genom. Proteom. 2014, 11, 201–213. [Google Scholar]
- Meyerson, M.; Counter, C.M.; Eaton, E.N.; Ellisen, L.W.; Steiner, P.; Caddle, S.D.; Ziaugra, L.; Beijersbergen, R.L.; Davidoff, M.J.; Liu, Q.; et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997, 90, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Jie, M.M.; Chang, X.; Zeng, S.; Liu, C.; Liao, G.B.; Wu, Y.R.; Liu, C.H.; Hu, C.J.; Yang, S.M.; Li, X.Z. Diverse regulatory manners of human telomerase reverse transcriptase. Cell Commun. Signal. 2019, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Müller, M.; Pak, H.S.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Perreault, C. Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell. Mol. Life Sci. 2018, 75, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Abd-Aziz, N.; Poh, C.L. Development of Peptide-Based Vaccines for Cancer. J. Oncol. 2022, 2022, 9749363. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Nakagawa, H.; Tamai, T.; Kitahara, M.; Fushimi, K.; Nio, K.; Terashima, T.; Iida, N.; Arai, K.; Yamashita, T.; et al. Peptide vaccine-treated, long-term surviving cancer patients harbor self-renewing tumor-specific CD8+ T cells. Nat. Commun. 2022, 13, 3123. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Tagliamonte, M. Peptide-based vaccine for cancer therapies. Front. Immunol. 2023, 14, 1210044. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, N.O.; Jaramillo, M.; Mansour, H.M.; Sun, B. Therapeutic Cancer Vaccines—Antigen Discovery and Adjuvant Delivery Platforms. Pharmaceutics 2022, 14, 1448. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, J.; Wang, L.; Liu, B. Personalized neoantigen vaccination with synthetic long peptides: Recent advances and future perspectives. Theranostics 2020, 10, 6011–6023. [Google Scholar] [CrossRef] [PubMed]
- Corradin, G.; Kajava, A.V.; Verdini, A. Long synthetic peptides for the production of vaccines and drugs: A technological platform coming of age. Sci. Transl. Med. 2010, 2, 50rv3. [Google Scholar] [CrossRef] [PubMed]
- Facciolà, A.; Visalli, G.; Laganà, A.; Di Pietro, A. An Overview of Vaccine Adjuvants: Current Evidence and Future Perspectives. Vaccines 2022, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Cai, Y.; Jiang, Y.; He, X.; Wei, Y.; Yu, Y.; Tian, X. Vaccine adjuvants: Mechanisms and platforms. Signal Transduct. Target. Ther. 2023, 8, 283. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Cavalluzzo, B.; Mauriello, A.; Ragone, C.; Buonaguro, F.M.; Tornesello, M.L.; Buonaguro, L. Molecular mimicry and cancer vaccine development. Mol. Cancer 2023, 22, 75. [Google Scholar] [CrossRef]
- Fikes, J.D.; Sette, A. Design of multi-epitope, analogue-based cancer vaccines. Expert Opin. Biol. Ther. 2003, 3, 985–993. [Google Scholar] [CrossRef]
- Mauerer Zirlik, K.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Cytotoxic T cells generated against heteroclitic peptides kill primary tumor cells independent of the binding affinity of the native tumor antigen peptide. Blood 2006, 108, 3865–3870. [Google Scholar] [CrossRef]
- Cavalluzzo, B.; Ragone, C.; Mauriello, A.; Petrizzo, A.; Manolio, C.; Caporale, A.; Vitagliano, L.; Ruvo, M.; Buonaguro, L.; Tagliamonte, M. Identification and characterization of heteroclitic peptides in TCR-binding positions with improved HLA-binding efficacy. J. Transl. Med. 2021, 19, 89. [Google Scholar] [CrossRef]
- Arbelaez, C.A.; Estrada, J.; Gessner, M.A.; Glaus, C.; Morales, A.B.; Mohn, D.; Phee, H.; Lipford, J.R.; Johnston, J.A. A nanoparticle vaccine that targets neoantigen peptides to lymphoid tissues elicits robust antitumor T cell responses. NPJ Vaccines 2020, 5, 106. [Google Scholar] [CrossRef] [PubMed]
- Varypataki, E.M.; Silva, A.L.; Barnier-Quer, C.; Collin, N.; Ossendorp, F.; Jiskoot, W. Synthetic long peptide-based vaccine formulations for induction of cell mediated immunity: A comparative study of cationic liposomes and PLGA nanoparticles. J. Control. Release 2016, 226, 98–106. [Google Scholar] [CrossRef]
- Gao, S.; Yang, D.; Fang, Y.; Lin, X.; Jin, X.; Wang, Q.; Wang, X.; Ke, L.; Shi, K. Engineering nanoparticles for targeted remodeling of the tumor microenvironment to improve cancer immunotherapy. Theranostics 2019, 9, 126–151. [Google Scholar] [CrossRef] [PubMed]
- Grippin, A.J.; Sayour, E.J.; Mitchell, D.A. Translational nanoparticle engineering for cancer vaccines. Oncoimmunology 2017, 6, e1290036. [Google Scholar] [CrossRef]
- Seo, N.; Akiyoshi, K.; Shiku, H. Exosome-mediated regulation of tumor immunology. Cancer Sci. 2018, 109, 2998–3004. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Sanders, B.; Koldijk, M.; Schuitemaker, H. Inactivated Viral Vaccines. Vaccine Analysis: Strategies, Principles, and Control; Springer Nature: New York, NY, USA, 2015; ISBN 9783662450246. [Google Scholar]
- Fan, Y.; Bai, T.; Tian, Y.; Zhou, B.; Wang, Y.; Yang, L. H2o2-inactivated salmonella typhimurium re88 strain as a new cancer vaccine carrier: Evaluation in a mouse model of cancer. Drug Des. Dev. Ther. 2021, 15, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Jones, J.O.; Andino, R. Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 2010, 28, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, B.; Chauchet, X.; Wang, Y.; Polack, B.; Le Gouëllec, A. Live-attenuated bacteria as a cancer vaccine vector. Expert Rev. Vaccines 2013, 12, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Morein, B.; Helenius, A.; Simons, K.; Pettersson, R.; Kääriäinen, L.; Schirrmacher, V. Effective subunit vaccines against an enveloped animal virusories. Nature 1978, 276, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.L.; Impens, F. Immunopeptidomics for next-generation bacterial vaccine development. Trends Microbiol. 2021, 29, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D. Cancer Vaccines. Nat. Med. 1998, 4, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, S.A.; Lorincz, R.; Boucher, P.; Curiel, D.T. Adenoviral vector vaccine platforms in the SARS-CoV-2 pandemic. NPJ Vaccines 2021, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- de Pinho Favaro, M.T.; Atienza-Garriga, J.; Martínez-Torró, C.; Parladé, E.; Vázquez, E.; Corchero, J.L.; Ferrer-Miralles, N.; Villaverde, A. Recombinant vaccines in 2022: A perspective from the cell factory. Microb. Cell Factories 2022, 21, 203. [Google Scholar] [CrossRef] [PubMed]
- Travieso, T.; Li, J.; Mahesh, S.; Mello, J.D.F.R.E.; Blasi, M. The use of viral vectors in vaccine development. NPJ Vaccines 2022, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Ragothaman, M.; Yoo, S.Y. Engineered Phage-Based Cancer Vaccines: Current Advances and Future Directions. Vaccines 2023, 11, 919. [Google Scholar] [CrossRef] [PubMed]
- Pranchevicius, M.S.; Vieira, T.R. Production of recombinant immunotherapeutics for anticancer treatment: The role of bioengineering. Bioengineered 2013, 4, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Atkin-Smith, G.K.; Duan, M.; Chen, W.; Poon, I.K.H. The induction and consequences of Influenza A virus-induced cell death. Cell Death Dis. 2018, 9, 1002. [Google Scholar] [CrossRef] [PubMed]
- Imre, G. Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID-19. In The COVID-19 Resource Centre Is Hosted on Elsevier Connect, the Company’s Public News and Information; 2020; Available online: https://cir.nii.ac.jp/crid/1370576118741734285 (accessed on 23 February 2024).
- Yoo, J.S.; Sasaki, M.; Cho, S.X.; Kasuga, Y.; Zhu, B.; Ouda, R.; Orba, Y.; de Figueiredo, P.; Sawa, H.; Kobayashi, K.S. SARS-CoV-2 inhibits induction of the MHC class I pathway by targeting the STAT1-IRF1-NLRC5 axis. Nat. Commun. 2021, 12, 6602. [Google Scholar] [CrossRef] [PubMed]
- Pishesha, N.; Harmand, T.J.; Rothlauf, P.W.; Praest, P.; Alexander, R.K.; Van den Doel, R.; Liebeskind, M.J.; Vakaki, M.A.; McCaul, N.; Wijne, C.; et al. A class II MHC-targeted vaccine elicits immunity against SARS-CoV-2 and its variants. Proc. Natl. Acad. Sci. USA 2021, 118, e2116147118. [Google Scholar] [CrossRef] [PubMed]
- Minev, B.R.; McFarland, B.J.; Spiess, P.J.; Rosenberg, S.A.; Restifo, N.P. Insertion Signal Sequence Fused to Minimal Peptides Elicits Specific CD8+ T-Cell Responses and Prolongs Survival of Thymoma-bearing Mice. Cancer Res. 1994, 54, 4155–4161. [Google Scholar] [PubMed]
- Olson, E.; Ceccarelli, T.; Raghavan, M. Endo-lysosomal assembly variations among human leukocyte antigen class I (HLA class I) allotypes. Elife 2023, 12, e79144. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhong, Q.; Tian, T.; Dubin, K.; Athale, S.K.; Kupper, T.S. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat. Med. 2010, 16, 224–227. [Google Scholar] [CrossRef]
- Chiuppesi, F.; Salazar, A.; Contreras, H.; Nguyen, V.H.; Martinez, J. Development of a Synthetic Poxvirus-Based SARS-CoV-2 Vaccine. bioRxiv 2020. [Google Scholar] [CrossRef]
- Esposito, I.; Cicconi, P.; D’Alise, A.M.; Brown, A.; Esposito, M.; Swadling, L.; Holst, P.J.; Bassi, M.R.; Stornaiuolo, M.; Mori, F.; et al. MHC Class II Invariant Chain-Adjuvanted Viral Vectored Vaccines Enhances T Cell Responses in Humans. Sci. Transl. Med. 2020, 12, eaaz7715. [Google Scholar] [CrossRef]
- Fiander, A.; Nimako, M.; Man, S.; Wilkinson, G.W.G.; Boursnell, M.E.G.; Rutherford, E.; Hickling, J.K.; Inglis, S.C.; Evans, A.S.; Adams, M.; et al. A recombinant vaccinia virus encoding human papillomavirus types 16 and 18, E6 and E7 proteins as immunotherapy for cervical cancer. Lancet 1996, 347, 1523–1527. [Google Scholar]
- Wang, W.; Xu, H.; Ye, Q.; Tao, F.; Wheeldon, I.; Yuan, A.; Hu, Y.; Wu, J. Systemic immune responses to irradiated tumours via the transport of antigens to the tumour periphery by injected flagellate bacteria. Nat. Biomed. Eng. 2022, 6, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Keenan, B.P.; Jaffee, E.M. Whole cell vaccines—Past progress and future strategies. Semin. Oncol. 2012, 39, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Bencherif, S.A.; Sands, R.W.; Ali, O.A.; Li, W.A.; Lewin, S.A.; Braschler, T.M.; Shih, T.Y.; Verbeke, C.S.; Bhatta, D.; Dranoff, G.; et al. Injectable cryogel-based whole-cell cancer vaccines. Nat. Commun. 2015, 6, 7556. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.L.; Benencia, F.; Coukos, G. Whole tumor antigen vaccines. Semin. Immunol. 2010, 22, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Warfel, J.M.; Zimmerman, L.I.; Merkel, T.J. Acellular pertussis vaccines protect against disease butfail to prevent infection and transmission ina nonhuman primate model. Proc. Natl. Acad. Sci. USA 2014, 111, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Alghounaim, M.; Alsaffar, Z.; Alfraij, A.; Bin-Hasan, S.; Hussain, E. Whole-Cell and Acellular Pertussis Vaccine: Reflections on Efficacy. Med. Princ. Pract. 2022, 31, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Stoitzner, P.; Romani, N.; Rademacher, C.; Probst, H.C.; Mahnke, K. Antigen targeting to dendritic cells: Still a place in future immunotherapy? Eur. J. Immunol. 2022, 52, 1909–1924. [Google Scholar] [CrossRef]
