
Disruption of Transmembrane Phosphatidylserine Asymmetry by HIV-1 Incorporated SERINC5 Is Not Responsible for Virus Restriction

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Chemicals, Lipids, and Plasmids
2.2. Virus Production, Purification, and Western Blotting
2.3. Large Unilamellar Vesicle Preparation and Surface Immobilization
2.4. Fluorescence Imaging of Surface-Immobilized Pseudoviruses and Lipid Vesicles
2.5. Image Analysis for AnxV Quantification, Lipid Order, and Immunofluorescence Correlation
2.6. Effects of Methyl-α-Cyclodextrin Treatment of Pseudoviruses
2.7. Statistical Analysis
3. Results
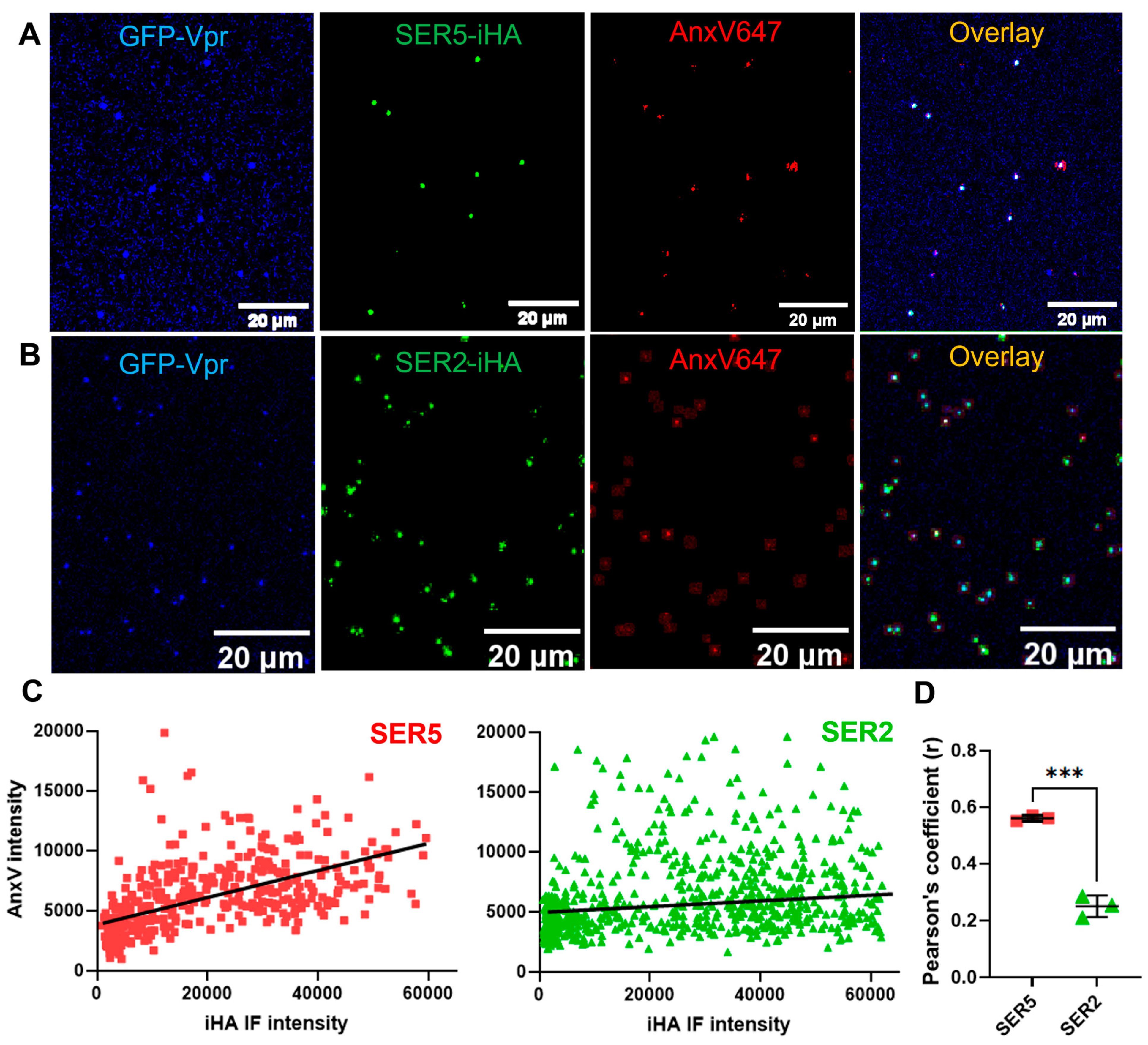
3.1. Quantitative Analyses of PS Signal on Single Pseudoviruses Demonstrates A Specific Increase in PS Signal upon SER5 Incorporation
3.2. The External PS Signal Correlates with SER5 Incorporation into Virions
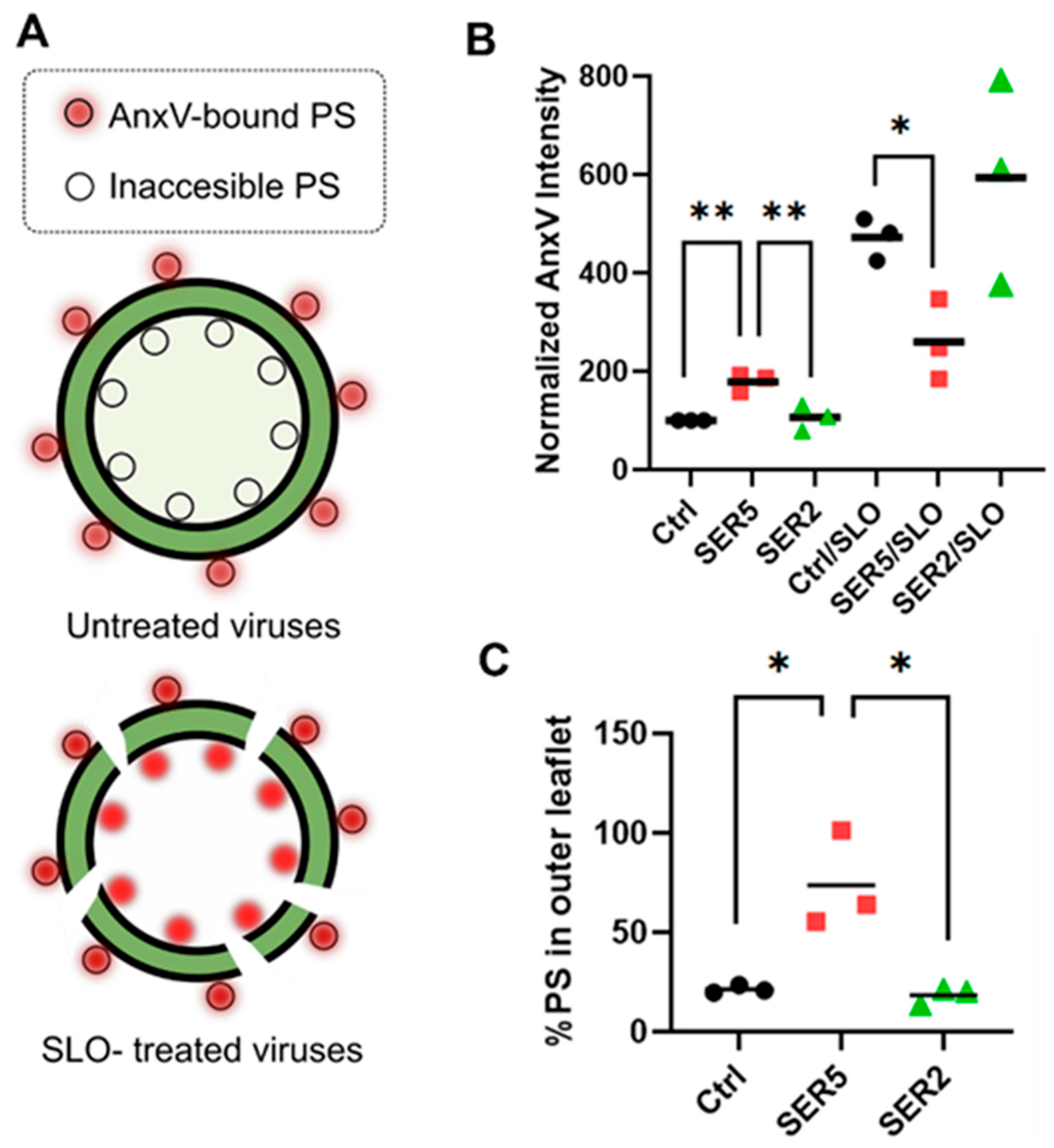
3.3. SER5 Incorporation Disrupts the Asymmetric Trans-Membrane PS Distribution
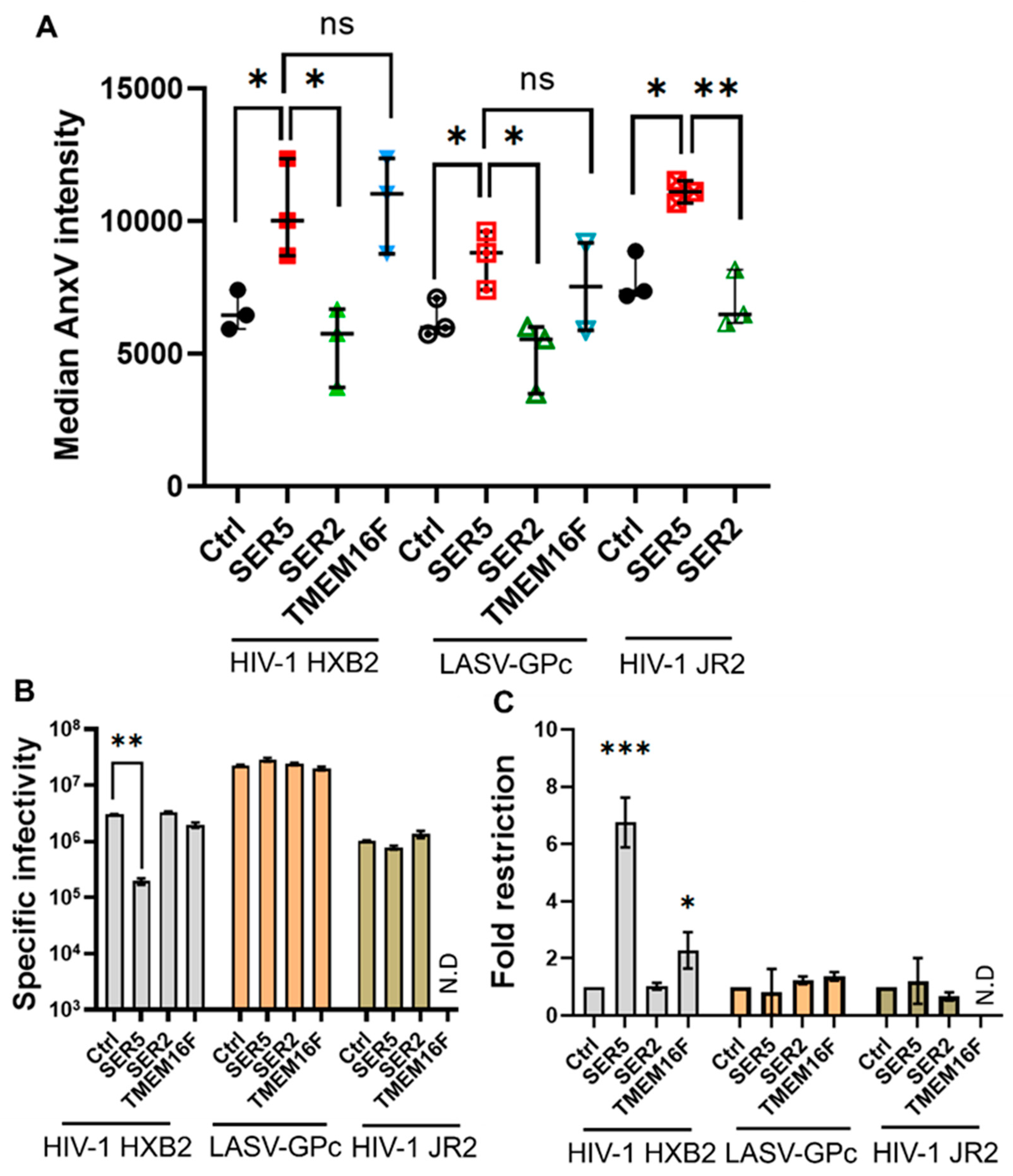
3.4. Enhanced PS Exposure on SER5 Virions Does Not Correlate Strongly with Specific Infectivity
3.5. SER5 Restricts Infectivity of Viruses Produced by Cells with Constitutively High Levels of Externalized PS
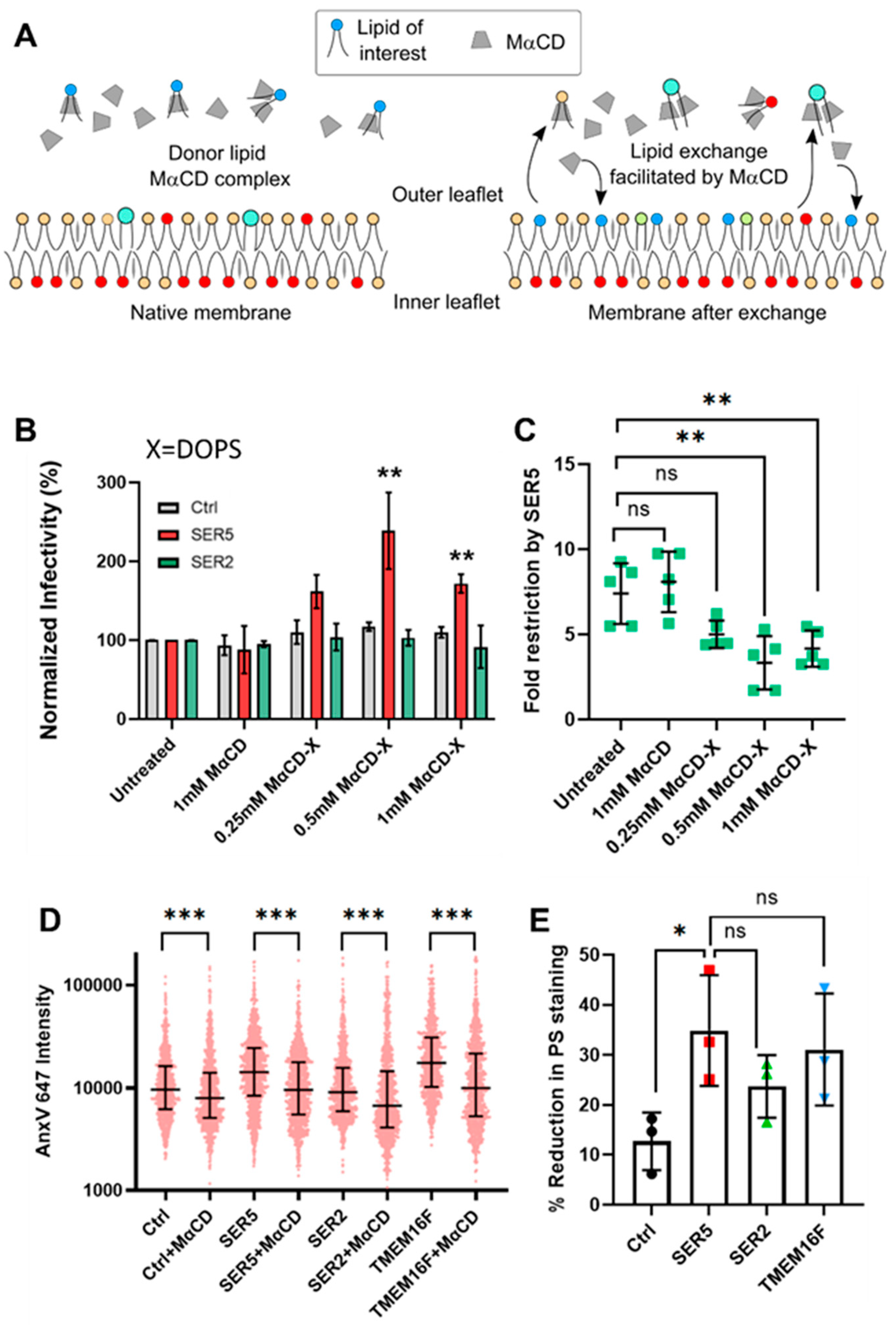
3.6. Manipulation of Viral Lipid Composition Reveals That SER5 Restriction Is Unrelated to External PS Content of the Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef] [PubMed]
- Alli-Balogun, G.O.; Levine, T.P. Fungal Ice2p is in the same superfamily as SERINCs, restriction factors for HIV and other viruses. Proteins 2021, 89, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.F.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218. [Google Scholar] [CrossRef]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Enriquez, G.V.; Escoto-Delgadillo, M.; Vazquez-Valls, E.; Torres-Mendoza, B.M. SERINC as a Restriction Factor to Inhibit Viral Infectivity and the Interaction with HIV. J. Immunol. Res. 2017, 2017, 1548905. [Google Scholar] [CrossRef] [PubMed]
- Firrito, C.; Bertelli, C.; Vanzo, T.; Chande, A.; Pizzato, M. SERINC5 as a New Restriction Factor for Human Immunodeficiency Virus and Murine Leukemia Virus. Annu. Rev. Virol. 2018, 5, 323–340. [Google Scholar] [CrossRef]
- Timilsina, U.; Umthong, S.; Lynch, B.; Stablewski, A.; Stavrou, S. SERINC5 Potently Restricts Retrovirus Infection. Mbio 2020, 11, e00588-20. [Google Scholar] [CrossRef]
