

The Emerging Role of Ubiquitin-Specific Protease 36 (USP36) in Cancer and Beyond

 ,
,  and
and 
Abstract
:1. Introduction
2. The Structure of USP36
3. Biological Function of USP36
3.1. Classical Deubiquitination Activity of USP36
3.2. The Functions of USP36 in Nucleolar Protein SUMOylation
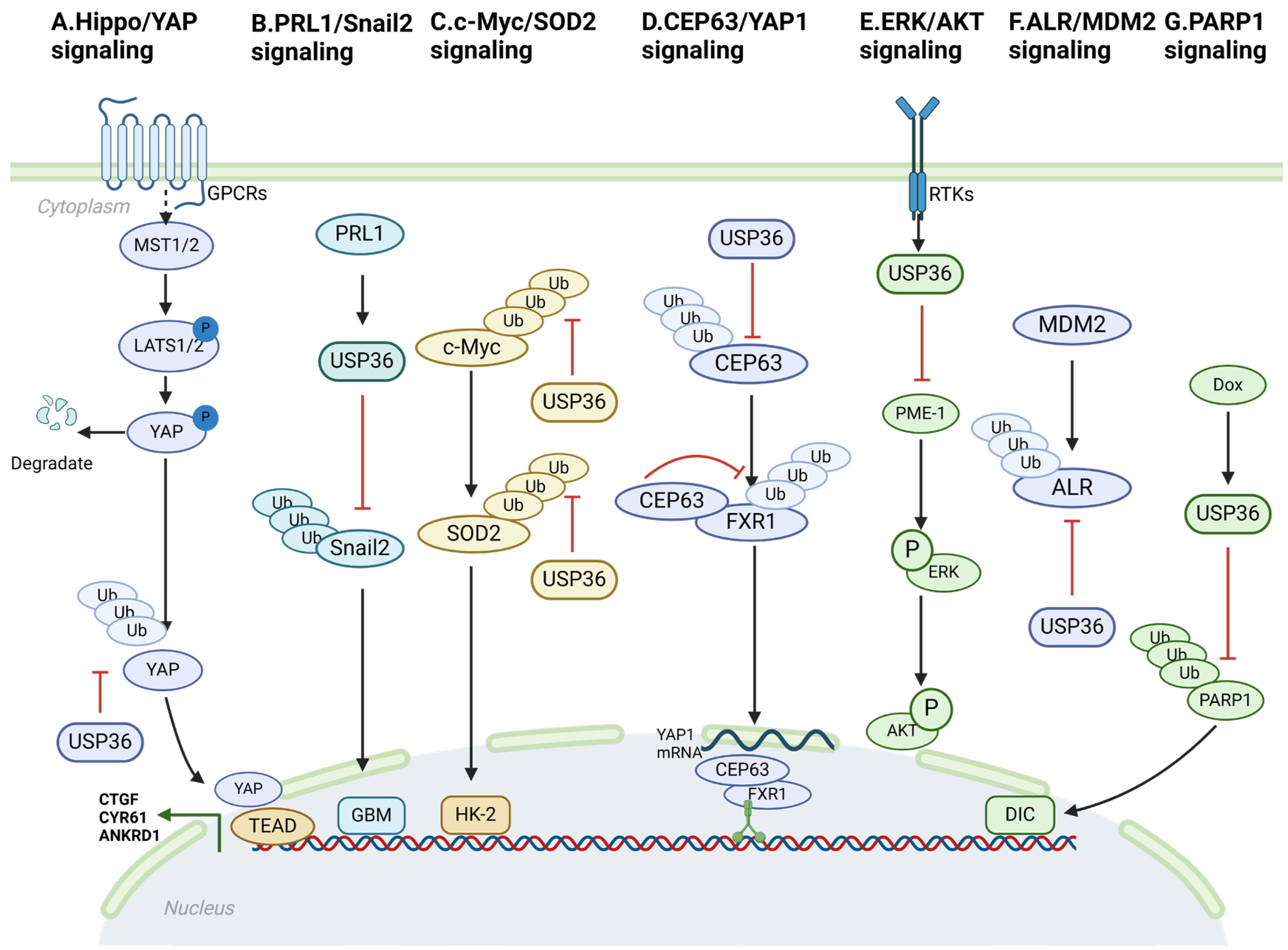
4. USP36-Mediated Signaling Pathway
4.1. Hippo/YAP Signaling
4.2. PRL1/Snail2 Signaling
4.3. c-Myc/SOD2 and c-Myc/Fbw7g Signaling
4.4. CEP63/YAP1 Signaling
4.5. ERK/AKT Signaling
4.6. ALR/MDM2 Signaling
4.7. USP36/PARP1 Signaling
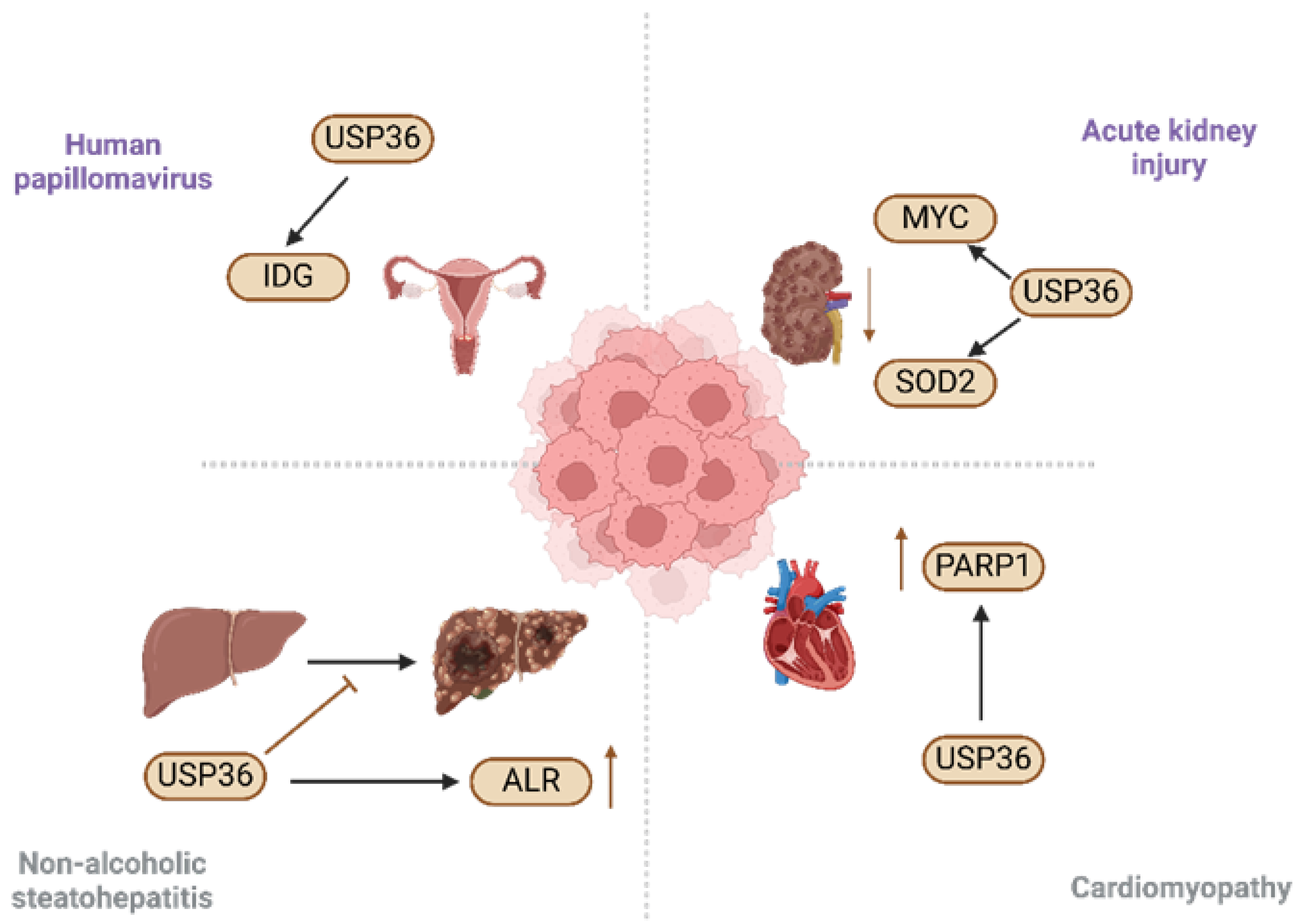
5. Role of USP36 in Diseases
5.1. Acute Kidney Injury
5.2. Non-Alcoholic Steatohepatitis
5.3. Human Papillomavirus
5.4. Cardiomyopathy
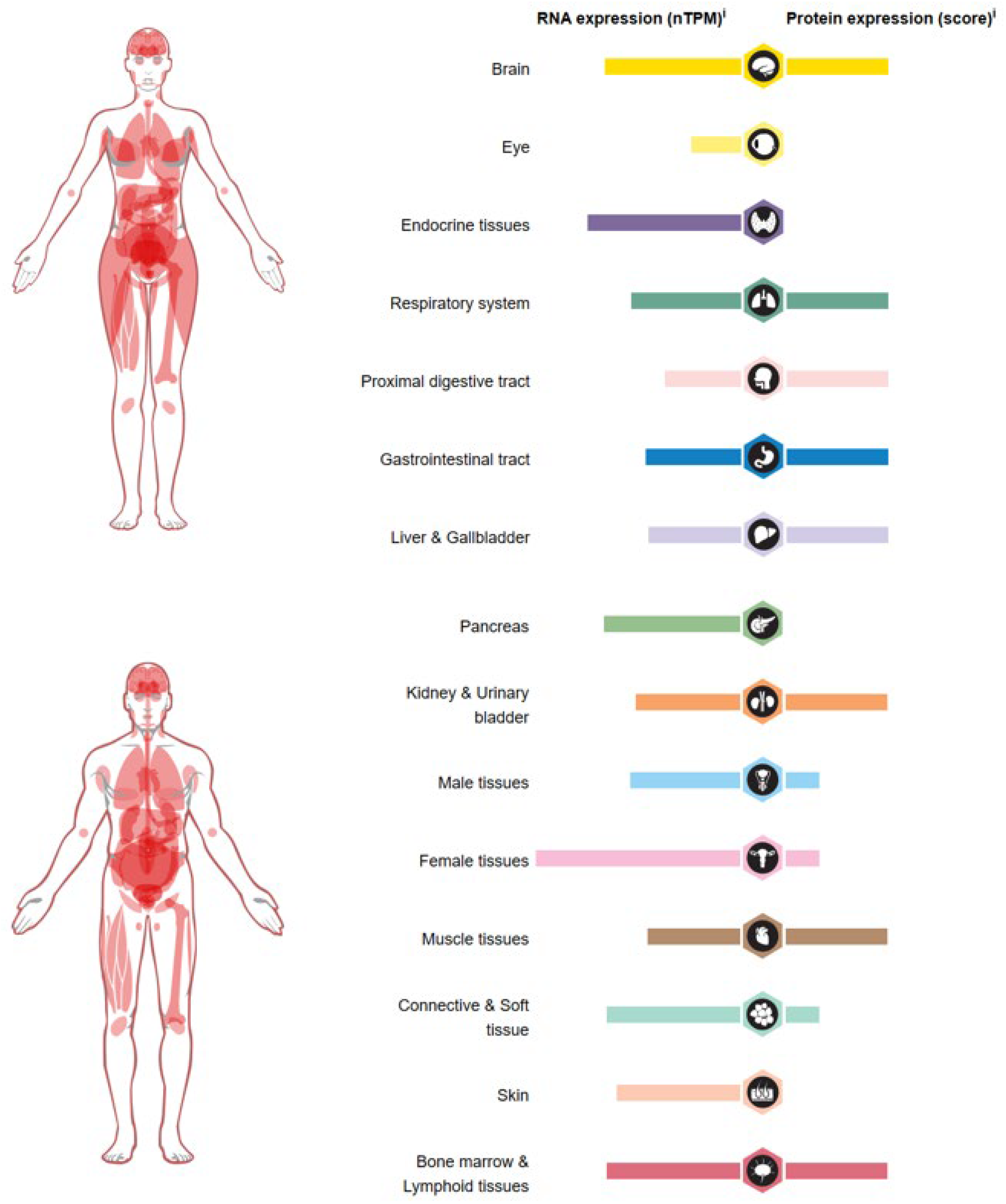
6. Role of USP36 in Cancer
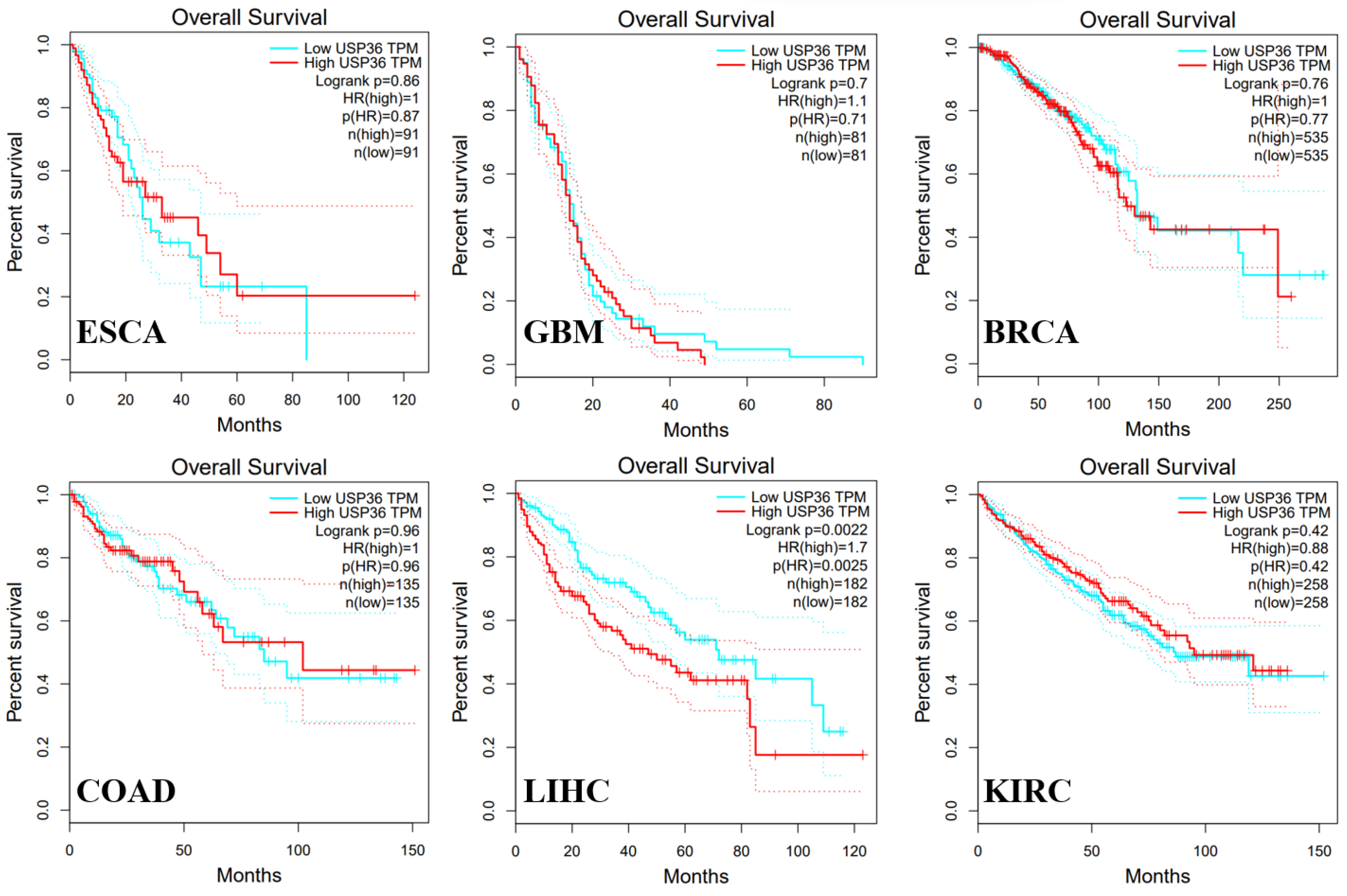
6.1. Esophageal Carcinoma
6.2. Glioblastoma
6.3. Hepatocellular Carcinoma
6.4. Colorectal Cancer
6.5. Breast Cancer
6.6. T Cell Lymphoma
7. USP36 as a Target for Cancer Therapy
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dewson, G.; Eichhorn, P.J.A.; Komander, D. Deubiquitinases in cancer. Nat. Rev. Cancer 2023, 23, 842–862. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.M.; Armstrong, L.A.; Kulathu, Y. Deubiquitinases: From mechanisms to their inhibition by small molecules. Mol. Cell 2022, 82, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.Y.; Li, C.Y.; Chen, R.Y.; Shi, J.J.; Liu, Y.J.; Lu, J.F.; Yang, G.J.; Chen, J. The emerging role of deubiquitylating enzyme USP21 as a potential therapeutic target in cancer. Bioorg. Chem. 2024, 10, 107400. [Google Scholar] [CrossRef]
- Ashton-Beaucage, D.; Lemieux, C.; Udell, C.M.; Sahmi, M.; Rochette, S.; Therrien, M. The Deubiquitinase USP47 Stabilizes MAPK by Counteracting the Function of the N-end Rule ligase POE/UBR4 in Drosophila. PLoS Biol. 2016, 14, e1002539. [Google Scholar] [CrossRef] [PubMed]
- Jolly, L.A.; Kumar, R.; Penzes, P.; Piper, M.; Gecz, J. The DUB Club: Deubiquitinating Enzymes and Neurodevelopmental Disorders. Biol. Psychiatry 2022, 92, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Kitamura, N.; Komada, M. Nucleophosmin/B23 regulates ubiquitin dynamics in nucleoli by recruiting deubiquitylating enzyme USP36. J. Biol. Chem. 2009, 284, 27918–27923. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Matsumoto, M.; Inada, T.; Yamamoto, A.; Nakayama, K.I.; Kitamura, N.; Komada, M. Nucleolar structure and function are regulated by the deubiquitylating enzyme USP36. J. Cell Sci. 2009, 122, 678–686. [Google Scholar] [CrossRef]