- Liu, K. Dendritic Cells. Encycl. Cell Biol. 2016, 3, 741–749. [Google Scholar] [CrossRef]
- Dong, W.; Du, J.; Shen, H.; Gao, D.; Li, Z.; Wang, G.; Mu, X.; Liu, Q. Administration of embryonic stem cells generates effective antitumor immunity in mice with minor and heavy tumor load. Cancer Immunol. Immunother. 2010, 59, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Telli, M.L.; Wu, J.C. Induced pluripotent stem cell-based cancer vaccines. Front. Immunol. 2019, 10, 460435. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Bolton, E.M.; Pocock, S.; Sharples, L.D.; Pedersen, R.A.; Bradley, J.A. Banking on human embryonic stem cells: Estimating the number of donor cell lines needed for HLA matching. Lancet 2005, 366, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.J.; Brook, F.A.; Gardner, R.L.; Cobbold, S.P.; Waldmann, H.; Fairchild, P.J. Embryonic stem cell-derived tissues are immunogenic but their inherent immune privilege promotes the induction of tolerance. Proc. Natl. Acad. Sci. USA 2007, 104, 20920–20925. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Yu, C.Y.; Chen, M.J.; Chou, S.H.; Chiang, M.S.; Chou, W.H.; Ko, B.S.; Huang, H.P.; Kuo, H.C.; Ho, H.N. Characteristic expression of major histocompatibility complex and immune privilege genes in human pluripotent stem cells and their derivatives. Cell Transplant. 2015, 24, 845–864. [Google Scholar] [CrossRef] [PubMed]
- Drukker, M.; Katz, G.; Urbach, A.; Schuldiner, M.; Markel, G.; Itskovitz-Eldor, J.; Reubinoff, B.; Mandelboim, O.; Benvenisty, N. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9864–9869. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.; Gong, L.; Yu, D.; An, C.; Bunpetch, V.; Dai, J.; Huang, H.; Zou, X.; Ouyang, H.; et al. The Plasticity of Mesenchymal Stem Cells in Regulating Surface HLA-I. iScience 2019, 15, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Mehler, V.J.; Burns, C.J.; Stauss, H.; Francis, R.J.; Moore, M.L. Human iPSC-Derived Neural Crest Stem Cells Exhibit Low Immunogenicity. Mol. Ther.-Methods Clin. Dev. 2020, 16, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Álvarez, B.; Rodriguez, R.M.; Calvanese, V.; Blanco-Gelaz, M.A.; Suhr, S.T.; Ortega, F.; Otero, J.; Cibelli, J.B.; Moore, H.; Fraga, M.F.; et al. Epigenetic mechanisms regulate MHC and antigen processing molecules in human embryonic and induced pluripotent stem cells. PLoS ONE 2010, 5, e10192. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Lin, X.; Cui, D.; Cui, C.; Li, H.; Xiao, L. Functional disruption of human leukocyte antigen II in human embryonic stem cell. Biol. Res. 2015, 48, 59. [Google Scholar] [CrossRef] [PubMed]
- Drukker, M.; Katchman, H.; Katz, G.; Even-Tov Friedman, S.; Shezen, E.; Hornstein, E.; Mandelboim, O.; Reisner, Y.; Benvenisty, N. Human Embryonic Stem Cells and Their Differentiated Derivatives Are Less Susceptible to Immune Rejection Than Adult Cells. Stem Cells 2006, 24, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Paskin, N. Toward unique identifiers. Proc. IEEE 1999, 87, 1208–1227. [Google Scholar] [CrossRef]
- Bifari, F. Immunological properties of embryonic and adult stem cells. World J. Stem Cells 2010, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.L.; Coukos, G.; Kandalaft, L.E. Whole tumor antigen vaccines: Where are we? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, H.; Xu, R.H.; Liu, B.; Li, Z. Vaccination with human pluripotent stem cells generates a broad spectrum of immunological and clinical responses against colon cancer. Stem Cells 2009, 27, 3103–3111. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Chen, X.H.; Chang, X.H.; Ye, X.; Li, Y.; Cui, H. Human embryonic stem cells—A potential vaccine for ovarian cancer. Asian Pac. J. Cancer Prev. 2012, 13, 4295–4300. [Google Scholar] [CrossRef] [PubMed]
- Gordeeva, O.; Khaydukov, S. Tumorigenic and Differentiation Potentials of Embryonic Stem Cells Depend on TGF β Family Signaling: Lessons from Teratocarcinoma Cells Stimulated to Differentiate with Retinoic Acid. Stem Cells Int. 2017, 2017, 7284872. [Google Scholar] [CrossRef] [PubMed]
- Lezmi, E.; Benvenisty, N. The Tumorigenic Potential of Human Pluripotent Stem Cells. Stem Cells Transl. Med. 2022, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference maps of human es and ips cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, Y. Embryonic stem cell and induced pluripotent stem cell: An epigenetic perspective. Cell Res. 2013, 23, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Efrat, S. Epigenetic Memory: Lessons From iPS Cells Derived From Human β Cells. Front. Endocrinol. 2021, 11, 614234. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Takasawa, K.; Okamura, K.; Arai, Y.; Sekiya, A.; Akutsu, H.; Umezawa, A. Identification of an epigenetic signature in human induced pluripotent stem cells using a linear machine learning model. Hum. Cell 2021, 34, 99–110. [Google Scholar] [CrossRef]
- de Boni, L.; Gasparoni, G.; Haubenreich, C.; Tierling, S.; Schmitt, I.; Peitz, M.; Koch, P.; Walter, J.; Wüllner, U.; Brüstle, O. DNA methylation alterations in iPSC- and hESC-derived neurons: Potential implications for neurological disease modeling. Clin. Epigenetics 2018, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, M.S.; Strano, A.; Guan, K. Human Induced Pluripotent Stem Cells: From Cell Origin, Genomic Stability, and Epigenetic Memory to Translational Medicine. Stem Cells 2022, 40, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Inui, S.; Minami, K.; Ito, E.; Imaizumi, H.; Mori, S.; Koizumi, M.; Fukushima, S.; Miyagawa, S.; Sawa, Y.; Matsuura, N. Irradiation strongly reduces tumorigenesis of human induced pluripotent stem cells. J. Radiat. Res. 2017, 58, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Ito, E.; Minami, K.; Harada, A.; Mochizuki-Oda, N.; Sawa, Y.; Miyagawa, S. Elimination of residual undifferentiated induced pluripotent stem cells (iPSCs) using irradiation for safe clinical applications of iPSC-derived cardiomyocytes. Biochem. Biophys. Res. Commun. 2021, 574, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, H.; Pourfathollah, A.A.; Nikougoftar Zarif, M.; Shahbazfar, A.A. Irradiation enhances susceptibility of tumor cells to the antitumor effects of TNF-α activated adipose derived mesenchymal stem cells in breast cancer model. Sci. Rep. 2016, 6, 28433. [Google Scholar] [CrossRef] [PubMed]
- Kooreman, N.G.; Kim, Y.; de Almeida, P.E.; Termglinchan, V.; Diecke, S.; Shao, N.Y.; Wei, T.T.; Yi, H.; Dey, D.; Nelakanti, R.; et al. Autologous iPSC-Based Vaccines Elicit Anti-tumor Responses In Vivo. Cell Stem Cell 2018, 22, 501–513.e7. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, X.; Liu, L.; Zhou, M.; Zhou, G.; Liu, T. Development of the T-ALLiPSC-based therapeutic cancer vaccines for T-cell acute lymphoblastic leukemia. Med. Oncol. 2022, 39, 200. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, T.; Hou, B.; Huang, X. An iPSC-derived exosome-pulsed dendritic cell vaccine boosts antitumor immunity in melanoma. Mol. Ther. 2023, 31, 2376–2390. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, L.; Zhao, T.; Zhang, J. A nanotherapeutic system for gastric cancer suppression by synergistic chemotherapy and immunotherapy based on iPSCs and DCs exosomes. Cancer Immunol. Immunother. 2023, 72, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, F.; Razmi, M.; Tajik, F.; Zöller, M.; Dehghan Manshadi, M.; Mahdavinezhad, F.; Tiyuri, A.; Ghods, R.; Madjd, Z. Efficacy of Whole Cancer Stem Cell-Based Vaccines: A Systematic Review of Preclinical and Clinical Studies. Stem Cells 2023, 41, 207–232. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.S.; Miller, M.S.; Thirawatananond, P.; Douglass, J.; Wright, K.M.; Hsiue, E.H.C.; Mog, B.J.; Aytenfisu, T.Y.; Murphy, M.B.; Aitana Azurmendi, P.; et al. Structural engineering of chimeric antigen receptors targeting HLA-restricted neoantigens. Nat. Commun. 2021, 12, 5271. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther.-Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Böll, B.; Schellongowski, P.; Valade, S.; Metaxa, V.; Azoulay, E.; von Bergwelt-Baildon, M. Critical care management of chimeric antigen receptor T-cell therapy recipients. CA Cancer J. Clin. 2022, 72, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Syed, F.; El Fakih, R.; Alahmari, A.D.; Osman Ali, A.S.; Aljurf, M. Chimeric Antigen Receptor Structure and Manufacturing of Clinical Grade CAR Engineered Cells using Different Bioreactors. Hematol. Oncol. Stem Cell Ther. 2022, 15, 137–152. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.A.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.; Munshi, P.N.; Casulo, C.; Maloney, D.G.; de Vos, S.; et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): A single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022, 23, 91–103. [Google Scholar] [CrossRef]
- Mishra, A.; Maiti, R.; Mohan, P.; Gupta, P. Antigen loss following CAR-T cell therapy: Mechanisms, implications, and potential solutions. Eur. J. Haematol. 2023, 112, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Vago, L.; Mutis, T. Tumour Escape from CAR-T Cells. In The EBMT/EHA CAR-T Cell Handbook; Springer: Cham, Germany, 2022; pp. 15–22. [Google Scholar] [CrossRef]
- Ma, L.; Hostetler, A.; Morgan, D.M.; Maiorino, L.; Sulkaj, I.; Whittaker, C.A.; Neeser, A.; Pires, I.S.; Yousefpour, P.; Gregory, J.; et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. Cell 2023, 186, 3148–3165.e20. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Murphy, T.L.; Murphy, K.M. Dendritic cells in cancer immunology. Cell. Mol. Immunol. 2022, 19, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell. Mol. Immunol. 2023, 20, 432–447. [Google Scholar] [CrossRef]
- Katakam, A.K.; Brightbill, H.; Franci, C.; Kung, C.; Nunez, V.; Jones, C.; Peng, I.; Jeet, S.; Wu, L.C.; Mellman, I.; et al. Dendritic cells require NIK for CD40-dependent cross-priming of CD8+ T cells. Proc. Natl. Acad. Sci. USA 2015, 112, 14664–14669. [Google Scholar] [CrossRef] [PubMed]
