

BUB1 Inhibition Sensitizes TNBC Cell Lines to Chemotherapy and Radiotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Co-Expression Studies Using the cBioPortal Database
2.2. Chemicals
2.3. Cell Lines and Cell Culture
2.4. Drug Treatment and Radiation
2.5. Cell Growth and Viability
2.6. Clonogenic Survival Assay
3. Results
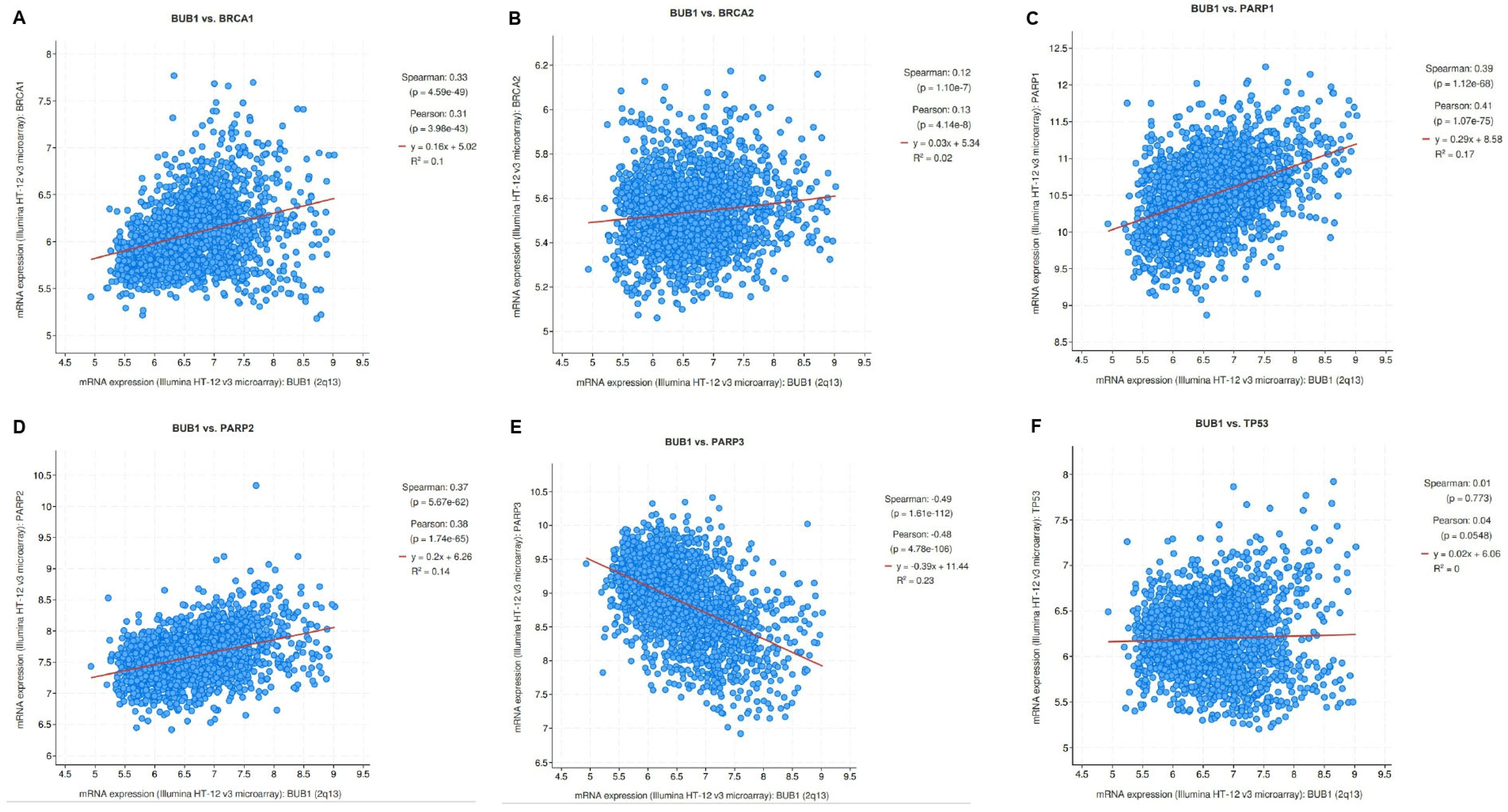
3.1. Correlation of BUB1 mRNA Expression with BRCA1/2, PARP1/2/3, and TP53 in Breast Cancer
3.2. Antiproliferative Effects of Single Agents Olaparib, Cisplatin, and Paclitaxel in TNBC Cell Lines
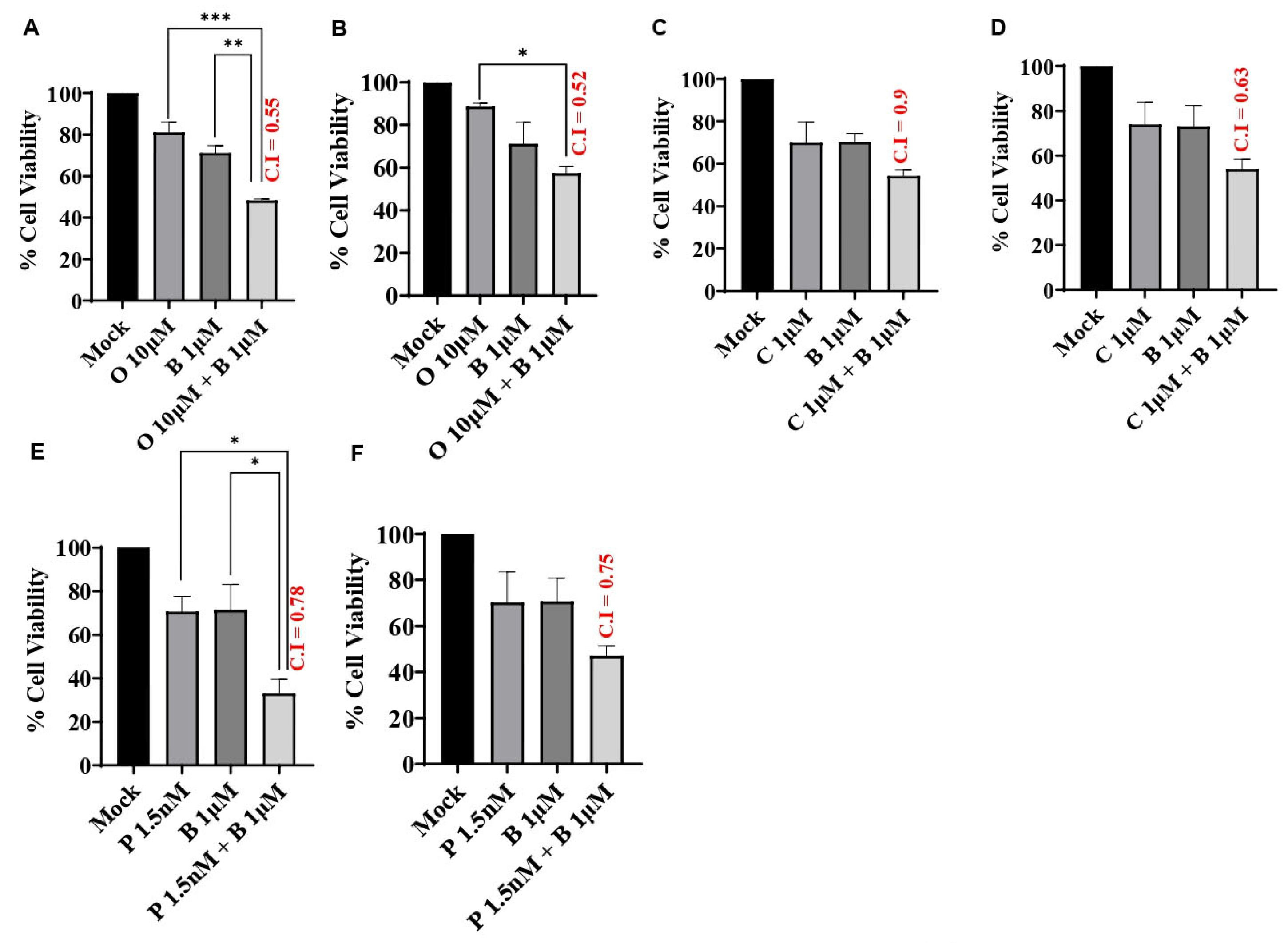
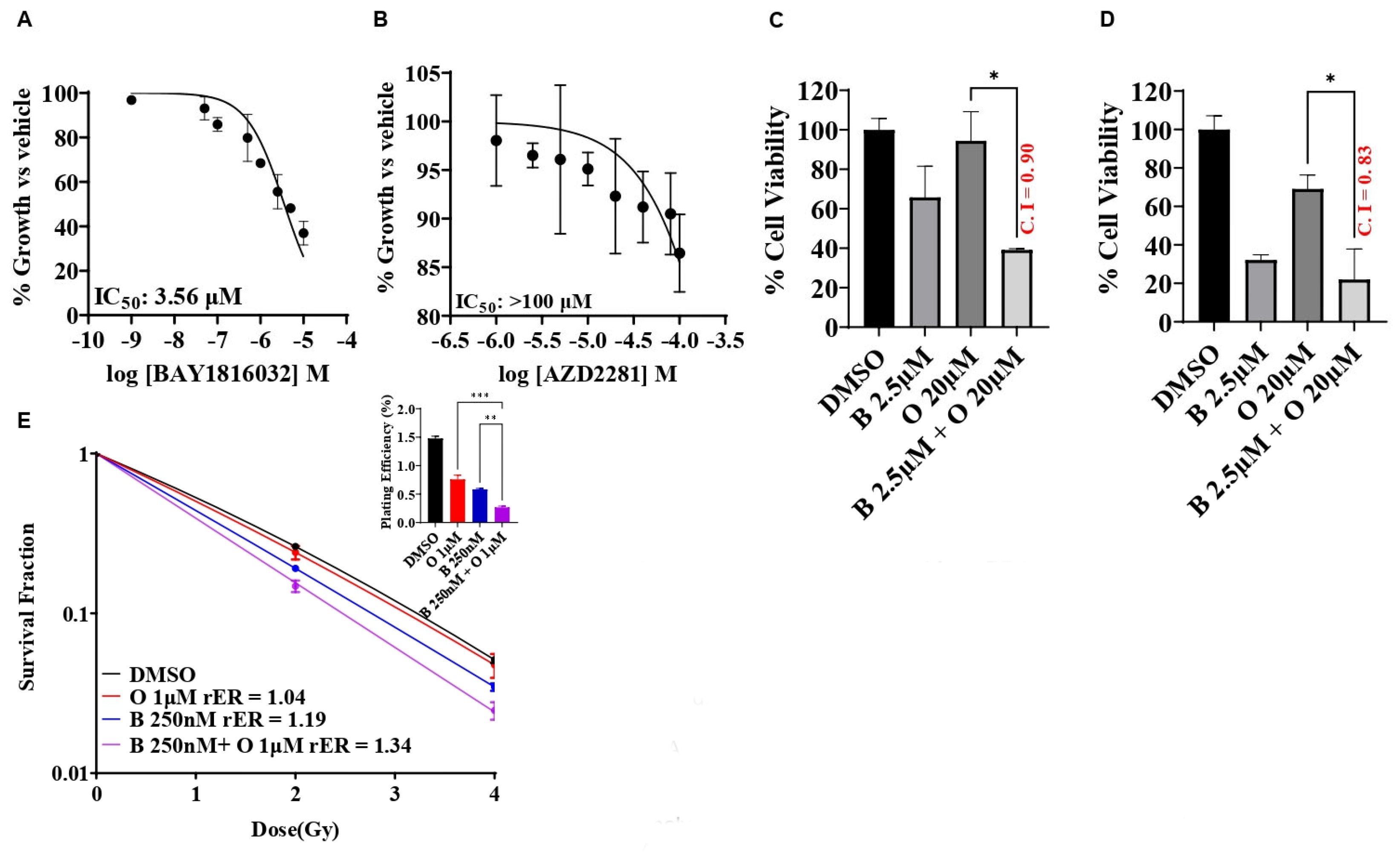
3.3. BUB1 Inhibitor Synergistically Sensitizes TNBC to Olaparib, Cisplatin, and Paclitaxel
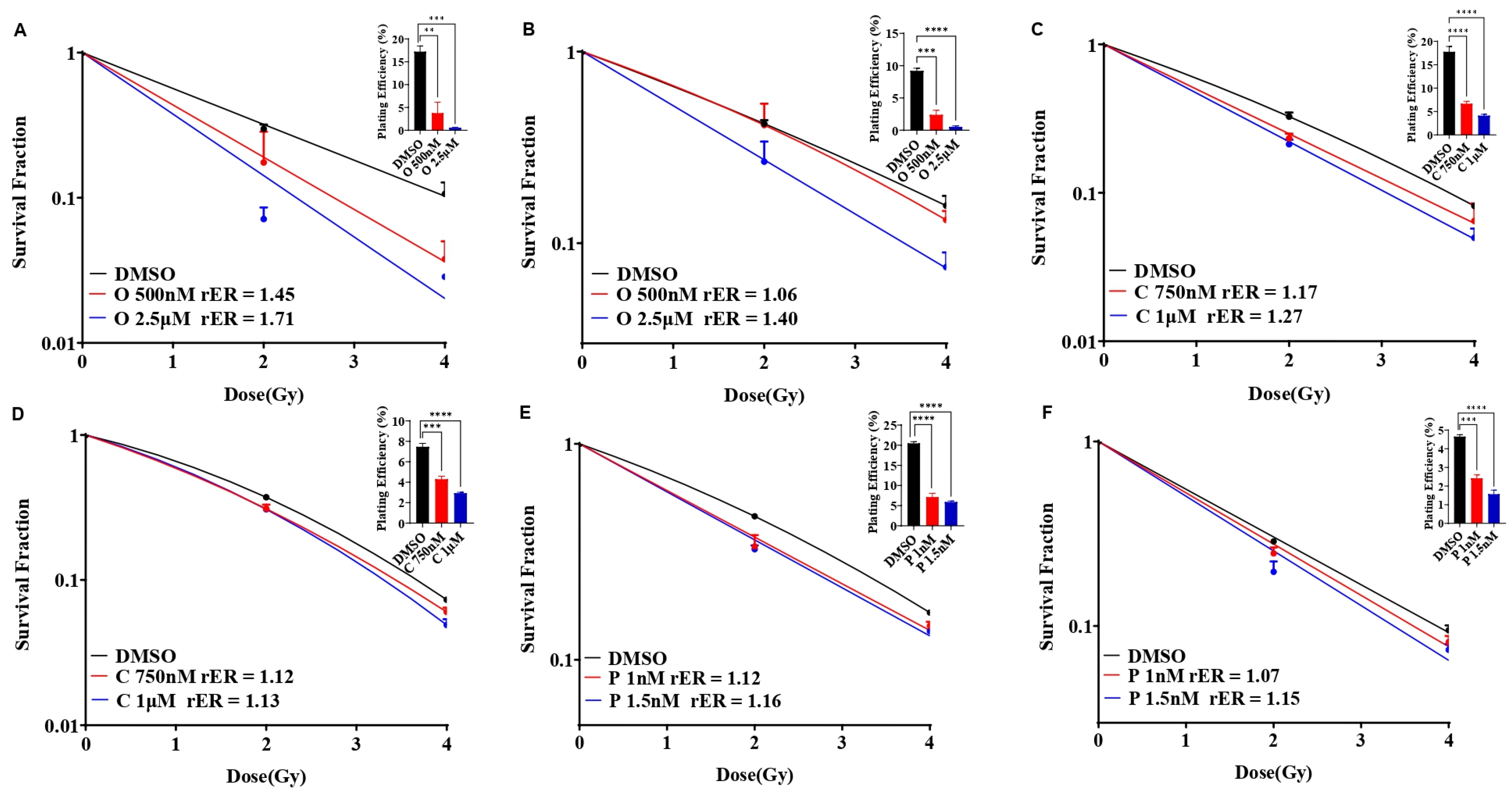
3.4. Olaparib, Cisplatin, and Paclitaxel Differentially Radiosensitize SUM159 and MDA-MB-231 Cell Lines
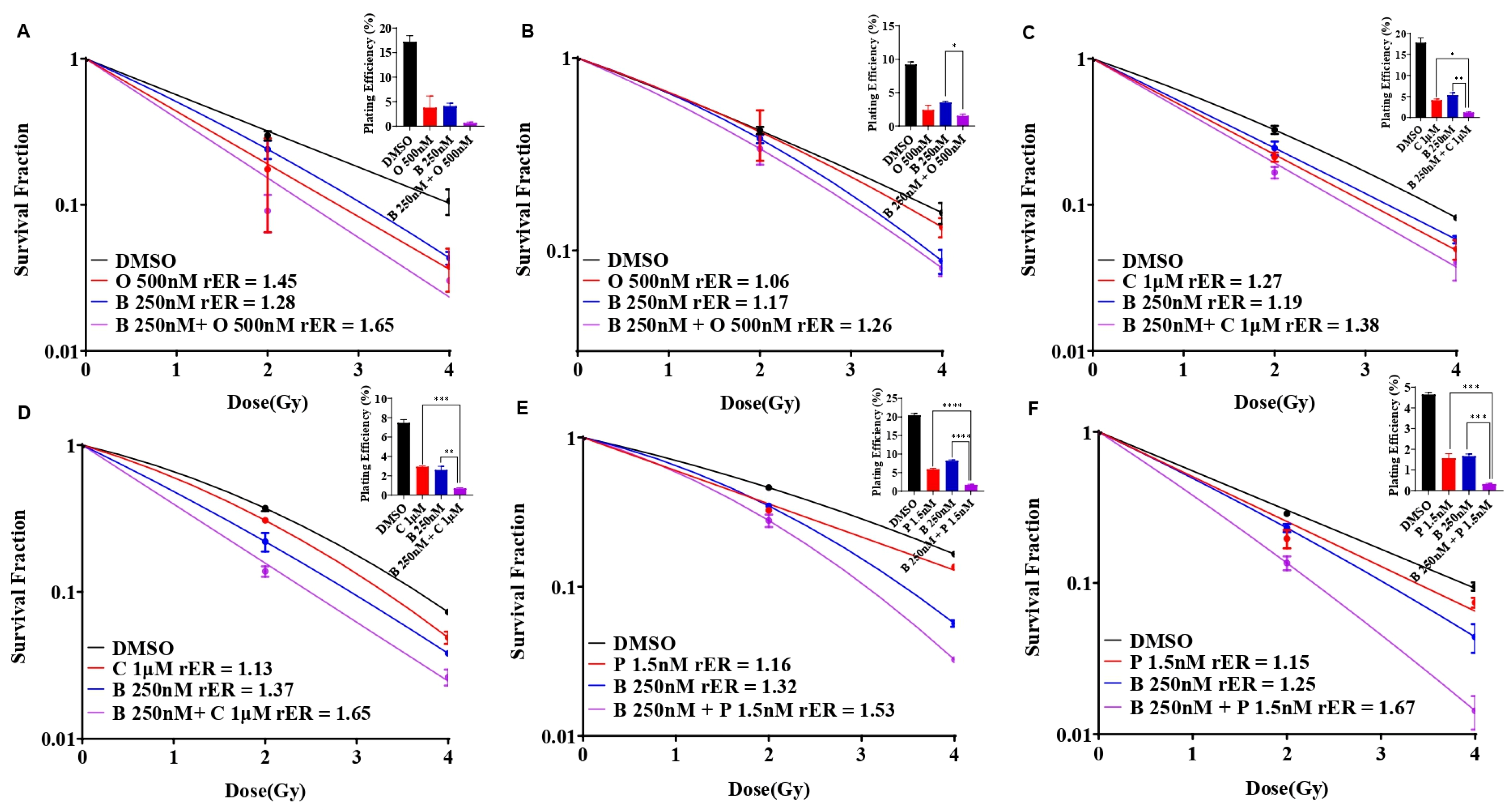
3.5. BUB1 Inhibitor Enhances Radiation Sensitization by Olaparib, Cisplatin, and Paclitaxel in SUM159 and MDA-MB-231 Cell Lines
3.6. BUB1 Inhibitor BAY1816032 Sensitizes BRCA Mutant TNBC Cell Line to PARP Inhibitor and Radiation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Wu, Y.; Ma, F.; Kaklamani, V.; Xu, B. Advances in medical treatment of breast cancer in 2022. Cancer Innov. 