- Xu, S.F.; Zheng, Z.C.; Pathak, J.L.; Cheng, H.Y.; Zhou, Z.L.; Chen, Y.P.; Wu, Q.Y.; Wang, L.J.; Zeng, M.T.; Wu, L.H. The Emerging Role of the Serine Incorporator Protein Family in Regulating Viral Infection. Front. Cell Dev. Biol. 2022, 10, 856468. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Bajak, K.; Fackler, O.T. Beyond Impairment of Virion Infectivity: New Activities of the Anti-HIV Host Cell Factor SERINC5. Viruses 2024, 16, 284. [Google Scholar] [CrossRef]
- Schulte, B.; Selyutina, A.; Opp, S.; Herschhorn, A.; Sodroski, J.G.; Pizzato, M.; Diaz-Griffero, F. Localization to detergent-resistant membranes and HIV-1 core entry inhibition correlate with HIV-1 restriction by SERINC5. Virology 2018, 515, 52–65. [Google Scholar] [CrossRef]
- Lai, K.K.; Munro, J.B.; Shi, G.L.; Majdoul, S.; Compton, A.A.; Rein, A. Restriction of Influenza A Virus by SERINC5. Mbio 2022, 13, e02923-22. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, H.; Zhang, J.; Yang, J.; Bai, L.; Zheng, B.S.; Zheng, T.H.; Wang, Y.C.; Li, J.H.; Zhang, W.Y. SERINC5 Inhibits the Secretion of Complete and Genome-Free Hepatitis B Virions Through Interfering With the Glycosylation of the HBV Envelope. Front. Microbiol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Timilsina, U.; Umthong, S.; Ivey, E.B.; Waxman, B.; Stavrou, S. SARS-CoV-2 ORF7a potently inhibits the antiviral effect of the host factor SERINC5. Nat. Commun. 2022, 13, 2935. [Google Scholar] [CrossRef]
- Li, S.N.; Ahmad, I.; Shi, J.; Wang, B.; Yu, C.Q.; Zhang, L.X.; Zheng, Y.H. Murine Leukemia Virus Glycosylated Gag Reduces Murine SERINC5 Protein Expression at Steady-State Levels via the Endosome/Lysosome Pathway to Counteract SERINC5 Antiretroviral Activity. J. Virol. 2019, 93, e01651-18. [Google Scholar] [CrossRef]
- Ahmad, I.; Li, S.N.; Li, R.R.; Chai, Q.Q.; Zhang, L.X.; Wang, B.; Yu, C.Q.; Zheng, Y.H. The retroviral accessory proteins S2, Nef, and glycoMA use similar mechanisms for antagonizing the host restriction factor SERINC5. J. Biol. Chem. 2019, 294, 7013–7024. [Google Scholar] [CrossRef] [PubMed]
- Ahi, Y.S.; Zhang, S.; Thappeta, Y.; Denman, A.; Feizpour, A.; Gummuluru, S.; Reinhard, B.; Muriaux, D.; Fivash, M.J.; Rein, A. Functional Interplay Between Murine Leukemia Virus Glycogag, Serinc5, and Surface Glycoprotein Governs Virus Entry, with Opposite Effects on Gammaretroviral and Ebolavirus Glycoproteins. Mbio 2016, 7, e01985-16. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.W.; Usami, Y.; Wu, Y.F.; Göttlinger, H. A Long Cytoplasmic Loop Governs the Sensitivity of the Anti-viral Host Protein SERINC5 to HIV-1 Nef. Cell Rep. 2018, 22, 869–875. [Google Scholar] [CrossRef]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef]