- Manh, T.P.V.; Alexandre, Y.; Baranek, T.; Crozat, K.; Dalod, M. Plasmacytoid, conventional, and monocyte-derived dendritic cells undergo a profound and convergent genetic reprogramming during their maturation. Eur. J. Immunol. 2013, 43, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Smith, J.L.; Caspary, G.; Brasel, K.; Pettit, D.; Maraskovsky, E.; Maliszewski, C.R. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Theisen, D. The role of cDC1s in vivo: CD8 T cell priming through cross-presentation. F1000Research 2017, 6, 98. [Google Scholar] [CrossRef]
- Ferris, S.T.; Durai, V.; Wu, R.; Theisen, D.J.; Ward, J.P.; Bern, M.D.; Iv, J.T.D.; Bagadia, P.; Liu, T.; Carlos, G.; et al. cDC1 prime and are licensed by CD4 T cells to induce anti-tumour immunity. Nature 2021, 584, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Segura, E. Cross-dressed cDC1s instruct T cells in allorecognition. Immunol. Cell Biol. 2020, 98, 520–523. [Google Scholar] [CrossRef]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 2020, 52, 1039–1056.e9. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Ikegawa, M.; Kawai, T. Antigen Presentation in the Lung. Front. Immunol. 2022, 13, 860915. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Do Kim, K.; Lee, H.K. The role of dendritic cells in tumor microenvironments and their uses as therapeutic targets. BMB Rep. 2021, 54, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Komori, S.; Kotani, T.; Murata, Y.; Matozaki, T. The Role of Type-2 Conventional Dendritic Cells in the Regulation of Tumor Immunity. Cancers 2022, 14, 1976. [Google Scholar] [CrossRef] [PubMed]
- Ugur, M.; Mueller, S.N. T cell and dendritic cell interactions in lymphoid organs: More than just being in the right place at the right time. Immunol. Rev. 2019, 289, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, X.; Cheng, Y.; Cao, X. Dendritic cell migration in inflammation and immunity. Cell. Mol. Immunol. 2021, 18, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, C.A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589.e8. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallée, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.e24. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Peng, P.; Loschko, J.; Feng, L.; Pham, P.; Cui, W.; Lee, K.P.; Krug, A.B.; Jiang, A. Plasmacytoid dendritic cells cross-prime naive CD8 T cells by transferring antigen to conventional dendritic cells through exosomes. Proc. Natl. Acad. Sci. USA 2020, 117, 23730–23741. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kasahara, S.; Jhingran, A.; Tosini, N.L.; Zhai, B.; Aufiero, M.A.; Mills, K.A.M.; Gjonbalaj, M.; Espinosa, V.; Rivera, A.; et al. During Aspergillus Infection, Monocyte-Derived DCs, Neutrophils, and Plasmacytoid DCs Enhance Innate Immune Defense through CXCR3-Dependent Crosstalk. Cell Host Microbe 2020, 28, 104–116.e4. [Google Scholar] [CrossRef] [PubMed]
- Marzaioli, V.; Canavan, M.; Floudas, A.; Wade, S.C.; Low, C.; Veale, D.J.; Fearon, U. Monocyte-Derived Dendritic Cell Differentiation in Inflammatory Arthritis Is Regulated by the JAK/STAT Axis via NADPH Oxidase Regulation. Front. Immunol. 2020, 11, 1406. [Google Scholar] [CrossRef] [PubMed]
- Boulet, S.; Daudelin, J.F.; Odagiu, L.; Pelletier, A.N.; Yun, T.J.; Lesage, S.; Cheong, C.; Labrecque, N. The orphan nuclear receptor NR4A3 controls the differentiation of monocyte-derived dendritic cells following microbial stimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 15150–15159. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.L.; Dalod, M. The Quest for Faithful In Vitro Models of Human Dendritic Cells Types. Mol. Immunol. 2020, 123, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Bruzzì, S.; Heymann, F.; Liepelt, A.; Krenkel, O.; Toscani, A.; Ramavath, N.N.; Cotella, D.; Albano, E.; Tacke, F. CX3CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation. Cells 2019, 8, 1099. [Google Scholar] [CrossRef] [PubMed]
- Leblanc-Hotte, A.; Audiger, C.; Chabot-Roy, G.; Lombard-Vadnais, F.; Delisle, J.S.; Peter, Y.A.; Lesage, S. Immature and mature bone marrow-derived dendritic cells exhibit distinct intracellular mechanical properties. Sci. Rep. 2023, 13, 1967. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.S.; El-Sukkari, D.; Villadangos, J.A. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood 2004, 103, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Kim, J. Properties of immature and mature dendritic cells: Phenotype, morphology, phagocytosis, and migration. RSC Adv. 2019, 9, 11230–11238. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S.; Chen, L.M.; Kroschewski, R.; Ebersold, M.; Turley, S.; Trombetta, S.; Galán, J.E.; Mellman, I. Developmental control of endocytosis in dendritic cells by Cdc42. Cell 2000, 102, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, J.A.; Schnorrer, P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 2007, 7, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiang, Y.; Xin, V.W.; Wang, X.W.; Peng, X.C.; Liu, X.Q.; Wang, D.; Li, N.; Cheng, J.T.; Lyv, Y.N.; et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020, 13, 107. [Google Scholar] [CrossRef] [PubMed]
- Platt, C.D.; Ma, J.K.; Chalouni, C.; Ebersold, M.; Bou-Reslan, H.; Carano, R.A.D.; Mellman, I.; Delamarre, L. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc. Natl. Acad. Sci. USA 2010, 107, 4287–4292. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.B.; Alsarraj, M.; Gladue, R.P.; Bedian, V.; Antonia, S.J. An agonist antibody specific for CD40 induces dendritic cell maturation and promotes autologous anti-tumour T-cell responses in an in vitro mixed autologous tumour cell/lymph node cell model. Scand. J. Immunol. 2007, 65, 479–486. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Ravetch, J.V. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell 2015, 161, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Nino-Castro, A.C.; Schultze, J.L. Regulatory dendritic cells: There is more than just immune activation. Front. Immunol. 2012, 3, 274. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 2007, 7, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, Ö.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Alloatti, A.; Kotsias, F.; Magalhaes, J.G.; Amigorena, S. Dendritic cell maturation and cross-presentation: Timing matters! Immunol. Rev. 2016, 272, 97–108. [Google Scholar] [CrossRef] [PubMed]
- van der Wel, N.N.; Sugita, M.; Fluitsma, D.M.; Cao, X.; Schreibelt, G.; Brenner, M.B.; Peters, P.J. CD1 and Major Histocompatibility Complex II Molecules Follow a Different Course during Dendritic Cell Maturation. Mol. Biol. Cell 2003, 14, 3378–3388. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, J.A.; Schnorrer, P.; Wilson, N.S. Control of MHC class II antigen presentation in dendritic cells: A balance between creative and destructive forces. Immunol. Rev. 2005, 207, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Hubo, M.; Trinschek, B.; Kryczanowsky, F.; Tuettenberg, A.; Steinbrink, K.; Jonuleit, H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front. Immunol. 2013, 4, 82. [Google Scholar] [CrossRef] [PubMed]
- Carenza, C.; Calcaterra, F.; Oriolo, F.; Di Vito, C.; Ubezio, M.; Della Porta, M.G.; Mavilio, D.; Della Bella, S. Costimulatory molecules and immune checkpoints are differentially expressed on different subsets of dendritic cells. Front. Immunol. 2019, 10, 1325. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Rad, A.N.; Grosenbach, D.W.; Sabzevari, H.; Gómez Yafal, A.; Gritz, L.; Schlom, J. Enhanced activation of T cells by dendritic cells engineered to hyperexpress a triad of costimulatory molecules. J. Natl. Cancer Inst. 2000, 92, 1228–1239. [Google Scholar] [CrossRef]
- Liu, X.; Xia, X.; Wang, X.; Zhou, J.; Sung, L.A.; Long, J.; Geng, X.; Zeng, Z.; Yao, W. Tropomodulin1 Expression Increases Upon Maturation in Dendritic Cells and Promotes Their Maturation and Immune Functions. Front. Immunol. 2021, 11, 587441. [Google Scholar] [CrossRef] [PubMed]
- Tiberio, L.; Del Prete, A.; Schioppa, T.; Sozio, F.; Bosisio, D.; Sozzani, S. Chemokine and chemotactic signals in dendritic cell migration review-article. Cell. Mol. Immunol. 2018, 15, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J.; Yang, D.; Biragyn, A.; Howard, O.M.Z.; Plotz, P. Chemokine receptors on dendritic cells promote autoimmune reactions. Arthritis Res. 2002, 4 (Suppl. S3), 183–188. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Pascual, V.; Palucka, A.K. Autoimmunity through cytokine-induced dendritic cell activation. Immunity 2004, 20, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Ebner, S.; Ratzinger, G.; Krösbacher, B.; Schmuth, M.; Weiss, A.; Reider, D.; Kroczek, R.A.; Herold, M.; Heufler, C.; Fritsch, P.; et al. Production of IL-12 by Human Monocyte-Derived Dendritic Cells Is Optimal When the Stimulus Is Given at the Onset of Maturation, and Is Further Enhanced by IL-4. J. Immunol. 2001, 166, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.B.; Guerra, J.; Firek, A.; Langridge, W.H.R. Extracellular vimentin modulates human dendritic cell activation. Mol. Immunol. 2018, 104, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-R.; Muckersie, E.; Robertson, M.; Xu, H.; Liversidge, J.; Forrester, J.V. Secretion of interleukin-10 or interleukin-12 by LPS-activated dendritic cells is critically dependent on time of stimulus relative to initiation of purified DC culture. J. Leukoc. Biol. 2002, 72, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Erra Díaz, F.; Ochoa, V.; Merlotti, A.; Dantas, E.; Mazzitelli, I.; Gonzalez Polo, V.; Sabatté, J.; Amigorena, S.; Segura, E.; Geffner, J. Extracellular Acidosis and mTOR Inhibition Drive the Differentiation of Human Monocyte-Derived Dendritic Cells. Cell Rep. 2020, 31, 107613. [Google Scholar] [CrossRef] [PubMed]
- Boudier, A.; Aubert-Pouëssel, A.; Louis-Plence, P.; Gérardin, C.; Jorgensen, C.; Devoisselle, J.M.; Bégu, S. The control of dendritic cell maturation by pH-sensitive polyion complex micelles. Biomaterials 2009, 30, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hanč, P.; Schulz, O.; Fischbach, H.; Martin, S.R.; Kjær, S.; Reis e Sousa, C. A pH- and ionic strength-dependent conformational change in the neck region regulates DNGR-1 function in dendritic cells. EMBO J. 2016, 35, 2484–2497. [Google Scholar] [CrossRef]