- Taillebourg, E.; Gregoire, I.; Viargues, P.; Jacomin, A.-C.; Thevenon, D.; Faure, M.; Fauvarque, M.-O. The deubiquitinating enzyme USP36 controls selective autophagy activation by ubiquitinated proteins. Autophagy 2012, 8, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-M.; Zhang, Y.Z.; Ramakrishna, S.; Lim, C.T. Electrospinning and mechanical characterization of gelatin nanofibers. Polymer 2004, 45, 5361–5368. [Google Scholar] [CrossRef]
- Richardson, L.A.; Reed, B.J.; Charette, J.M.; Freed, E.F.; Fredrickson, E.K.; Locke, M.N.; Baserga, S.J.; Gardner, R.G. A conserved deubiquitinating enzyme controls cell growth by regulating RNA polymerase I stability. Cell Rep. 2012, 2, 372–385. [Google Scholar] [CrossRef]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Sun, X.X.; Chen, Y.; Li, Y.; Wang, X.; Dai, R.S.; Zhu, H.M.; Klimek, J.; David, L.; Fedorov, L.M.; et al. The deubiquitinase USP36 promotes snoRNP group SUMOylation and is essential for ribosome biogenesis. EMBO Rep. 2021, 22, e50684. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef]
- Panse, V.G.; Kressler, D.; Pauli, A.; Petfalski, E.; Gnädig, M.; Tollervey, D.; Hurt, E. Formation and nuclear export of preribosomes are functionally linked to the small-ubiquitin-related modifier pathway. Traffic 2006, 7, 1311–1321. [Google Scholar] [CrossRef]
- Finkbeiner, E.; Haindl, M.; Muller, S. The SUMO system controls nucleolar partitioning of a novel mammalian ribosome biogenesis complex. Embo J 2011, 30, 1067–1078. [Google Scholar] [CrossRef]
- Raman, N.; Weir, E.; Müller, S. The AAA ATPase MDN1 Acts as a SUMO-Targeted Regulator in Mammalian Pre-ribosome Remodeling. Mol. Cell 2016, 64, 607–615. [Google Scholar] [CrossRef]
- Filippopoulou, C.; Thomé, C.C.; Perdikari, S.; Ntini, E.; Simos, G.; Bohnsack, K.E.; Chachami, G. Hypoxia-driven deSUMOylation of EXOSC10 promotes adaptive changes in the transcriptome profile. Cell Mol. Life Sci. 2024, 81, 58. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Dai, R.S.; Savage, J.C.; Shinde, U.; Klimek, J.; David, L.L.; Young, E.A.; Hafner, M.; Sears, R.C.; et al. The ubiquitin-specific protease USP36 SUMOylates EXOSC10 and promotes the nucleolar RNA exosome function in rRNA processing. Nucleic Acids Res. 2023, 51, 3934–3949. [Google Scholar] [CrossRef]
- Li, Y.; Carey, T.S.; Feng, C.H.; Zhu, H.-M.; Sun, X.-X.; Dai, M.-S. The Ubiquitin-specific Protease USP36 Associates with the Microprocessor Complex and Regulates miRNA Biogenesis by SUMOylating DGCR8. Cancer Res. Commun. 2023, 3, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes. Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012, 143, 1319–1329.e1311. [Google Scholar] [CrossRef]
- Plouffe, S.W.; Hong, A.W.; Guan, K.L. Disease implications of the Hippo/YAP pathway. Trends Mol. Med. 2015, 21, 212–222. [Google Scholar] [CrossRef]