- Haider, T.; Snetkov, X.; Jolly, C. HIV envelope tail truncation confers resistance to SERINC5 restriction. Proc. Natl. Acad. Sci. USA 2021, 118, e2101450118. [Google Scholar] [CrossRef]
- Beitari, S.; Ding, S.L.; Pan, Q.H.; Finzi, A.; Liang, C. Effect of HIV-1 Env on SERINC5 Antagonism. J. Virol. 2017, 91, e02214-16. [Google Scholar] [CrossRef]
- Featherstone, A.; Aiken, C. SERINC5 Inhibits HIV-1 Infectivity by Altering the Conformation of gp120 on HIV-1 Particles. J. Virol. 2020, 94, e00594-20. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.E.; Kiessling, V.; Pornillos, O.; White, J.M.; Ganser-Pornillos, B.K.; Tamm, L.K. HIV-cell membrane fusion intermediates are restricted by Serincs as revealed by cryo-electron and TIRF microscopy. J. Biol. Chem. 2020, 295, 15183–15195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Shi, J.; Qiu, X.S.; Chai, Q.Q.; Frabutt, D.A.; Schwartz, R.C.; Zheng, Y.H. CD4 Expression and Env Conformation Are Critical for HIV-1 Restriction by SERINC5. J. Virol. 2019, 93, e00544-19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Sood, C.; Marin, M.; Aaron, J.; Chew, T.L.; Salaita, K.; Melikyan, G.B. 3D Superresolution Studies of the Effect of SERINC5 on Env Glycoprotein Distribution on HIV-1 Particles. Biophys. J. 2020, 118, 151a. [Google Scholar] [CrossRef]
- Ward, A.E.; Sokovikova, D.; Waxham, M.N.; Heberle, F.A.; Levental, I.; Levental, K.R.; Kiessling, V.; White, J.M.; Tamm, L.K. Serinc5 Restricts HIV Membrane Fusion by Altering Lipid Order and Heterogeneity in the Viral Membrane. ACS Infect. Dis. 2023, 9, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Trautz, B.; Wiedemann, H.; Lüchtenborg, C.; Pierini, V.; Kranich, J.; Glass, B.; Kräusslich, H.G.; Brocker, T.; Pizzato, M.; Ruggieri, A.; et al. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J. Biol. Chem. 2017, 292, 13702–13713. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, G.; Chen, Y.C.; Marin, M.; Wu, H.; Melikyan, G.B. SERINC5-Mediated Restriction of HIV-1 Infectivity Correlates with Resistance to Cholesterol Extraction but Not with Lipid Order of Viral Membrane. Viruses 2022, 14, 1636. [Google Scholar] [CrossRef]