2023, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zagami, P.; Carey, L.A. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Niwińska, A.; Murawska, M.; Pieńkowski, T. Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Med. Oncol. 2013, 30, 388. [Google Scholar] [CrossRef] [PubMed]
- Newton, E.E.; Mueller, L.E.; Treadwell, S.M.; Morris, C.A.; Machado, H.L. Molecular Targets of Triple-Negative Breast Cancer: Where Do We Stand? Cancers 2022, 14, 482. [Google Scholar] [CrossRef]
- Corrales-Guerrero, S.; Cui, T.; Castro-Aceituno, V.; Yang, L.; Nair, S.; Feng, H.; Venere, M.; Yoon, S.; DeWees, T.; Shen, C.; et al. Inhibition of RRM2 radiosensitizes glioblastoma and uncovers synthetic lethality in combination with targeting CHK1. Cancer Lett. 2023, 570, 216308. [Google Scholar] [CrossRef] [PubMed]
- Pesch, A.M.; Chandler, B.C.; Michmerhuizen, A.R.; Carter, H.M.; Hirsh, N.H.; Wilder-Romans, K.; Liu, M.; Ward, T.; Ritter, C.L.; Nino, C.A.; et al. Bcl-xL inhibition radiosensitizes PIK3CA/PTEN wild-type triple negative breast cancers with low Mcl-1 expression. Cancer Res. Commun. 2022, 2, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.A.; Sugiman-Marangos, S.; VanderVere-Carozza, P.; Wagner, M.; Turchi, J.; Lees-Miller, S.P.; Junop, M.S.; Meek, K. Unraveling the complexities of DNA-dependent protein kinase autophosphorylation. Mol. Cell. Biol. 2014, 34, 2162–2175. [Google Scholar] [CrossRef]