- Zhou, A.; Yu, H.; Liu, J.; Zheng, J.; Jia, Y.; Wu, B.; Xiang, L. Role of Hippo-YAP Signaling in Osseointegration by Regulating Osteogenesis, Angiogenesis, and Osteoimmunology. Front. Cell Dev. Biol. 2020, 8, 780. [Google Scholar] [CrossRef]
- Zhang, W.; Luo, J.; Xiao, Z.; Zang, Y.; Li, X.; Zhou, Y.; Zhou, J.; Tian, Z.; Zhu, J.; Zhao, X. USP36 facilitates esophageal squamous carcinoma progression via stabilizing YAP. Cell Death Dis. 2022, 13, 1021. [Google Scholar] [CrossRef]
- Bai, Y.; Zhou, H.-M.; Zhang, L.; Dong, Y.; Zeng, Q.; Shou, W.; Zhang, Z.-Y. Role of phosphatase of regenerating liver 1 (PRL1) in spermatogenesis. Sci. Rep. 2016, 6, 34211. [Google Scholar] [CrossRef]
- Bai, Y.; Luo, Y.; Liu, S.; Zhang, L.; Shen, K.; Dong, Y.; Walls, C.D.; Quilliam, L.A.; Wells, C.D.; Cao, Y.; et al. PRL-1 Protein Promotes ERK1/2 and RhoA Protein Activation through a Non-canonical Interaction with the Src Homology 3 Domain of p115 Rho GTPase-activating Protein*. J. Biol. Chem. 2011, 286, 42316–42324. [Google Scholar] [CrossRef]
- Assani, G.; Zhou, Y. Effect of modulation of epithelial-mesenchymal transition regulators Snail1 and Snail2 on cancer cell radiosensitivity by targeting of the cell cycle, cell apoptosis and cell migration/invasion. Oncol. Lett. 2019, 17, 23–30. [Google Scholar] [CrossRef]
- Meng, J.; Ai, X.; Lei, Y.; Zhong, W.; Qian, B.; Qiao, K.; Wang, X.; Zhou, B.; Wang, H.; Huai, L.; et al. USP5 promotes epithelial-mesenchymal transition by stabilizing SLUG in hepatocellular carcinoma. Theranostics 2019, 9, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Min, L.; Liu, C.; Chen, S.; Gao, C.; Ma, J.; Wu, X.; Li, W.; Wu, L.; Zhu, L. RNF8 Promotes Epithelial-Mesenchymal Transition in Lung Cancer Cells via Stabilization of Slug. Mol. Cancer Res. 2020, 18, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Cai, X.; Xu, K.; Song, S.; Xiao, Z.; Hou, Y.; Qi, X.; Liu, F.; Chen, Y.; Yang, H.; et al. PRL1 Promotes Glioblastoma Invasion and Tumorigenesis via Activating USP36-Mediated Snail2 Deubiquitination. Front. Oncol. 2021, 11, 795633. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.Y.; Watson, J.D.; Yan, P.S.; Barsyte-Lovejoy, D.; Khosravi, F.; Wong, W.W.; Farnham, P.J.; Huang, T.H.; Penn, L.Z. Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 2003, 13, 882–886. [Google Scholar] [CrossRef]
- Bieda, M.; Xu, X.; Singer, M.A.; Green, R.; Farnham, P.J. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006, 16, 595–605. [Google Scholar] [CrossRef]