- Leonhardt, S.A.; Purdy, M.D.; Grover, J.R.; Yang, Z.W.; Poulos, S.; Mcintire, W.E.; Tatham, E.A.; Erramilli, S.K.; Nosol, K.; Lai, K.K.; et al. Antiviral HIV-1 SERINC restriction factors disrupt virus membrane asymmetry. Nat. Commun. 2023, 14, 4368. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Fukuda, M.; Kudo, T.; Matsuzaki, N.; Azuma, T.; Sekine, K.; Endo, H.; Handat, T. Flip-Flop of Phospholipids in Vesicles: Kinetic Analysis with Time-Resolved Small-Angle Neutron Scattering. J. Phys. Chem. B 2009, 113, 6745–6748. [Google Scholar] [CrossRef]
- Rothman, J.E.; Tsai, D.K.; Dawidowicz, E.A.; Lenard, J. Transbilayer Phospholipid Asymmetry and Its Maintenance in Membrane of Influenza-Virus. Biochemistry 1976, 15, 2361–2370. [Google Scholar] [CrossRef]
- Shaw, J.M.; Moore, N.F.; Patzer, E.J.; Correafreire, M.C.; Wagner, R.R.; Thompson, T.E. Compositional Asymmetry and Transmembrane Movement of Phosphatidylcholine in Vesicular Stomatitis-Virus Membranes. Biochemistry 1979, 18, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Simons, K.; Denkamp, J.A.F.O.; Vandeenen, L.L.M. Phospholipid Asymmetry in Semliki Forest Virus Grown on Baby Hamster-Kidney (Bhk-21) Cells. Biochemistry 1981, 20, 1974–1981. [Google Scholar] [CrossRef] [PubMed]
- Urbancic, I.; Brun, J.; Shrestha, D.; Waithe, D.; Eggeling, C.; Chojnacki, J. Lipid Composition but Not Curvature Is the Determinant Factor for the Low Molecular Mobility Observed on the Membrane of Virus-Like Vesicles. Viruses 2018, 10, 415. [Google Scholar] [CrossRef] [PubMed]
- Bryer, A.J.; Reddy, T.; Lyman, E.; Perilla, J.R. Full scale structural, mechanical and dynamical properties of HIV-1 liposomes. PLoS Comput. Biol. 2022, 18, e1009781. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, J.; Waithe, D.; Carravilla, P.; Huarte, N.; Galiani, S.; Enderlein, J.; Eggeling, C. Envelope glycoprotein mobility on HIV-1 particles depends on the virus maturation state. Nat. Commun. 2017, 8, 545. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Reyes Ballista, J.M.; Miazgowicz, K.L.; Acciani, M.D.; Jimenez, A.R.; Belloli, R.S.; Havranek, K.E.; Brindley, M.A. Chikungunya virus entry and infectivity is primarily facilitated through cell line dependent attachment factors in mammalian and mosquito cells. Front. Cell Dev. Biol. 2023, 11, 1085913. [Google Scholar] [CrossRef]