- Lee, H.; Park, H.; Yu, H.S.; Na, K.; Oh, K.T.; Lee, E.S. Dendritic cell-targeted ph-responsive extracellular vesicles for anticancer vaccination. Pharmaceutics 2019, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 Controls Phagosomal pH to Regulate Antigen Processing during Crosspresentation by Dendritic Cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Pérez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Mahmud, A.R.; Siddiquee, M.F.-R.-; Shahriar, A.; Biswas, P.; Shimul, M.E.K.; Ahmed, S.Z.; Ema, T.I.; Rahman, N.; Khan, M.A.; et al. Role of T cells in cancer immunotherapy: Opportunities and challenges. Cancer Pathog. Ther. 2023, 1, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Park, J.J.; Peng, L.; Yang, Q.; Chow, R.D.; Dong, M.B.; Lam, S.Z.; Guo, J.; Tang, E.; Zhang, Y.; et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022, 34, 595–614.e14. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Haining, W.N.; Schumacher, T.N.M. When Cancer Cells Become the Enablers of an Antitumor Immune Response. Cancer Discov. 2022, 12, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Al Moussawy, M.; Abdelsamed, H.A. Non-cytotoxic functions of CD8 T cells: “repentance of a serial killer”. Front. Immunol. 2022, 13, 1001129. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Zhou, L.; Wang, B.; Sun, X.; Zhang, Q.; Chen, H.; Wan, N.; Ye, H.; Wu, X.; Sun, D.; et al. Targeting cathepsin B by cycloastragenol enhances antitumor immunity of CD8 T cells via inhibiting MHC-I degradation. J. Immunother. Cancer 2022, 10, e004874. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Wang, Q.; Korner, H.; Zhang, L.; Wei, W. Molecular mechanisms of T cells activation by dendritic cells in autoimmune diseases. Front. Pharmacol. 2018, 9, 642. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Vesely, M.D. Stimulating T Cells against Cancer with Agonist Immunostimulatory Monoclonal Antibodies. Int. Rev. Cell Mol. Biol. 2019, 342, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Shi, Y.; Haymaker, C.L.; Naing, A.; Ciliberto, G.; Hajjar, J. T-cell agonists in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000966. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Dallas, B. Flies Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Weyand, C.M. T-cell co-stimulatory pathways in autoimmunity. Arthritis Res. Ther. 2008, 10, S3. [Google Scholar] [CrossRef] [PubMed]
- Goleva, E.; Lyubchenko, T.; Kraehenbuehl, L.; Lacouture, M.E.; Leung, D.Y.M.; Kern, J.A. Our current understanding of checkpoint inhibitor therapy in cancer immunotherapy. Ann. Allergy Asthma Immunol. 2021, 126, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Mccallion, O.; Bilici, M.; Hester, J.; Issa, F. Regulatory T-cell therapy approaches. Clin. Exp. Immunol. 2023, 211, 96–107. [Google Scholar] [CrossRef]
- Dieckmann, D.; Plottner, H.; Berchtold, S.; Berger, T.; Schuler, G. Ex vivo isolation and characterization of CD4+CD25+ T cells with regulatory properties from human blood. J. Exp. Med. 2001, 193, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rocamora-Reverte, L.; Melzer, F.L.; Würzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front. Immunol. 2021, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cheng, X.; Zhang, L.; Lu, X.; Chaudhary, S.; Teng, R.; Frederickson, C.; Champion, M.M.; Zhao, R.; Cheng, L.; et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 10094–10099. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Li, K.; Ni, Y.; Liang, X.; Zhao, X. Myeloid-Derived Suppressor Cells: Implications in the Resistance of Malignant Tumors to T Cell-Based Immunotherapy. Front. Cell Dev. Biol. 2021, 9, 707198. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Dario, A.A.; Vignali, L.W.C.; Creg, J.W. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Yan, S.; Kotschenreuther, K.; Deng, S.; Kofler, D.M. Regulatory T cells in rheumatoid arthritis: Functions, development, regulation, and therapeutic potential. Cell. Mol. Life Sci. 2022, 79, 533. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, S.; Gao, X.; Zhou, J.; Zhu, X.; Wang, D.; Cai, Y.; Li, F.; Yang, Q.; Gu, X.; et al. Interleukin-6-mediated CCR9+ interleukin-17-producing regulatory T cells polarization increases the severity of necrotizing enterocolitis. EBioMedicine 2019, 44, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Gough, M.J.; Crittenden, M.R. The paradox of radiation and T cells in tumors. Neoplasia 2022, 31, 100808. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Cui, B. Effect of radiotherapy on T cell and PD-1 / PD-L1 blocking therapy in tumor microenvironment. Hum. Vaccines Immunother. 2021, 17, 1555–1567. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Estévez, D.; Sánchez, R.; Tejada, R.E.; Parra-López, C. Chemotherapy and radiation therapy elicits tumor specific T cell responses in a breast cancer patient. BMC Cancer 2016, 16, 104257. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, I.J.; Bernal-Estévez, D.A.; Llano-León, M.; Bonilla, C.E.; Parra-López, C.A. Neoadjuvant chemotherapy modulates exhaustion of T cells in breast cancer patients. PLoS ONE 2023, 18, e0280851. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, Q.; Huang, X.; Koo, S.; Kong, N.; Tao, W. mRNA-based cancer therapeutics. Nat. Rev. Cancer 2023, 23, 526–543. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, R.R.; Boyer, J.D.; Ugen, K.E.; Lacy, K.E.; Gluckman, S.J.; Bagarazzi, M.L.; Chattergoon, M.A.; Baine, Y.; Higgins, T.J.; Ciccarelli, R.B.; et al. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: Safety and host response. J. Infect. Dis. 1998, 178, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z. DNA Vaccine. Adv. Genet. 2005, 54, 257–289. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 146. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.K.; Ottensmeier, C.H.; Johnson, P.; Zhu, D.; Buchan, S.L.; McCann, K.J.; Roddick, J.S.; King, A.T.; McNicholl, F.; Savelyeva, N.; et al. DNA vaccines to attack cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 14646–14652. [Google Scholar] [CrossRef] [PubMed]
- Jorritsma, S.H.T.; Gowans, E.J.; Grubor-Bauk, B.; Wijesundara, D.K. Delivery methods to increase cellular uptake and immunogenicity of DNA vaccines. Vaccine 2016, 34, 5488–5494. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Luo, Y.; Lo, J.F.; Kaplan, C.D.; Mizutani, M.; Mizutani, N.; Lee, J.D.; Primus, F.J.; Becker, J.C.; Xiang, R.; et al. DNA-based vaccines activate innate and adaptive antitumor immunity by engaging the NKG2D receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 10846–10851. [Google Scholar] [CrossRef]
- Rezaei, T.; Davoudian, E.; Khalili, S.; Amini, M.; Hejazi, M.; de la Guardia, M.; Mokhtarzadeh, A. Strategies in DNA vaccine for melanoma cancer. Pigment Cell Melanoma Res. 2021, 34, 869–891. [Google Scholar] [CrossRef] [PubMed]
- Sudowe, S.; Dominitzki, S.; Montermann, E.; Bros, M.; Grabbe, S.; Reske-Kunz, A.B. Uptake and presentation of exogenous antigen and presentation of endogenously produced antigen by skin dendritic cells represent equivalent pathways for the priming of cellular immune responses following biolistic DNA immunization. Immunology 2009, 128, e193–e205. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. CpG motifs: The active ingredient in bacterial extracts? Nat. Med. 2003, 9, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant (author manuscript). Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type i interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Kutzler, M.A.; Weiner, D.B. DNA vaccines: Ready for prime time? Nat. Rev. Genet. 2008, 9, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Faurez, F.; Dory, D.; Le Moigne, V.; Gravier, R.; Jestin, A. Biosafety of DNA vaccines: New generation of DNA vectors and current knowledge on the fate of plasmids after injection. Vaccine 2010, 28, 3888–3895. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Vanvarenberg, K.; Préat, V.; Vandermeulen, G. Codon-Optimized P1A-Encoding DNA Vaccine: Toward a Therapeutic Vaccination against P815 Mastocytoma. Nucleic Acids-Mol. Ther. 2017, 8, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Stachyra, A.; Redkiewicz, P.; Kosson, P.; Protasiuk, A.; Góra-Sochacka, A.; Kudla, G.; Sirko, A. Codon optimization of antigen coding sequences improves the immune potential of DNA vaccines against avian influenza virus H5N1 in mice and chickens. Virol. J. 2016, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Ferrall, L.; Gaillard, S.; Wang, C.; Chi, W.Y.; Huang, C.H.; Roden, R.B.S.; Wu, T.C.; Chang, Y.N.; Hung, C.F. Development of dna vaccine targeting e6 and e7 proteins of human papillomavirus 16 (Hpv16) and hpv18 for immunotherapy in combination with recombinant vaccinia boost and pd-1 antibody. mBio 2021, 12, 10–1128. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, L.; Lin, A.; Xu, C.; Li, Z.; Liu, K.; Liu, B.; Ma, X.; Zhao, F.; Jiang, H.; et al. Algorithm for Optimized mRNA Design Improves Stability and Immunogenicity. Nature 2023, 621, 396–403. [Google Scholar] [CrossRef]
- Ferraro, B.; Morrow, M.P.; Hutnick, N.A.; Shin, T.H.; Lucke, C.E.; Weiner, D.B. Clinical applications of DNA vaccines: Current progress. Clin. Infect. Dis. 2011, 53, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Jeang, J.; Yang, A.; Wu, T.C.; Hung, C.F. DNA vaccine for cancer immunotherapy. Hum. Vaccines Immunother. 2014, 10, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Target, A.M.; Reference, B.F. Molecular mechanisms for enhanced DNA vaccine immunogenicity. Expert Rev. Vaccines 2016, 15, 313–329. [Google Scholar] [CrossRef]
- Saade, F.; Petrovsky, N. Technologies for enhanced efficacy of DNA vaccines. Expert Rev. Vaccines 2012, 11, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Ledgerwood, J.E.; Pierson, T.C.; Hubka, S.A.; Desai, N.; Rucker, S.; Gordon, I.J.; Enama, M.E.; Nelson, S.; Nason, M.; Gu, W.; et al. A west nile virus DNA vaccine utilizing a modified promoter induces neutralizing antibody in younger and older healthy adults in a phase I clinical trial. J. Infect. Dis. 2011, 203, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Garrod, T.J.; Grubor-Bauk, B.; Gargett, T.; Li, Y.; Miller, D.S.; Yu, W.; Major, L.; Burrell, C.J.; Wesselingh, S.; Suhrbier, A.; et al. DNA vaccines encoding membrane-bound or secreted forms of heat shock protein 70 exhibit improved potency. Eur. J. Immunol. 2014, 44, 1992–2002. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, W.; Xu, Q.; Liu, Y.; Liu, H.; Guo, R.; Chiou, C.J.; Gao, K.; Jin, B.; Chen, C.; et al. Adjuvant DNA vaccine pNMM promotes enhanced specific immunity and anti-tumor effects. Hum. Vaccines Immunother. 2023, 19, 2202127. [Google Scholar] [CrossRef] [PubMed]