- Liu, Q.; Sheng, W.; Ma, Y.; Zhen, J.; Roy, S.; Alvira Jafar, C.; Xin, W.; Wan, Q. USP36 protects proximal tubule cells from ischemic injury by stabilizing c-Myc and SOD2. Biochem. Biophys. Res. Commun. 2019, 513, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Grim, J.E.; Eisenman, R.N.; Clurman, B.E. A Nucleolar Isoform of the Fbw7 Ubiquitin Ligase Regulates c-Myc and Cell Size. Curr. Biol. 2004, 14, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 2006, 16, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Lutterbach, B.; Hann, S.R. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol. Cell Biol. 1994, 14, 5510–5522. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes. Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-X.; Sears, R.C.; Dai, M.-S. Deubiquitinating c-Myc: USP36 steps up in the nucleolus. Cell Cycle 2015, 14, 3786–3793. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.X.; He, X.; Yin, L.; Komada, M.; Sears, R.C.; Dai, M.S. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2015, 112, 3734–3739. [Google Scholar] [CrossRef]
- Fraile, J.M.; Campos-Iglesias, D.; Rodríguez, F.; Astudillo, A.; Vilarrasa-Blasi, R.; Verdaguer-Dot, N.; Prado, M.A.; Paulo, J.A.; Gygi, S.P.; Martín-Subero, J.I.; et al. Loss of the deubiquitinase USP36 destabilizes the RNA helicase DHX33 and causes preimplantation lethality in mice. J. Biol. Chem. 2018, 293, 2183–2194. [Google Scholar] [CrossRef]
- Thevenon, D.; Engel, E.; Avet-Rochex, A.; Gottar, M.; Bergeret, E.; Tricoire, H.; Benaud, C.; Baudier, J.; Taillebourg, E.; Fauvarque, M.-O. The Drosophila ubiquitin-specific protease dUSP36/Scny targets IMD to prevent constitutive immune signaling. Cell Host Microbe 2009, 64, 309–320. [Google Scholar] [CrossRef]
- Löffler, H.; Fechter, A.; Matuszewska, M.; Saffrich, R.; Mistrik, M.; Marhold, J.; Hornung, C.; Westermann, F.; Bartek, J.; Krämer, A. Cep63 recruits Cdk1 to the centrosome: Implications for regulation of mitotic entry, centrosome amplification, and genome maintenance. Cancer Res. 2011, 71, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Sir, J.H.; Barr, A.R.; Nicholas, A.K.; Carvalho, O.P.; Khurshid, M.; Sossick, A.; Reichelt, S.; D’Santos, C.; Woods, C.G.; Gergely, F. A primary microcephaly protein complex forms a ring around parental centrioles. Nat. Genet. 2011, 43, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, D.; Kodani, A.; Gonzalez, D.M.; Mancias, J.D.; Mochida, G.H.; Vagnoni, C.; Johnson, J.; Krogan, N.; Harper, J.W.; Reiter, J.F.; et al. Microcephaly Proteins Wdr62 and Aspm Define a Mother Centriole Complex Regulating Centriole Biogenesis, Apical Complex, and Cell Fate. Neuron 2016, 92, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, C.; Novack, J.P.; Reed, J.C.; Matsuzawa, S.I. K63 ubiquitination in immune signaling. Trends Immunol. 2022, 43, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Perrotti, D.; Neviani, P. Protein phosphatase 2A: A target for anticancer therapy. Lancet Oncol. 2013, 14, e229–e238. [Google Scholar] [CrossRef] [PubMed]