- Callahan, M.K.; Popernack, P.M.; Tsutsui, S.; Truong, L.; Schlegel, R.A.; Henderson, A.J. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J. Immunol. 2003, 170, 4840–4845. [Google Scholar] [CrossRef]
- Zaitseva, E.; Zaitsev, E.; Melikov, K.; Arakelyan, A.; Marin, M.; Villasmil, R.; Margolis, L.B.; Melikyan, G.B.; Chernomordik, L.V. Fusion Stage of HIV-1 Entry Depends on Virus-Induced Cell Surface Exposure of Phosphatidylserine. Cell Host Microbe 2017, 22, 99. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Malim, M.H.; Hauber, J.; Fenrick, R.; Cullen, B.R. Immunodeficiency Virus Rev Trans-Activator Modulates the Expression of the Viral Regulatory Genes. Nature 1988, 335, 181–183. [Google Scholar] [CrossRef]
- Sood, C.; Francis, A.C.; Desai, T.M.; Melikyan, G.B. An improved labeling strategy enables automated detection of single-virus fusion and assessment of HIV-1 protease activity in single virions. J. Biol. Chem. 2017, 292, 20196–20207. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010, 468, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Marin, M.; Kim, J.H.; Desai, T.M.; Melikyan, G.B. Distinct Requirements for HIV-Cell Fusion and HIV-mediated Cell-Cell Fusion. J. Biol. Chem. 2015, 290, 6558–6573. [Google Scholar] [CrossRef]
- Hammonds, J.; Chen, X.M.; Zhang, X.G.; Lee, F.; Spearman, P. Advances in methods for the production, purification, and characterization of HIV-1 Gag-Env pseudovirion vaccines. Vaccine 2007, 25, 8036–8048. [Google Scholar] [CrossRef]
- Stamou, D.; Duschl, C.; Delamarche, E.; Vogel, H. Self-assembled microarrays of attoliter molecular vessels. Angew. Chem. Int. Ed. 2003, 42, 5580–5583. [Google Scholar] [CrossRef] [PubMed]
- Lorizate, M.; Brügger, B.; Akiyama, H.; Glass, B.; Müller, B.; Anderluh, G.; Wieland, F.T.; Kräusslich, H.G. Probing HIV-1 Membrane Liquid Order by Laurdan Staining Reveals Producer Cell-dependent Differences. J. Biol. Chem. 2009, 284, 22238–22247. [Google Scholar] [CrossRef]
- de Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffensnakken, H.; Reutelingsperger, C. A Novel Assay for Apoptosis—Flow Cytometric Detection of Phosphatidylserine Expression on Early Apoptotic Cells Using Fluorescein-Labeled Annexin-V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- van Engeland, M.; Nieland, L.J.W.; Ramaekers, F.C.S.; Schutte, B.; Reutelingsperger, C.P.M. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 1998, 31, 1–9. [Google Scholar] [CrossRef]