- Krieg, P.A.; Melton, D.A. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984, 12, 7057–7070. [Google Scholar] [CrossRef] [PubMed]
- Boczkowski, B.D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic Cells Pulsed with RNA Are Potent Antigen—Presenting Cells In Vitro and In Vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Lévy, J.-P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Peng, X.; Yang, Y.; Chen, Q.; Liu, J.; Tan, J.; Lou, C.; Liao, Z. mRNA vaccine in cancer therapy: Current advance and future outlook. Clin. Transl. Med. 2023, 13, e1384. [Google Scholar] [CrossRef]
- Duan, L.; Wang, Q.; Zhang, C.; Yang, D.; Zhang, X. Potentialities and Challenges of mRNA Vaccine in Cancer Immunotherapy. Front. Immunol. 2022, 13, 923647. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Greener, M. mRNA anti-cancer vaccine research gains momentum. Prescriber 2023, 34, 5–8. [Google Scholar] [CrossRef]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Jiang, L.; Liao, S.; Wu, F.; Yang, G.; Hou, L.; Liu, L. Vaccines’ New Era—RNA Vaccine. Viruses 2023, 15, 1760. [Google Scholar] [CrossRef] [PubMed]
- Szabó, G.T.; Mahiny, A.J.; Vlatkovic, I. COVID-19 mRNA vaccines: Platforms and current developments. Mol. Ther. 2022, 30, 1850–1868. [Google Scholar] [CrossRef] [PubMed]
- Perenkov, A.D.; Sergeeva, A.D.; Vedunova, M.V. In Vitro Transcribed RNA-Based Platform Vaccines: Past, Present, and Future. Vaccines 2023, 11, 1600. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 794528. [Google Scholar] [CrossRef] [PubMed]
- Tockary, T.A.; Abbasi, S.; Matsui-Masai, M.; Hayashi, A.; Yoshinaga, N.; Boonstra, E.; Wang, Z.; Fukushima, S.; Kataoka, K.; Uchida, S. Comb-structured mRNA vaccine tethered with short double-stranded RNA adjuvants maximizes cellular immunity for cancer treatment. Proc. Natl. Acad. Sci. USA 2023, 120, 2017. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and challenges in the delivery of mrna-based vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Pei, J.; Xu, S.; Liu, J.; Yu, J. Recent advances in mRNA cancer vaccines: Meeting challenges and embracing opportunities. Front. Immunol. 2023, 14, 1246682. [Google Scholar] [CrossRef]
- Kim, S.C.; Sekhon, S.S.; Shin, W.R.; Ahn, G.; Cho, B.K.; Ahn, J.Y.; Kim, Y.H. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency. Mol. Cell. Toxicol. 2022, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Wang, X. RNA modification in mRNA cancer vaccines. Clin. Exp. Med. 2023, 23, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Ogurtsov, A.Y.; Spiridonov, N.A. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res. 2006, 34, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Xu, Z.; Miao, L.; Huang, L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol. Ther. 2018, 26, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther.-Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bernard, M.C.; Bazin, E.; Petiot, N.; Lemdani, K.; Commandeur, S.; Verdelet, C.; Margot, S.; Perkov, V.; Ripoll, M.; Garinot, M.; et al. The impact of nucleoside base modification in mRNA vaccine is influenced by the chemistry of its lipid nanoparticle delivery system. Mol. Ther.-Nucleic Acids 2023, 32, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Hu, Q. Advances in saRNA Vaccine Research against Emerging/Re-Emerging Viruses. Vaccines 2023, 11, 1142. [Google Scholar] [CrossRef] [PubMed]
- Deviatkin, A.A.; Simonov, R.A.; Trutneva, K.A.; Maznina, A.A.; Soroka, A.B.; Kogan, A.A.; Feoktistova, S.G.; Khavina, E.M.; Mityaeva, O.N.; Volchkov, P.Y. Cap-Independent Circular mRNA Translation Efficiency. Vaccines 2023, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Berlanda Scorza, F.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W.; et al. Self-Amplifying mRNA Vaccines; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; Volume 89. [Google Scholar]
- Schmidt, C.; Schnierle, B.S. Self-Amplifying RNA Vaccine Candidates: Alternative Platforms for mRNA Vaccine Development. Pathogens 2023, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Voigt, E.A.; Gerhardt, A.; Hanson, D.; Jennewein, M.F.; Battisti, P.; Reed, S.; Singh, J.; Mohamath, R.; Bakken, J.; Beaver, S.; et al. A self-amplifying RNA vaccine against COVID-19 with long-term room-temperature stability. NPJ Vaccines 2022, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Haefner, E.; Gerbeth, J.; Beissert, T.; Sahin, U.; Perkovic, M.; Schnierle, B.S. A taRNA vaccine candidate induces a specific immune response that protects mice against Chikungunya virus infections. Mol. Ther.-Nucleic Acids 2022, 28, 743–754. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, H.; Lu, S.S.; Jones, K.A.; Govind, A.P.; Jeyifous, O.; Simmons, C.Q.; Tabatabaei, N.; Green, W.N.; Holder, J.L.; et al. RNA-based translation activators for targeted gene upregulation. Nat. Commun. 2023, 14, 6827. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.; Wu, Y.; Lian, J. Circular RNA vaccine in disease prevention and treatment. Signal Transduct. Target. Ther. 2023, 8, 341. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Zhou, S.; Dain, L.; Mei, L.; Zhu, G. Circular RNA: An emerging frontier in RNA therapeutic targets, RNA therapeutics, and mRNA vaccines. J. Control. Release 2022, 348, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, S.K.; Belk, J.A.; Amaya, L.; Li, Z.; Cardenas, A.; Abe, B.T.; Chen, C.K.; Wender, P.A.; Chang, H.Y. Engineering circular RNA for enhanced protein production. Nat. Biotechnol. 2023, 41, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Fotin-Mleczek, M.; Roth, G.; Becker, C.; Dam, T.C.; Verdurmen, W.P.R.; Brock, R.; Probst, J.; Schlake, T. Protein expression from exogenous mRNA: Uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 2011, 8, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Del Toro Runzer, C.; Anand, S.; Mota, C.; Moroni, L.; Plank, C.; van Griensven, M.; Balmayor, E.R. Cellular uptake of modified mRNA occurs via caveolae-mediated endocytosis, yielding high protein expression in slow-dividing cells. Mol. Ther.-Nucleic Acids 2023, 32, 960–979. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Zhao, X.; Song, X.R. Ex vivo pulsed dendritic cell vaccination against cancer. Acta Pharmacol. Sin. 2020, 41, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Ni, H.; Scales, D.; Dude, A.; Capodici, J.; McGibney, K.; Abdool, A.; Isaacs, S.N.; Cannon, G.; Karikó, K. HIV Gag mRNA Transfection of Dendritic Cells (DC) Delivers Encoded Antigen to MHC Class I and II Molecules, Causes DC Maturation, and Induces a Potent Human In Vitro Primary Immune Response. J. Immunol. 2000, 165, 4710–4717. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhu, Y.; He, J.; Sun, X. Path towards mRNA delivery for cancer immunotherapy from bench to bedside. Theranostics 2024, 14, 96–115. [Google Scholar] [CrossRef]
- Tateshita, N.; Miura, N.; Tanaka, H.; Masuda, T.; Ohtsuki, S.; Tange, K.; Nakai, Y.; Yoshioka, H.; Akita, H. Development of a lipoplex-type mRNA carrier composed of an ionizable lipid with a vitamin E scaffold and the KALA peptide for use as an ex vivo dendritic cell-based cancer vaccine. J. Control. Release 2019, 310, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Borch, T.H.; Engell-Noerregaard, L.; Zeeberg Iversen, T.; Ellebaek, E.; Met, Ö.; Hansen, M.; Andersen, M.H.; thor Straten, P.; Svane, I.M. mRNA-transfected dendritic cell vaccine in combination with metronomic cyclophosphamide as treatment for patients with advanced malignant melanoma. Oncoimmunology 2016, 5, e1207842. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Nair, S.K. RNA-transfected dendritic cells in cancer immunotherapy. J. Clin. Investig. 2000, 106, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Dörrie, J.; Schaft, N.; Schuler, G.; Schuler-Thurner, B. Therapeutic cancer vaccination with ex vivo rna-transfected dendritic cells—An update. Pharmaceutics 2020, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines 2014, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles from Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef] [PubMed]
- Jewell, C.M.; Bustamante López, S.C.; Irvine, D.J. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc. Natl. Acad. Sci. USA 2011, 108, 15745–15750. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Gao, W.; Alber, S.; Trichel, A.; Murphey-Corb, M.; Watkins, S.C.; Gambotto, A.; Barratt-Boyes, S.M. Adenovirus-Transduced Dendritic Cells Injected into Skin or Lymph Node Prime Potent Simian Immunodeficiency Virus-Specific T Cell Immunity in Monkeys. J. Immunol. 2003, 171, 6875–6882. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Sayegh, N.; Graciotti, M.; Kandalaft, L.E. Electroporation as a method of choice to generate genetically modified dendritic cell cancer vaccines. Curr. Opin. Biotechnol. 2020, 65, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishikawa, M.; Kobayashi, N.; Takakura, Y. Gene silencing in primary and metastatic tumors by small interfering RNA delivery in mice: Quantitative analysis using melanoma cells expressing firefly and sea pansy luciferases. J. Control. Release 2005, 105, 332–343. [Google Scholar] [CrossRef]
- Steitz, J.; Britten, C.M.; Wölfel, T.; Tüting, T. Effective induction of anti-melanoma immunity following genetic vaccination with synthetic mRNA coding for the fusion protein EGFP.TRP2. Cancer Immunol. Immunother. 2006, 55, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Mandl, C.W.; Aberle, J.H.; Aberle, S.W.; Holzmann, H.; Allison, S.L.; Heinz, F.X. In vitro-synthesized infectious RNA as an attenuated live vaccine in a flavivirus model. Nat. Med. 1998, 4, 1438–1440. [Google Scholar] [CrossRef]
- Bolhassani, A.; Khavari, A.; Oraf, Z. Electroporation—Advantages and Drawbacks for Delivery of Drug, Gene and Vaccine. In Application of Nanotechnology in Drug Delivery; InTech: Rijeka, Croatia, 2014; pp. 369–397. [Google Scholar] [CrossRef]
- Choi, Y.S.; Lee, M.Y.; David, A.E.; Park, Y.S. Nanoparticles for gene delivery: Therapeutic and toxic effects. Mol. Cell. Toxicol. 2014, 10, 1–8. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy—An overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef] [PubMed]