- Arriazu, E.; Pippa, R.; Odero, M.D. Protein Phosphatase 2A as a Therapeutic Target in Acute Myeloid Leukemia. Front. Oncol. 2016, 6, 78. [Google Scholar] [CrossRef]
- Kim, S.Y.; Choi, J.; Lee, D.H.; Park, J.H.; Hwang, Y.J.; Baek, K.H. PME-1 is regulated by USP36 in ERK and Akt signaling pathways. FEBS Lett. 2018, 592, 1575–1588. [Google Scholar] [CrossRef]
- Zheng, M.; Ai, Z.; Guo, Y.; Chen, Y.; Xie, P.; An, W. Imbalance in ALR ubiquitination accelerates the progression of nonalcoholic steatohepatitis to hepatocellular carcinoma. Oncogene 2023, 42, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Wu, L.; Wang, L.; Du, Y.; Zhang, Y.; Ren, J. Mitochondrial quality control mechanisms as therapeutic targets in doxorubicin-induced cardiotoxicity. Trends Pharmacol. Sci. 2023, 44, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jiang, Z.; Kan, J.; Jiang, X.; Pan, C.; You, S.; Chang, R.; Zhang, J.; Yang, H.; Zhu, L.; et al. USP36-mediated PARP1 deubiquitination in doxorubicin-induced cardiomyopathy. Cell Signal 2024, 117, 111070. [Google Scholar] [CrossRef] [PubMed]
- Iden, M.; Tsaih, S.W.; Huang, Y.W.; Liu, P.; Xiao, M.; Flister, M.J.; Rader, J.S. Multi-omics mapping of human papillomavirus integration sites illuminates novel cervical cancer target genes. Br. J. Cancer 2021, 125, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Ympa, Y.P.; Sakr, Y.; Reinhart, K.; Vincent, J.L. Has mortality from acute renal failure decreased? A systematic review of the literature. Am. J. Med. 2005, 118, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Amdur, R.L.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011, 79, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Hultström, M.; Becirovic-Agic, M.; Jönsson, S. Comparison of acute kidney injury of different etiology reveals in-common mechanisms of tissue damage. Physiol. Genom. 2018, 50, 127–141. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- LaBrecque, D.R.; Pesch, L.A. Preparation and partial characterization of hepatic regenerative stimulator substance (SS) from rat liver. J. Physiol. 1975, 248, 273–284. [Google Scholar] [CrossRef]
- Lane, D.P.; Hall, P.A. MDM2--arbiter of p53’s destruction. Trends Biochem. Sci. 1997, 22, 372–374. [Google Scholar] [CrossRef]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Shen, J.; Liu, J.; Han, K.; Liang, L.; Gao, Y. Gene Signature and Prognostic Value of Ubiquitin-Specific Proteases Members in Hepatocellular Carcinoma and Explored the Immunological Role of USP36. Front. Biosci. 2022, 27, 190. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Cao, C.H.; Han, K.; Lv, Y.R.; Ma, X.D.; Cao, J.H.; Chen, J.W.; Li, S.; Lin, J.L.; Fang, Y.J.; et al. CEP63 upregulates YAP1 to promote colorectal cancer progression through stabilizing RNA binding protein FXR1. Oncogene 2022, 41, 4433–4445. [Google Scholar] [CrossRef]