- Carravilla, P.; Chojnacki, J.; Rujas, E.; Insausti, S.; Largo, E.; Waithe, D.; Apellaniz, B.; Sicard, T.; Julien, J.P.; Eggeling, C.; et al. Molecular recognition of the native HIV-1 MPER revealed by STED microscopy of single virions. Nat. Commun. 2019, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.; Bagatolli, L.A.; Echabe, I.; Arrondo, J.L.R.; Argarana, C.E.; Cantor, C.R.; Fidelio, G.D. Interaction of biotin with streptavidin—Thermostability and conformational changes upon binding. J. Biol. Chem. 1997, 272, 11288–11294. [Google Scholar] [CrossRef]
- Kirschman, J.; Marin, M.; Chen, Y.C.; Chen, J.H.; Herschhorn, A.; Smith, A.B.; Melikyan, G.B. SERINC5 Restricts HIV-1 Infectivity by Promoting Conformational Changes and Accelerating Functional Inactivation of Env. Viruses 2022, 14, 1388. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef]
- Walev, I.; Palmer, M.; Valeva, A.; Weller, U.; Bhakdi, S. Binding, Oligomerization, and Pore Formation by Streptolysin-O in Erythrocytes and Fibroblast Membranes—Detection of Nonlytic Polymers. Infect. Immun. 1995, 63, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, K.; Satoh, R.; Danbara, H.; Futaesaku, Y. A Ring-Shaped Structure with a Crown Formed by Streptolysin-O on the Erythrocyte-Membrane. J. Bacteriol. 1993, 175, 5953–5961. [Google Scholar] [CrossRef]
- Huber, R.; Romisch, J.; Paques, E.P. The Crystal and Molecular-Structure of Human Annexin-V, an Anticoagulant Protein That Binds to Calcium and Membranes. EMBO J. 1990, 9, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Pennington, H.N.; Lee, J. Lassa virus glycoprotein complex review: Insights into its unique fusion machinery. Biosci. Rep. 2022, 42, BSR20211930. [Google Scholar] [CrossRef]
- Suddala, K.C.; Lee, C.C.; Meraner, P.; Marin, M.; Markosyan, R.M.; Desai, T.M.; Cohen, F.S.; Brass, A.L.; Melikyan, G.B. Interferon-induced transmembrane protein 3 blocks fusion of sensitive but not resistant viruses by partitioning into virus-carrying endosomes. PLoS Pathog. 2019, 15, e1007532. [Google Scholar] [CrossRef]
- Segawa, K.; Suzuki, J.; Nagata, S. Constitutive exposure of phosphatidylserine on viable cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19246–19251. [Google Scholar] [CrossRef]