- Riecken, K.; Głów, D.; Fehse, B. How to package and SEND mRNA: A novel “humanized” vector system based on endogenous retroviruses. Signal Transduct. Target. Ther. 2021, 6, 384. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liang, B.; Wang, W.; Li, L.; Feng, N.; Zhao, Y.; Wang, T.; Yan, F.; Yang, S.; Xia, X. Viral vectored vaccines: Design, development, preventive and therapeutic applications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 149. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lim, J.; Wang, C.P.J.; Han, J.H.; Shin, H.E.; Kim, S.N.; Jeong, D.; Lee, S.H.; Chun, B.H.; Park, C.G.; et al. Lipid nanoparticle-based mRNA delivery systems for cancer immunotherapy. Nano Converg. 2023, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Strelkova Petersen, D.M.; Chaudhary, N.; Arral, M.L.; Weiss, R.M.; Whitehead, K.A. The mixing method used to formulate lipid nanoparticles affects mRNA delivery efficacy and organ tropism. Eur. J. Pharm. Biopharm. 2023, 192, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Lin, Y.; Wei, T.; Cheng, Q. Lipid Nanoparticle (LNP) Enables mRNA Delivery for Cancer Therapy. Adv. Mater. 2023, 35, e2303261. [Google Scholar] [CrossRef]
- Chander, N.; Basha, G.; Yan Cheng, M.H.; Witzigmann, D.; Cullis, P.R. Lipid nanoparticle mRNA systems containing high levels of sphingomyelin engender higher protein expression in hepatic and extra-hepatic tissues. Mol. Ther.-Methods Clin. Dev. 2023, 30, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Kordalivand, N.; Tondini, E.; Lau, C.Y.J.; Vermonden, T.; Mastrobattista, E.; Hennink, W.E.; Ossendorp, F.; van Nostrum, C.F. Cationic synthetic long peptides-loaded nanogels: An efficient therapeutic vaccine formulation for induction of T-cell responses. J. Control. Release 2019, 315, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Haug, B.E.; Camilio, K.A.; Eliassen, L.T.; Stensen, W.; Svendsen, J.S.; Berg, K.; Mortensen, B.; Serin, G.; Mirjolet, J.F.; Bichat, F.; et al. Discovery of a 9-mer Cationic Peptide (LTX-315) as a Potential First in Class Oncolytic Peptide. J. Med. Chem. 2016, 59, 2918–2927. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, A.; Rajendrakumar, S.K.; Cherukula, K.; Park, M.S.; Padmanaban, S.; Vasukuty, A.; Mohanty, A.; Lee, J.Y.; Bae, W.K.; Park, I.K. A sugar modified amphiphilic cationic nano-adjuvant ceased tumor immune suppression and rejuvenated peptide vaccine induced antitumor immunity in cervical cancer. Biomater. Sci. 2023, 11, 1853–1866. [Google Scholar] [CrossRef] [PubMed]
- Heuts, J.; Jiskoot, W.; Ossendorp, F.; Maaden, K. Van Der Cationic Nanoparticle-Based Cancer Vaccines. Pharmaceutics 2021, 13, 596. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.N.; Li, M.; Luo, Y.L.; Chen, Q.; Wang, L.; Zhang, H.B.; Shen, S.; Gu, Z.; Wang, J. Cationic lipid-assisted nanoparticles for delivery of mRNA cancer vaccine. Biomater. Sci. 2018, 6, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Le, T.M.D.; Yoon, A.R.; Thambi, T.; Yun, C.O. Polymeric Systems for Cancer Immunotherapy: A Review. Front. Immunol. 2022, 13, 826876. [Google Scholar] [CrossRef] [PubMed]
- Ben-Akiva, E.; Karlsson, J.; Hemmati, S.; Yu, H.; Tzeng, S.Y.; Pardoll, D.M.; Green, J.J. Biodegradable lipophilic polymeric mRNA nanoparticles for ligand-free targeting of splenic dendritic cells for cancer vaccination. Proc. Natl. Acad. Sci. USA 2023, 120, e2301606120. [Google Scholar] [CrossRef] [PubMed]
- Duinkerken, S.; Horrevorts, S.K.; Kalay, H.; Ambrosini, M.; Rutte, L.; de Gruijl, T.D.; Garcia-Vallejo, J.J.; van Kooyk, Y. Glyco-dendrimers as intradermal anti-tumor vaccine targeting multiple skin DC subsets. Theranostics 2019, 9, 5797–5809. [Google Scholar] [CrossRef]
- Granot-Matok, Y.; Ezra, A.; Ramishetti, S.; Sharma, P.; Naidu, G.S.; Benhar, I.; Peer, D. Lipid nanoparticles-loaded with toxin mRNA represents a new strategy for the treatment of solid tumors. Theranostics 2023, 13, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Alameh, M.G.; Tombácz, I.; Bettini, E.; Lederer, K.; Ndeupen, S.; Sittplangkoon, C.; Gaudette, B.T.; Soliman, O.Y.; Wilmore, J.R.; Pine, M.; et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 2021, 54, 287–2892. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeong, M.; Park, J.; Jung, H.; Lee, H. Immunogenicity of lipid nanoparticles and its impact on the efficacy of mRNA vaccines and therapeutics. Exp. Mol. Med. 2023, 55, 2085–2096. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Wang, Z.G.; Liu, S.L. Lipid Nanoparticles for mRNA Delivery to Enhance Cancer Immunotherapy. Molecules 2022, 27, 5607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; You, X.; Wang, X.; Cui, L.; Wang, Z.; Xu, F.; Li, M.; Yang, Z.; Liu, J.; Huang, P.; et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through toll-like receptor 4 signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2005191118. [Google Scholar] [CrossRef] [PubMed]
- Jackson Hoffman, B.A.; Pumford, E.A.; Enueme, A.I.; Fetah, K.L.; Friedl, O.M.; Kasko, A.M. Engineered macromolecular Toll-like receptor agents and assemblies. Trends Biotechnol. 2023, 41, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Mohamad Razif, M.I.; Nizar, N.; Zainal Abidin, N.H.; Muhammad Ali, S.N.; Wan Zarimi, W.N.N.; Khotib, J.; Susanti, D.; Mohd Jailani, M.T.; Taher, M. Emergence of mRNA vaccines in the management of cancer. Expert Rev. Vaccines 2023, 22, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, K.; Antognini, D.; Combes, A.; Paden, M.; Zakhary, B.; Ogino, M.; Maclaren, G.; Brodie, D. Clinical advances and ongoing trials of mRNA vaccines for cancer treatment. Lancet Oncol. 2022, 23, 450–458. [Google Scholar]
- Wang, Y.; Zhang, R.; Tang, L.; Yang, L. Nonviral Delivery Systems of mRNA Vaccines for Cancer Gene Therapy. Pharmaceutics 2022, 14, 512. [Google Scholar] [CrossRef] [PubMed]
- Ni, L. Advances in mRNA-Based Cancer Vaccines. Vaccines 2023, 11, 1599. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Lindgren, G.; Lin, A.; Thompson, E.A.; Ols, S.; Röhss, J.; John, S.; Hassett, K.; Yuzhakov, O.; Bahl, K.; et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Goedegebuure, P.; Mardis, E.R.; Ellis, M.J.C.; Zhang, X.; Herndon, J.M.; Fleming, T.P.; Carreno, B.M.; Hansen, T.H.; Gillanders, W.E. Cancer genome sequencing and its implications for personalized cancer vaccines. Cancers 2011, 3, 4191–4211. [Google Scholar] [CrossRef]
- Fritah, H.; Rovelli, R.; Chiang, C.L.L.; Kandalaft, L.E. The current clinical landscape of personalized cancer vaccines. Cancer Treat. Rev. 2022, 106, 102383. [Google Scholar] [CrossRef] [PubMed]
- Hawlina, S.; Zorec, R.; Chowdhury, H.H. Potential of Personalized Dendritic Cell-Based Immunohybridoma Vaccines to Treat Prostate Cancer. Life 2023, 13, 1498. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Lin, E.Z.; Anekoji, M.; Ichim, T.E.; Hu, J.; Marincola, F.M.; Jones, L.D.; Kesari, S.; Ashili, S. Advancing personalized medicine in brain cancer: Exploring the role of mRNA vaccines. J. Transl. Med. 2023, 21, 830. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, C.S.; Hsu, J.C.; Hosseini, I.; Shen, B.Q.; Rotte, A.; Twomey, P.; Girish, S.; Wu, B. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol. Ther. 2021, 29, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Perrinjaquet, M.; Richard Schlegel, C. Personalized neoantigen cancer vaccines: An analysis of the clinical and commercial potential of ongoing development programs. Drug Discov. Today 2023, 28, 103773. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Princiotta, M.F.; Bridon, D.; Martin, W.D.; Steinberg, G.D.; De Groot, A.S. Neoantigen-based personalized cancer vaccines: The emergence of precision cancer immunotherapy. Expert Rev. Vaccines 2022, 21, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Türeci, Ö.; Löwer, M.; Schrörs, B.; Lang, M.; Tadmor, A.; Sahin, U. Challenges towards the realization of individualized cancer vaccines. Nat. Biomed. Eng. 2018, 2, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Katsikis, P.D.; Ishii, K.J.; Schliehe, C. Challenges in developing personalized neoantigen cancer vaccines. Nat. Rev. Immunol. 2024, 24, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Ruggiero, N.; Steinberg, G.D.; Martin, W.D.; De Groot, A.S. Neoadjuvant personalized cancer vaccines: The final frontier? Expert Rev. Vaccines 2024, 23, 205–212. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, X.; Wang, S.Z.; Xu, Y.; Jia, H.R.; Zhu, Y.X.; Wu, S.Y.; Zhang, X.; Feng, H.H.; Gao, G.; et al. Turning tumor cells into microvesicles as personalized cancer vaccines for cancer prevention and treatment. Nano Today 2024, 55, 102219. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Amaya-Ramirez, D.; Martinez-Enriquez, L.C.; Parra-López, C. Usefulness of Docking and Molecular Dynamics in Selecting Tumor Neoantigens to Design Personalized Cancer Vaccines: A Proof of Concept. Vaccines 2023, 11, 1174. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Adotévi, O.; Vernerey, D.; Jacoulet, P.; Meurisse, A.; Laheurte, C.; Almotlak, H.; Jacquin, M.; Kaulek, V.; Boullerot, L.; Malfroy, M.; et al. Safety, Immunogenicity, and 1-Year Efficacy of Universal Cancer Peptide-Based Vaccine in Patients with Refractory Advanced Non-Small-Cell Lung Cancer: A Phase Ib/Phase IIa De-Escalation Study. J. Clin. Oncol. 2023, 41, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Kaumaya, P.T.P.; Guo, L.; Overholser, J.; Penichet, M.L.; Bekaii-Saab, T. Immunogenicity and antitumor efficacy of a novel human PD-1 B-cell vaccine (PD1-Vaxx) and combination immunotherapy with dual trastuzumab/pertuzumab-like HER-2 B-cell epitope vaccines (B-Vaxx) in a syngeneic mouse model. Oncoimmunology 2020, 9, 1818437. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.S.; Vreeland, T.J.; Hale, D.F.; Jackson, D.O.; Trappey, A.F. Evaluation of attenuated tumor antigens and the implications for peptide-based cancer vaccine development. J. Cancer 2017, 8, 1255–1262. [Google Scholar] [CrossRef]
- Purcell, A.W.; McCluskey, J.; Rossjohn, J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007, 6, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Wada, H.; Goto, R.; Osada, T.; Yamamura, K.; Fukaya, S.; Shimizu, A.; Okubo, M.; Minamiguchi, K.; Ikizawa, K.; et al. TAS0314, a novel multi-epitope long peptide vaccine, showed synergistic antitumor immunity with PD-1/PD-L1 blockade in HLA-A*2402 mice. Sci. Rep. 2020, 10, 17284. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.R.; McMahan, R.H.; Kemmler, C.B.; Kappler, J.W.; Slansky, J.E. Peptide vaccines prevent tumor growth by activating T cells that respond to native tumor antigens. Proc. Natl. Acad. Sci. USA 2010, 107, 4652–4657. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Admistration (FDA). 2021. Available online: www.fda.gov (accessed on 1 March 2024).