- Wu, H.; Jiao, Y.; Zhou, C.; Guo, X.; Wu, Z.; Lv, Q. miR-140-3p/usp36 axis mediates ubiquitination to regulate PKM2 and suppressed the malignant biological behavior of breast cancer through Warburg effect. Cell Cycle 2023, 22, 680–692. [Google Scholar] [CrossRef]
- Li, B.; Yan, J.; Phyu, T.; Fan, S.; Chung, T.H.; Mustafa, N.; Lin, B.; Wang, L.; Eichhorn, P.J.A.; Goh, B.C.; et al. MELK mediates the stability of EZH2 through site-specific phosphorylation in extranodal natural killer/T-cell lymphoma. Blood 2019, 134, 2046–2058. [Google Scholar] [CrossRef]
- Chang, G.; Xie, G.S.; Ma, L.; Li, P.; Li, L.; Richard, H.T. USP36 promotes tumorigenesis and drug sensitivity of glioblastoma by deubiquitinating and stabilizing ALKBH5. Neuro Oncol. 2023, 25, 841–853. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, R.; Zeng, H.; Zhang, S.; He, J. Annual report on status of cancer in China, 2011. Chin. J. Cancer Res. 2015, 27, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.D.; Liao, X.Y.; Chen, Y.B.; Huang, S.Y.; Xue, W.Q.; Li, F.F.; Ge, X.S.; Liu, D.Q.; Cai, Q.; Long, J.; et al. Genomic Characterization of Esophageal Squamous Cell Carcinoma Reveals Critical Genes Underlying Tumorigenesis and Poor Prognosis. Am. J. Hum. Genet. 2016, 98, 709–727. [Google Scholar] [CrossRef]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Lo Sardo, F.; Canu, V.; Maugeri-Saccà, M.; Strano, S.; Blandino, G. YAP and TAZ: Monocorial and bicorial transcriptional co-activators in human cancers. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188756. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Y.; Wang, W.; Wang, S.; Hou, J.; Zhang, A.; Lv, B.; Gao, C.; Yan, Z.; Pang, D.; et al. Regulation of Hippo/YAP signaling and Esophageal Squamous Carcinoma progression by an E3 ubiquitin ligase PARK2. Theranostics 2020, 10, 9443–9457. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, H.W.; Wang, S.; Fan, L.; Feng, S.; Cai, X.; Peng, C.; Wu, X.; Lu, J.; Chen, D.; et al. USP9X deubiquitinates ALDH1A3 and maintains mesenchymal identity in glioblastoma stem cells. J. Clin. Invest. 2019, 129, 2043–2055. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Patel, N.P.; Lyon, K.A.; Huang, J.H. The effect of race on the prognosis of the glioblastoma patient: A brief review. Neurol. Res. 2019, 41, 967–971. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e596. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 561–577.e522. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Cao, J.; Lei, X.; Shi, Y.; Wu, L. Multi-omics data identified TP53 and LRP1B as key regulatory gene related to immune phenotypes via EPCAM in HCC. Cancer Med. 2022, 11, 2145–2158. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Shan, Q.; Zhan, Q.; Ye, Q.; Liu, P.; Xu, S.; He, X.; Ma, J.; Xiang, J.; Jiang, G.; et al. USP22 promotes hypoxia-induced hepatocellular carcinoma stemness by a HIF1α/USP22 positive feedback loop upon TP53 inactivation. Gut 2020, 69, 1322–1334. [Google Scholar] [CrossRef]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, F.; Ma, R.; Zhang, L.; Du, G.; Niu, D.; Yin, D. Cep63 knockout inhibits the malignant phenotypes of papillary thyroid cancer cell line TPC-1. Oncol. Rep. 2021, 46, 8150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ren, P.; Xu, D.; Liu, X.; Liu, Z.; Zhang, C.; Li, Y.; Wang, L.; Du, X.; Xing, B. Human UTP14a promotes colorectal cancer progression by forming a positive regulation loop with c-Myc. Cancer Lett. 2019, 440–441, 106–115. [Google Scholar] [CrossRef]
- Zhong, X.D.; Chen, L.J.; Xu, X.Y.; Liu, Y.J.; Tao, F.; Zhu, M.H.; Li, C.Y.; Zhao, D.; Yang, G.J.; Chen, J. Berberine as a potential agent for breast cancer therapy. Front. Oncol. 2022, 12, 993775. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, P.; Yarani, R.; Valipour, E.; Kiani, S.; Hoseinkhani, Z.; Mansouri, K. Cell line-directed breast cancer research based on glucose metabolism status. Biomed. Pharmacother. 2022, 146, 112526. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.J.; Tao, F.; Zhong, H.J.; Yang, C.; Chen, J. Targeting PGAM1 in cancer: An emerging therapeutic opportunity. Eur. J. Med. Chem. 2022, 244, 114798. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Yan, J.; Ng, S.B.; Tay, J.L.; Lin, B.; Koh, T.L.; Tan, J.; Selvarajan, V.; Liu, S.C.; Bi, C.; Wang, S.; et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood 2013, 121, 4512–4520. [Google Scholar] [CrossRef] [PubMed]
- Adelaiye-Ogala, R.; Budka, J.; Damayanti, N.P.; Arrington, J.; Ferris, M.; Hsu, C.C.; Chintala, S.; Orillion, A.; Miles, K.M.; Shen, L.; et al. EZH2 Modifies Sunitinib Resistance in Renal Cell Carcinoma by Kinome Reprogramming. Cancer Res. 2017, 77, 6651–6666. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Z.; Cenciarini, M.E.; Proietti, C.J.; Amasino, M.; Hong, T.; Yang, M.; Liao, Y.; Chiang, H.C.; Kaklamani, V.G.; et al. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2-ERα-GREB1 Transcriptional Axis. Cancer Res. 2018, 78, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Banasavadi-Siddegowda, Y.; Mo, X.; Kim, S.H.; Mao, P.; Kig, C.; Nardini, D.; Sobol, R.W.; Chow, L.M.; Kornblum, H.I.; et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells 2013, 31, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cai, S.; Gu, T.; Song, F.; Xue, Y.; Sun, D. MiR-140-3p Impedes Gastric Cancer Progression and Metastasis by Regulating BCL2/BECN1-Mediated Autophagy. Onco Targets Ther. 2021, 14, 2879–2892. [Google Scholar] [CrossRef]