- Younan, P.; Iampietro, M.; Santos, R.I.; Ramanathan, P.; Popov, V.L.; Bukreyev, A. Role of Transmembrane Protein 16F in the Incorporation of Phosphatidylserine Into Budding Ebola Virus Virions. J. Infect. Dis. 2018, 218, S335–S345. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 Gag proteins: Diverse functions in the virus life cycle. Virology 1998, 251, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Ke, Z.L.; Zila, V.; Anders-Össwein, M.; Glass, B.; Mücksch, F.; Müller, R.; Schultz, C.; Müller, B.; Kräusslich, H.G.; et al. Maturation of the matrix and viral membrane of HIV-1. Science 2021, 373, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.B.; Hevroni, L.; Kol, N.; Eckert, D.M.; Tsvitov, M.; Kay, M.S.; Rousso, I. Virion stiffness regulates immature HIV-1 entry. Retrovirology 2013, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.; Soule, E.E.; Davidoff, K.S.; Daniels, S.I.; Naiman, N.E.; Yarchoan, R. Activity of Human Immunodeficiency Virus Type 1 Protease Inhibitors against the Initial Autocleavage in Gag-Pol Polyprotein Processing. Antimicrob. Agents Chemother. 2012, 56, 3620–3628. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Freed, E.O. HIV Type 1 Gag as a Target for Antiviral Therapy. AIDS Res. Hum. Retroviruses 2012, 28, 54–75. [Google Scholar] [CrossRef]
- Acciani, M.D.; Mendoza, M.F.L.; Havranek, K.E.; Duncan, A.M.; Iyer, H.; Linn, O.L.; Brindley, M.A. Ebola Virus Requires Phosphatidylserine Scrambling Activity for Efficient Budding and Optimal Infectivity. J. Virol. 2021, 95, e0116521. [Google Scholar] [CrossRef]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Tate, P.M.; Mastrodomenico, V.; Cunha, C.; McClure, J.; Barron, A.E.; Diamond, G.; Mounce, B.C.; Kirshenbaum, K. Peptidomimetic Oligomers Targeting Membrane Phosphatidylserine Exhibit Broad Antiviral Activity. ACS Infect. Dis. 2023, 9, 1508–1522. [Google Scholar] [CrossRef]
- Campbell, S.M.; Crowe, S.M.; Mak, J. Virion-associated cholesterol is critical for the maintenance of HIV-1 structure and infectivity. AIDS 2002, 16, 2253–2261. [Google Scholar] [CrossRef]
- Li, G.T.; Kim, J.; Huang, Z.; St Clair, J.R.; Brown, D.A.; London, E. Efficient replacement of plasma membrane outer leaflet phospholipids and sphingolipids in cells with exogenous lipids. Proc. Natl. Acad. Sci. USA 2016, 113, 14025–14030. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; London, E. Effect of Cyclodextrin and Membrane Lipid Structure upon Cyclodextrin-Lipid Interaction. Langmuir 2013, 29, 14631–14638. [Google Scholar] [CrossRef] [PubMed]
- Suresh, P.; London, E. Using cyclodextrin-induced lipid substitution to study membrane lipid and ordered membrane domain (raft) function in cells. BBA-Biomembranes 2022, 1864, 183774. [Google Scholar] [CrossRef]