- Stephens, A.J.; Burgess-Brown, N.A.; Jiang, S. Beyond Just Peptide Antigens: The Complex World of Peptide-Based Cancer Vaccines. Front. Immunol. 2021, 12, 696791. [Google Scholar] [CrossRef]
- bin Umair, M.; Akusa, F.N.; Kashif, H.; Seerat-e-Fatima, F.B.; Azhar, M.; Munir, I.; Ahmed, M.; Khalil, W.; Sharyar, H.; Rafique, S.; et al. Viruses as tools in gene therapy, vaccine development, and cancer treatment. Arch. Virol. 2022, 167, 1387–1404. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.; Kim, E.S. Oncolytic Viruses and Cancer Immunotherapy. Curr. Oncol. Rep. 2023, 25, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Feola, S.; Chiaro, J.; Cerullo, V. Integrating immunopeptidome analysis for the design and development of cancer vaccines. Semin. Immunol. 2023, 67, 101750. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Raina, N.; Pal, A.K.; Rani, R.; Sharma, A.; Gupta, M. Functional Nanomaterials and Nanocomposite in Cancer Vaccines; Elsevier Inc.: Amsterdam, The Netherlands, 2022; ISBN 9780128236864. [Google Scholar]
- Pandya, A.; Shah, Y.; Kothari, N.; Postwala, H.; Shah, A.; Parekh, P.; Chorawala, M.R. The Future of Cancer Immunotherapy: DNA Vaccines Leading the Way; Springer: Berlin/Heidelberg, Germany, 2023; Volume 40, ISBN 0123456789. [Google Scholar]
- Das, S.S.; Moitra, I.; Singh, S.K.; Verma, P.R.P.; Swain, S. DNA Vaccines for Cancer Treatment: Challenges and Promises; Elsevier Inc.: Amsterdam, The Netherlands, 2022; ISBN 9780128236864. [Google Scholar]
- Rinaldi, M.; Signori, E.; Rosati, P.; Cannelli, G.; Parrella, P.; Iannace, E.; Monego, G.; Ciafrè, S.A.; Farace, M.G.; Iurescia, S.; et al. Feasibilty of in utero DNA vaccination following naked gene transfer into pig fetal muscle: Transgene expression, immunity and safety. Vaccine 2006, 24, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Sun, C.; Chang, J.; Zhang, G.; Yin, X. mRNA Vaccine Development for Emerging Animal and Zoonotic Diseases. Viruses 2022, 14, 401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Tang, T.; Chen, Y.; Huang, X.; Liang, T. mRNA vaccines in disease prevention and treatment. Signal Transduct. Target. Ther. 2023, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Chehelgerdi, M.; Chehelgerdi, M. The Use of RNA-Based Treatments in the Field of Cancer Immunotherapy; BioMed Central: London, UK, 2023; Volume 22, ISBN 1294302301807. [Google Scholar]
- Wang, L.; Pegram, M.D.; Wu, J.C. Induced pluripotent stem cells as a novel cancer vaccine. Expert Opin. Biol. Ther. 2019, 19, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges, and prospects. Signal Transduct. Target. Ther. 2023, 8, 450. [Google Scholar] [CrossRef] [PubMed]
- Tahaghoghi-Hajghorbani, S.; Yazdani, M.; Nikpoor, A.R.; Hatamipour, M.; Ajami, A.; Jaafari, M.R.; Badiee, A.; Rafiei, A. Targeting the tumor microenvironment by liposomal Epacadostat in combination with liposomal gp100 vaccine. Sci. Rep. 2023, 13, 5802. [Google Scholar] [CrossRef] [PubMed]
- Twilhaar, M.K.N.; Czentner, L.; van Nostrum, C.F.; Storm, G.; Den Haan, J.M.M. Mimicking pathogens to augment the potency of liposomal cancer vaccines. Pharmaceutics 2021, 13, 954. [Google Scholar] [CrossRef] [PubMed]
- Alhamhoom, Y.; Kakinani, G.; Rahamathulla, M.; Ali, M.; Osmani, R.; Hani, U.; Yoonus Thajudeen, K.; Kiran Raj, G.; Gowda, D.V. Recent advances in the liposomal nanovesicles based immunotherapy in the treatment of cancer: A review. Saudi Pharm. J. 2023, 31, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Shi, H.; Fu, P.; Shi, P.; Yang, J. Liposomal Dendritic Cell Vaccine in Breast Cancer Immunotherapy. ACS Omega 2021, 6, 3991–3998. [Google Scholar] [CrossRef] [PubMed]
- Rommasi, F.; Esfandiari, N. Liposomal Nanomedicine: Applications for Drug Delivery in Cancer Therapy. Nanoscale Res. Lett. 2021, 16, 95. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Suita, Y.; Miriyala, S.; Dean, J.; Tapinos, N.; Shen, J. Advances in lipid-based nanoparticles for cancer chemoimmunotherapy. Pharmaceutics 2021, 13, 520. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Duan, S.; Ye, F.; Hou, X.; Li, X.; Zhao, J.; Yu, X.; Hu, Z.; Tang, Z.; Mo, F.; et al. The enhanced antitumor-specific immune response with mannose- and CpG-ODN-coated liposomes delivering TRP2 peptide. Theranostics 2018, 8, 1723–1739. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, H.; Zhang, T.; Wang, Y.; Qi, T.; Li, B.; Jiao, H. Lipid-mRNA nanoparticles landscape for cancer therapy. Front. Bioeng. Biotechnol. 2022, 10, 1053197. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Shi, K.; Jia, Y.P.; Hao, Y.; Peng, J.R.; Qian, Z.Y. Advanced biomaterials for cancer immunotherapy. Acta Pharmacol. Sin. 2020, 41, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.M.; Gattis, B.; Choi, M.R.; Zhang, B.; Gianneschi, N. Use of proteomimetic polymers for delivery of tumor antigens and adjuvants through formation of stable electrostatic complexes with small molecule STING agonists. J. Clin. Oncol. 2023, 41, 2583. [Google Scholar] [CrossRef]
- Verma, N.K.; Wong, B.H.S.; Poh, Z.S.; Udayakumar, A.; Verma, R.; Goh, R.K.J.; Duggan, S.P.; Shelat, V.G.; Chandy, K.G.; Grigoropoulos, N.F. Obstacles for T-lymphocytes in the tumour microenvironment: Therapeutic challenges, advances and opportunities beyond immune checkpoint. eBioMedicine 2022, 83, 104216. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, M.T.; Antignani, A.; Fitzgerald, D.J. Managing the TME to improve the efficacy of cancer therapy. Front. Immunol. 2022, 13, 954992. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, J.; Zhou, H.; Zeng, X.; Ruan, Z.; Pu, Z.; Jiang, X.; Matsui, A.; Zhu, L.; Amoozgar, Z.; et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat. Commun. 2022, 13, 758. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T. Personalized anti-cancer vaccine combining mRNA and immunotherapy tested in melanoma trial. Nat. Med. 2023, 29, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S. Vaccine Design: Methods and Protocols, Volume 3. Resources for Vaccine Development; Springer: New York, NY, USA, 2022; Volume 3, ISBN 9781071618912. [Google Scholar]