- Dou, D.; Ren, X.; Han, M.; Xu, X.; Ge, X.; Gu, Y.; Wang, X.; Zhao, S. Circ_0008039 supports breast cancer cell proliferation, migration, invasion, and glycolysis by regulating the miR-140-3p/SKA2 axis. Mol. Oncol. 2021, 15, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Yang, G.J.; Wang, W.; Ma, D.L.; Leung, C.H. Discovery of a tetrahydroisoquinoline-based CDK9-cyclin T1 protein-protein interaction inhibitor as an anti-proliferative and anti-migration agent against triple-negative breast cancer cells. Genes. Dis. 2022, 9, 1674–1688. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.S.; Yang, G.J.; Wang, W.; Leung, C.H.; Ma, D.L. Correction to: The design and development of covalent protein-protein interaction inhibitors for cancer treatment. J. Hematol. Oncol. 2020, 13, 102. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Leung, C.-H.; Zhang, J.-T.; Yang, G.-J.; Liu, H.; Han, Q.-B.; Ma, D.-L. Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors. Int. J. Mol. Sci. 2019, 20, 4445. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Functions | Substrates | Cancer Type |
|---|---|---|---|
| [31] | Facilitating ESCC progression via the Hippo/YAP axis | Hippo/YAP | Esophageal carcinoma |
| [77] | Deubiquitinating Snail2 and thus promoting glioblastoma invasion and tumorigenesis | Snail2 | Glioblastoma invasion |
| [73] | Synergizing with TP53 and promoting HCC progression | TP53 | Hepatocellular carcinoma |
| [74] | Stabilizing CEP63 by reducing its K48 ubiquitination and promoting colorectal cancer progression | CEP63 | Colorectal cancer |
| [75] | Deubiquitinating PKM2 to suppress the malignant phenotypes through the Warburg effect | PKM2 | Breast cancer |
| [76] | USP36 helps mediate the stabilization of EZH2 | MELK | T cell lymphoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, M.-Y.; Liu, Y.-J.; Shi, J.-J.; Chen, R.-Y.; Zhang, S.; Li, C.-Y.; Cao, J.-F.; Yang, G.-J.; Chen, J. The Emerging Role of Ubiquitin-Specific Protease 36 (USP36) in Cancer and Beyond. Biomolecules 2024, 14, 572. https://doi.org/10.3390/biom14050572
Niu M-Y, Liu Y-J, Shi J-J, Chen R-Y, Zhang S, Li C-Y, Cao J-F, Yang G-J, Chen J. The Emerging Role of Ubiquitin-Specific Protease 36 (USP36) in Cancer and Beyond. Biomolecules. 2024; 14(5):572. https://doi.org/10.3390/biom14050572
Chicago/Turabian StyleNiu, Meng-Yao, Yan-Jun Liu, Jin-Jin Shi, Ru-Yi Chen, Shun Zhang, Chang-Yun Li, Jia-Feng Cao, Guan-Jun Yang, and Jiong Chen. 2024. "The Emerging Role of Ubiquitin-Specific Protease 36 (USP36) in Cancer and Beyond" Biomolecules 14, no. 5: 572. https://doi.org/10.3390/biom14050572