- Lin, Y.C.; Chipot, C.; Scheuring, S. Annexin-V stabilizes membrane defects by inducing lipid phase transition. Nat. Commun. 2020, 11, 230. [Google Scholar] [CrossRef] [PubMed]
- Pye, V.E.; Rosa, A.; Bertelli, C.; Struwe, W.B.; Maslen, S.L.; Corey, R.; Liko, I.; Hassall, M.; Mattiuzzo, G.; Ballandras-Colas, A.; et al. A bipartite structural organization defines the SERINC family of HIV-1 restriction factors. Nat. Struct. Mol. Biol. 2020, 27, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 gag targeting to the plasma membrane. Mol. Biol. Cell 2004, 15, 122a–123a. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Zhu, Y.A.; Agostino, E.; Naing, L.; Hikichi, Y.; Soheilian, F.; Yoo, S.W.; Song, Y.; Zhang, P.J.; Slusher, B.S.; et al. Neutral sphingomyelinase 2 is required for HIV-1 maturation. Proc. Natl. Acad. Sci. USA 2023, 120, e2219475120. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Fleming, J.; St Clair, E.W.; Katinger, H.; Stiegler, G.; Kunert, R.; Robinson, J.; Scearce, R.M.; Plonk, K.; Staats, H.F.; et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 2005, 308, 1906–1908. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raghunath, G.; Abbott, E.H.; Marin, M.; Wu, H.; Reyes Ballista, J.M.; Brindley, M.A.; Melikyan, G.B. Disruption of Transmembrane Phosphatidylserine Asymmetry by HIV-1 Incorporated SERINC5 Is Not Responsible for Virus Restriction. Biomolecules 2024, 14, 570. https://doi.org/10.3390/biom14050570
Raghunath G, Abbott EH, Marin M, Wu H, Reyes Ballista JM, Brindley MA, Melikyan GB. Disruption of Transmembrane Phosphatidylserine Asymmetry by HIV-1 Incorporated SERINC5 Is Not Responsible for Virus Restriction. Biomolecules. 2024; 14(5):570. https://doi.org/10.3390/biom14050570
Chicago/Turabian StyleRaghunath, Gokul, Elizabeth H. Abbott, Mariana Marin, Hui Wu, Judith Mary Reyes Ballista, Melinda A. Brindley, and Gregory B. Melikyan. 2024. "Disruption of Transmembrane Phosphatidylserine Asymmetry by HIV-1 Incorporated SERINC5 Is Not Responsible for Virus Restriction" Biomolecules 14, no. 5: 570. https://doi.org/10.3390/biom14050570
APA StyleRaghunath, G., Abbott, E. H., Marin, M., Wu, H., Reyes Ballista, J. M., Brindley, M. A., & Melikyan, G. B. (2024). Disruption of Transmembrane Phosphatidylserine Asymmetry by HIV-1 Incorporated SERINC5 Is Not Responsible for Virus Restriction. Biomolecules, 14(5), 570. https://doi.org/10.3390/biom14050570