- Schmidt, J.; Guillaume, P.; Dojcinovic, D.; Karbach, J.; Coukos, G.; Luescher, I. In silico and cell-based analyses reveal strong divergence between prediction and observation of T-cell-recognized tumor antigen T-cell epitopes. J. Biol. Chem. 2017, 292, 11840–11849. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Su, G.H.; Ma, D.; Xiao, Y.; Shao, Z.M.; Jiang, Y.Z. Technological advances in cancer immunity: From immunogenomics to single-cell analysis and artificial intelligence. Signal Transduct. Target. Ther. 2021, 6, 312. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, X.; Zeng, S.; Ren, X.; Yan, Y.; Gong, Z. Applying artificial intelligence for cancer immunotherapy. Acta Pharm. Sin. B 2021, 11, 3393–3405. [Google Scholar] [CrossRef] [PubMed]
- Boehm, K.M.; Bhinder, B.; Raja, V.J.; Dephoure, N.; Elemento, O. Predicting peptide presentation by major histocompatibility complex class I using one million peptides. BMC Bioinform. 2019, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.; Padalhin, A. Reactive oxygen species overload: A review of plasma therapy and photobiomodulation for cancer treatment. Med. Lasers 2023, 12, 18–28. [Google Scholar] [CrossRef]
- Živanić, M.; Espona-Noguera, A.; Lin, A.; Canal, C. Current State of Cold Atmospheric Plasma and Cancer-Immunity Cycle: Therapeutic Relevance and Overcoming Clinical Limitations Using Hydrogels. Adv. Sci. 2023, 10, e2205803. [Google Scholar] [CrossRef] [PubMed]
- Labay, C.; Roldán, M.; Tampieri, F.; Stancampiano, A.; Bocanegra, P.E.; Ginebra, M.P.; Canal, C. Enhanced generation of reactive species by cold plasma in gelatin solutions for selective cancer cell death. ACS Appl. Mater. Interfaces 2020, 12, 47256–47269. [Google Scholar] [CrossRef] [PubMed]
- Murillo, D.; Huergo, C.; Gallego, B.; Rodríguez, R.; Tornín, J. Exploring the Use of Cold Atmospheric Plasma to Overcome Drug Resistance in Cancer. Biomedicines 2023, 11, 208. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.M.; Simms, M.S.; Mann, V.M.; Maitland, N.J.; O’connell, D.; Frame, F.M. Low-temperature plasma treatment induces DNA damage leading to necrotic cell death in primary prostate epithelial cells. Br. J. Cancer 2015, 112, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Gaur, N.; Kurita, H.; Oh, J.S.; Miyachika, S.; Ito, M.; Mizuno, A.; Cowin, A.J.; Allinson, S.; Short, R.D.; Szili, E.J. On cold atmospheric-pressure plasma jet induced DNA damage in cells. J. Phys. D Appl. Phys. 2021, 54, 035203. [Google Scholar] [CrossRef]
- Braný, D.; Dvorská, D.; Strnádel, J.; Matáková, T.; Halašová, E.; Škovierová, H. Effect of cold atmospheric plasma on epigenetic changes, dna damage and possibilities for its use in synergistic cancer therapy. Int. J. Mol. Sci. 2021, 22, 12252. [Google Scholar] [CrossRef] [PubMed]
- Kumar Dubey, S.; Dabholkar, N.; Narayan Pal, U.; Singhvi, G.; Kumar Sharma, N.; Puri, A.; Kesharwani, P. Emerging innovations in cold plasma therapy against cancer: A paradigm shift. Drug Discov. Today 2022, 27, 2425–2439. [Google Scholar] [CrossRef] [PubMed]
- Ruidiaz, M.E.; Cortes-Mateos, M.J.; Sandoval, S.; Martin, D.T.; Wang-Rodriguez, J.; Hasteh, F.; Wallace, A.; Vose, J.G.; Kummel, A.C.; Blair, S.L. Quantitative comparison of surgical margin histology following excision with traditional electrosurgery and a low-thermal-injury dissection device. J. Surg. Oncol. 2011, 104, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Pomicter, A.D.; Li, F.; Bhatt, S.; Chen, C.; Li, W.; Qi, M.; Huang, C.; Deininger, M.W.; Kong, M.G.; et al. Trident cold atmospheric plasma blocks three cancer survival pathways to overcome therapy resistance. Proc. Natl. Acad. Sci. USA 2021, 118, e2107220118. [Google Scholar] [CrossRef] [PubMed]
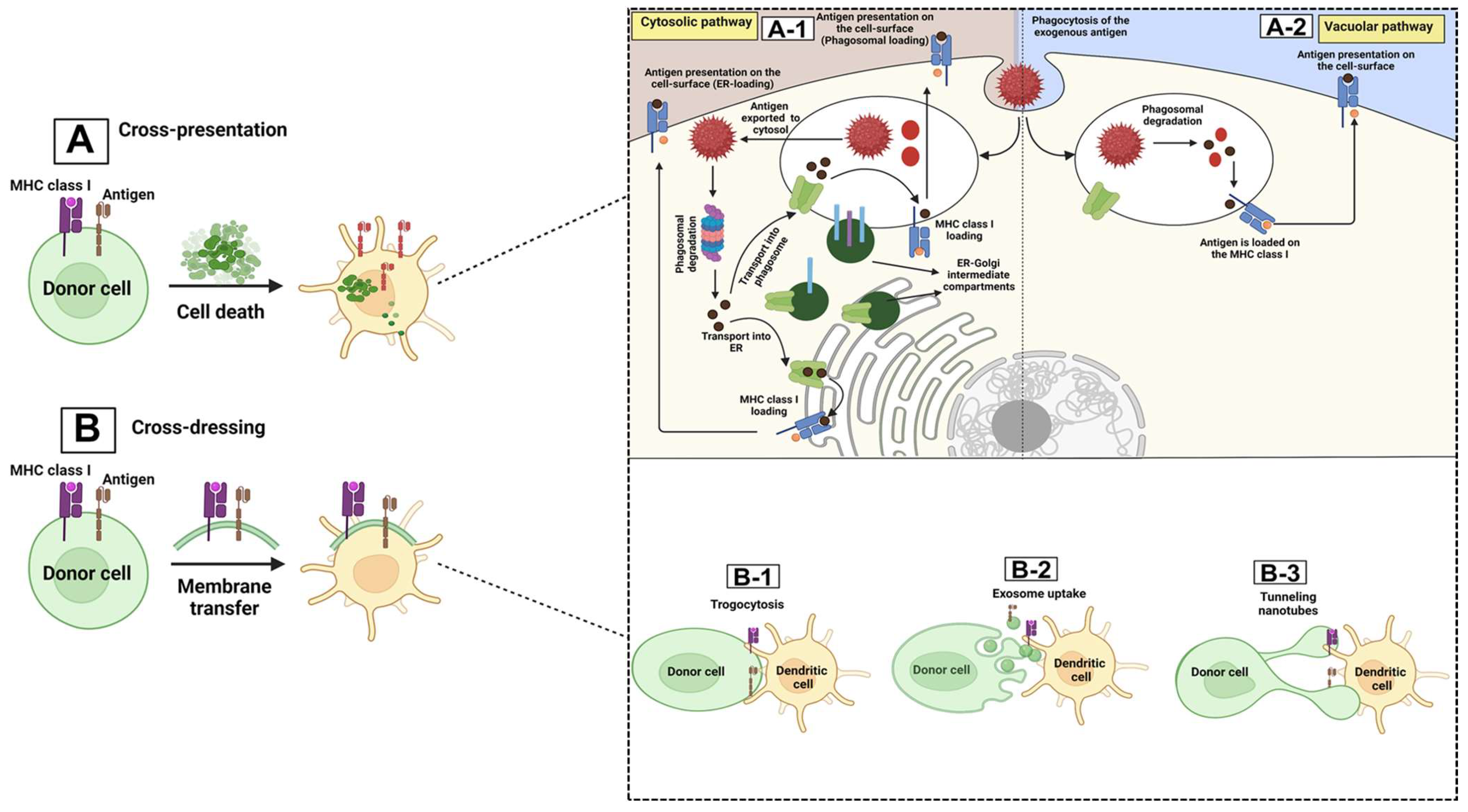
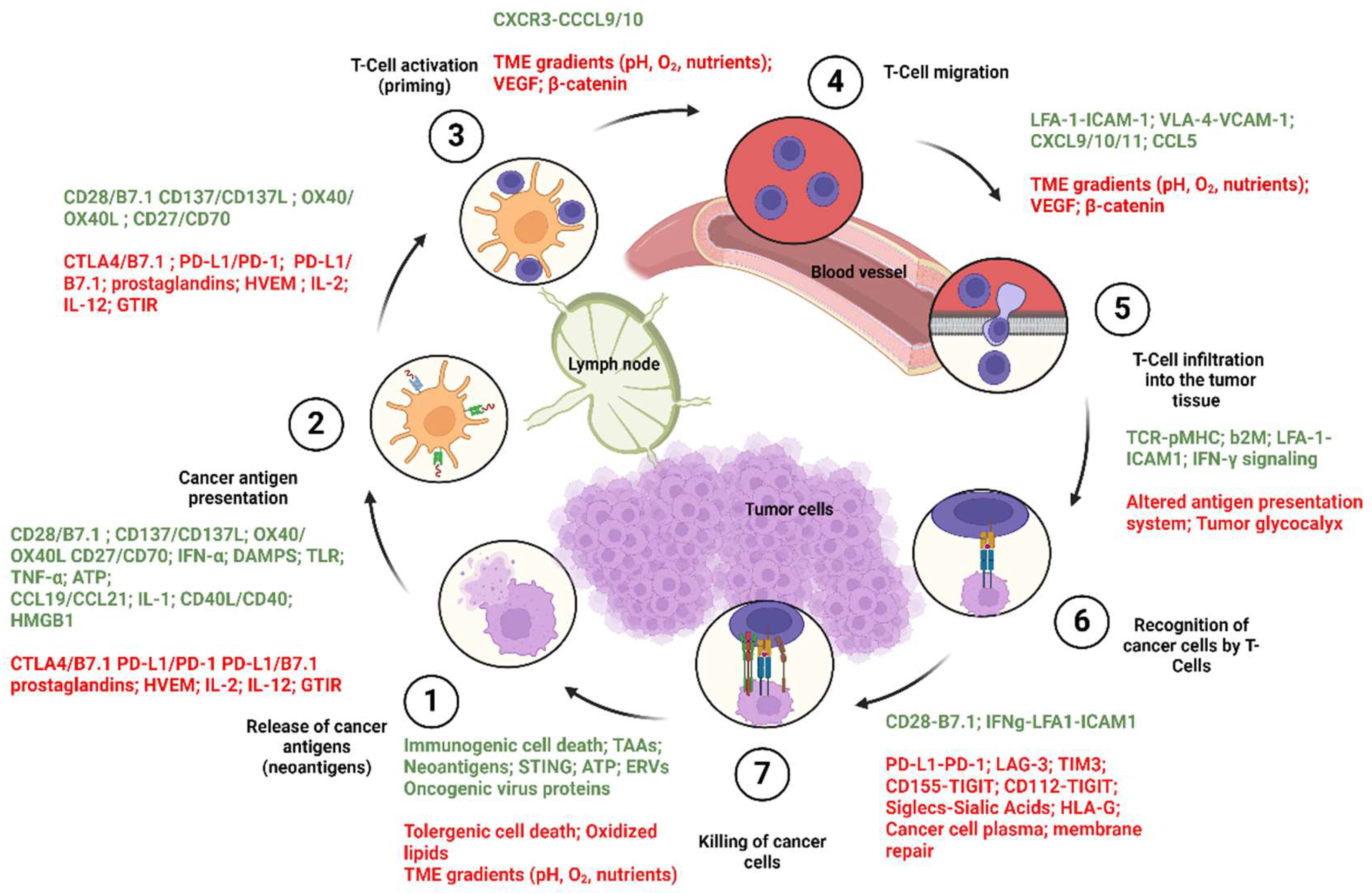
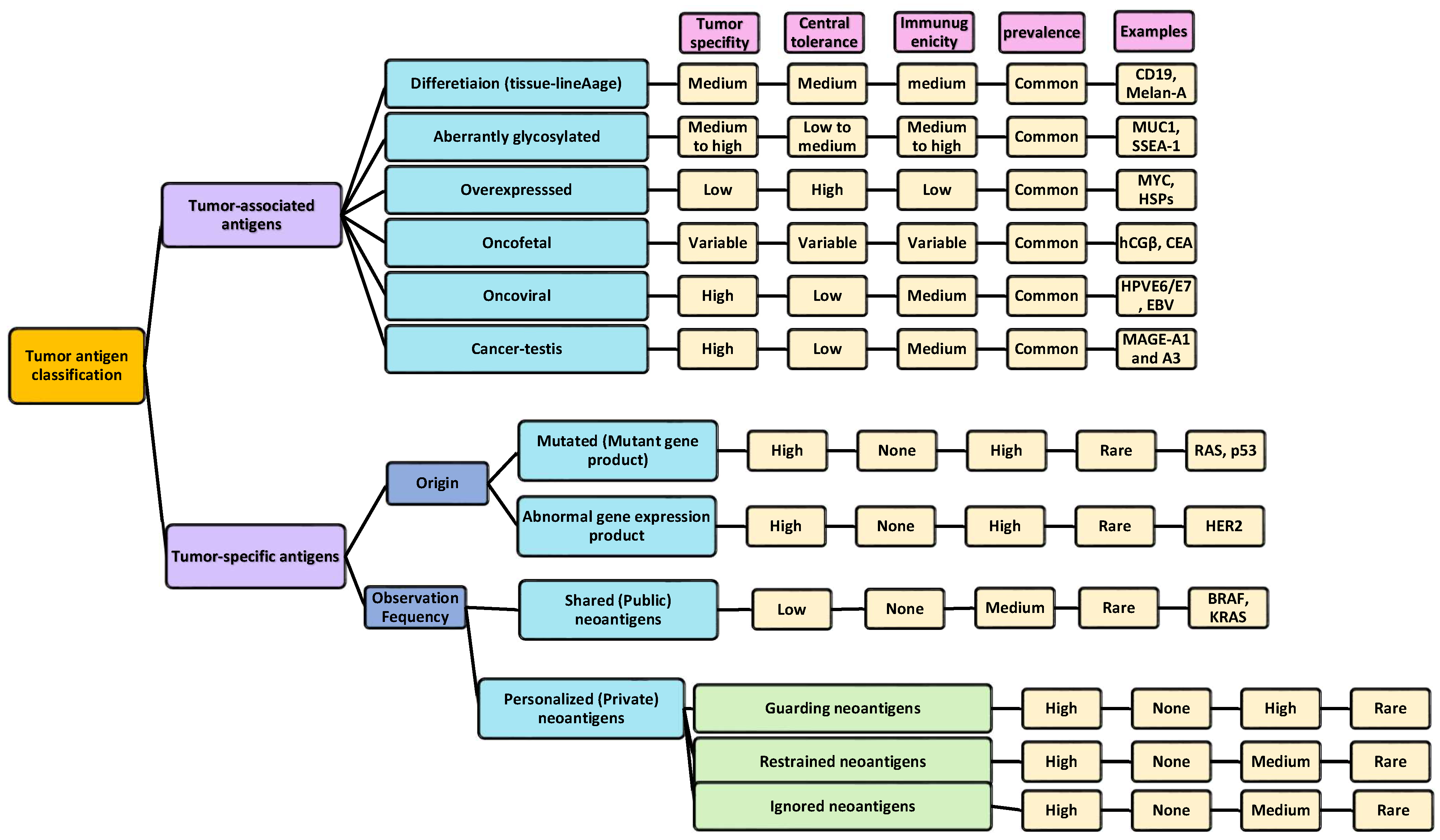

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advantages | Disadvantages | Status of Some of the Vaccines in Clinical Trials (2016–2023) | |
|---|---|---|---|
| Peptide vaccines |
|
| Glioblastoma/Glioma:
|
| Viral/bacterial-based vaccines |
|
| Pancreatic Cancer:
Gastric Cancer: Phase II (NCT04111172) Colorectal Cancer:
|
| DNA vaccines |
|
| Pancreatic Cancer:
|
| mRNA vaccines |
|
| Pancreatic Cancer:
|
| Cell-based vaccines (DCs, iPSCs, and CAR-T cells) |
|
| Pancreatic Cancer:
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheikhlary, S.; Lopez, D.H.; Moghimi, S.; Sun, B. Recent Findings on Therapeutic Cancer Vaccines: An Updated Review. Biomolecules 2024, 14, 503. https://doi.org/10.3390/biom14040503
Sheikhlary S, Lopez DH, Moghimi S, Sun B. Recent Findings on Therapeutic Cancer Vaccines: An Updated Review. Biomolecules. 2024; 14(4):503. https://doi.org/10.3390/biom14040503
Chicago/Turabian StyleSheikhlary, Sara, David Humberto Lopez, Sophia Moghimi, and Bo Sun. 2024. "Recent Findings on Therapeutic Cancer Vaccines: An Updated Review" Biomolecules 14, no. 4: 503. https://doi.org/10.3390/biom14040503
APA StyleSheikhlary, S., Lopez, D. H., Moghimi, S., & Sun, B. (2024). Recent Findings on Therapeutic Cancer Vaccines: An Updated Review. Biomolecules, 14(4), 503. https://doi.org/10.3390/biom14040503