- Jin, J.; Tao, Z.; Cao, J.; Li, T.; Hu, X. DNA damage response inhibitors: An avenue for TNBC treatment. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188521. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef]
- Morgan, M.A.; Parsels, L.A.; Maybaum, J.; Lawrence, T.S. Improving the efficacy of chemoradiation with targeted agents. Cancer Discov. 2014, 4, 280–291. [Google Scholar] [CrossRef]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef]
- Speers, C.; Tsimelzon, A.; Sexton, K.; Herrick, A.M.; Gutierrez, C.; Culhane, A.; Quackenbush, J.; Hilsenbeck, S.; Chang, J.; Brown, P. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin. Cancer Res. 2009, 15, 6327–6340. [Google Scholar] [CrossRef] [PubMed]
- Kausar, T.; Schreiber, J.S.; Karnak, D.; Parsels, L.A.; Parsels, J.D.; Davis, M.A.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015, 17, 757–766. [Google Scholar] [CrossRef]
- Tang, Z.Y.; Shu, H.J.; Oncel, D.; Chen, S.; Yu, H.T. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol. Cell 2004, 16, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Y.; Sun, Y.X.; Harley, S.E.; Zou, H.; Yu, H.T. Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proc. Natl. Acad. Sci. USA 2004, 101, 18012–18017. [Google Scholar] [CrossRef]
- Yu, H.; Tang, Z. Bub1 multitasking in mitosis. Cell Cycle 2005, 4, 262–265. [Google Scholar] [CrossRef]
- Williams, G.L.; Roberts, T.M.; Gjoerup, O.V. Bub1: Escapades in a cellular world. Cell Cycle 2007, 6, 1699–1704. [Google Scholar] [CrossRef]
- Zhu, L.J.; Pan, Y.; Chen, X.Y.; Hou, P.F. BUB1 promotes proliferation of liver cancer cells by activating SMAD2 phosphorylation. Oncol. Lett. 2020, 19, 3506–3512. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Xu, Y.; Woo, J.H.; Wang, Y.; Bae, Y.K.; Yoon, D.S.; Wersto, R.P.; Tully, E.; Wilsbach, K.; Gabrielson, E. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin. Cancer Res. 2006, 12, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Cicirò, Y.; Ragusa, D.; Sala, A. Expression of the checkpoint kinase BUB1 is a predictor of response to cancer therapies. Sci. Rep. 2024, 14, 4461. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; van’t Veer, L.; Lamb, J.; He, Y.D.; Mao, M.; Fine, B.M.; Bernards, R.; van de Vijver, M.; Deutsch, P.; Sachs, A.; et al. A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 2005, 65, 4059–4066. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Cai, J.H.; Wang, C.C.N. Identification of Key Prognostic Genes of Triple Negative Breast Cancer by LASSO-Based Machine Learning and Bioinformatics Analysis. Genes 2022, 13, 902. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Inamoto, T.; Tsujino, T.; Matsui, Y.; Konuma, T.; Nishimura, K.; Uchimoto, T.; Tsutsumi, T.; Matsunaga, T.; Maenosono, R.; et al. Increased BUB1B/BUBR1 expression contributes to aberrant DNA repair activity leading to resistance to DNA-damaging agents. Oncogene 2021, 40, 6210–6222. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Chen, W.-M.; Hassan, O.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Green, M.D.; Davis, A.J.; Speers, C.; et al. BUB1 regulates non-homologous end joining pathway to mediate radioresistance in triple-negative breast cancer. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Nyati, S.; Schinske-Sebolt, K.; Pitchiaya, S.; Chekhovskiy, K.; Chator, A.; Chaudhry, N.; Dosch, J.; Van Dort, M.E.; Varambally, S.; Kumar-Sinha, C.; et al. The kinase activity of the Ser/Thr kinase BUB1 promotes TGF-β signaling. Sci. Signal 2015, 8, ra1. [Google Scholar] [CrossRef]
- Li, M.; Duan, X.; Xiao, Y.; Yuan, M.; Zhao, Z.; Cui, X.; Wu, D.; Shi, J. BUB1 Is Identified as a Potential Therapeutic Target for Pancreatic Cancer Treatment. Front. Public Health 2022, 10, 900853. [Google Scholar] [CrossRef]
- Siemeister, G.; Mengel, A.; Fernández-Montalván, A.E.; Bone, W.; Schröder, J.; Zitzmann-Kolbe, S.; Briem, H.; Prechtl, S.; Holton, S.J.; Mönning, U.; et al. Inhibition of BUB1 Kinase by BAY 1816032 Sensitizes Tumor Cells toward Taxanes, ATR, and PARP Inhibitors In Vitro and In Vivo. Clin. Cancer Res. 2019, 25, 1404–1414. [Google Scholar] [CrossRef]
- Thoidingjam, S.; Sriramulu, S.; Hassan, O.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Gadgeel, S.; Nyati, S. BUB1 inhibition sensitizes lung cancer cell lines to radiotherapy and chemoradiotherapy. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Li, P.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Green, M.; Speers, C.; Nyati, S. Abstract 689: BUB1 interferes with the repair of radiation-induced DNA damage to radiosensitize triple-negative breast cancer. Cancer Res. 2024, 84, 689. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Padella, A.; Ghelli Luserna Di Rorà, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Kyndi, M.; Sorensen, F.B.; Knudsen, H.; Overgaard, M.; Nielsen, H.M.; Overgaard, J.; Danish Breast Cancer Cooperative, G. Estrogen receptor, progesterone receptor, HER-2, and response to postmastectomy radiotherapy in high-risk breast cancer: The Danish Breast Cancer Cooperative Group. J. Clin. Oncol. 2008, 26, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.P.; Harper, A.; Malcolm, J.; McAndrews, M.S.; Mockus, S.M.; Patterson, S.E.; Reynolds, T.; Baker, E.J.; Bult, C.J.; Chesler, E.J.; et al. Cisplatin-resistant triple-negative breast cancer subtypes: Multiple mechanisms of resistance. BMC Cancer 2019, 19, 1039. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Sun, L.; Gai, J.; Cao, Y.; Zhang, S. Exploring the resistance mechanism of triple-negative breast cancer to paclitaxel through the scRNA-seq analysis. PLoS ONE 2024, 19, e0297260. [Google Scholar] [CrossRef] [PubMed]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. J. Natl. Cancer Inst. 2018, 110, 855–862. [Google Scholar] [CrossRef]
- Langelier, M.F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014, 42, 7762–7775. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef]
- Li, J.; Qiu, J.; Han, J.; Li, X.; Jiang, Y. Tumor Microenvironment Characterization in Breast Cancer Identifies Prognostic Pathway Signatures. Genes 2022, 13, 1976. [Google Scholar] [CrossRef]
- Singh, D.D.; Yadav, D.K. TNBC: Potential Targeting of Multiple Receptors for a Therapeutic Breakthrough, Nanomedicine, and Immunotherapy. Biomedicines 2021, 9, 876. [Google Scholar] [CrossRef] [PubMed]
- Marijon, H.; Lee, D.H.; Ding, L.; Sun, H.; Gery, S.; de Gramont, A.; Koeffler, H.P. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed. Pharmacother. 2018, 99, 543–551. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Pesch, A.M.; Moubadder, L.; Chandler, B.C.; Wilder-Romans, K.; Cameron, M.; Olsen, E.; Thomas, D.G.; Zhang, A.; Hirsh, N.; et al. PARP1 Inhibition Radiosensitizes Models of Inflammatory Breast Cancer to Ionizing Radiation. Mol. Cancer Ther. 2019, 18, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Loap, P.; Loirat, D.; Berger, F.; Rodrigues, M.; Bazire, L.; Pierga, J.-Y.; Vincent-Salomon, A.; Laki, F.; Boudali, L.; Raizonville, L.; et al. Concurrent Olaparib and Radiotherapy in Patients With Triple-Negative Breast Cancer: The Phase 1 Olaparib and Radiation Therapy for Triple-Negative Breast Cancer Trial. JAMA Oncol. 2022, 8, 1802–1808. [Google Scholar] [CrossRef]
- Bellon, J.R.; Chen, Y.-H.; Rees, R.; Taghian, A.G.; Wong, J.S.; Punglia, R.S.; Shiloh, R.Y.; Warren, L.E.G.; Krishnan, M.S.; Phillips, J.; et al. A Phase 1 Dose-Escalation Trial of Radiation Therapy and Concurrent Cisplatin for Stage II and III Triple-Negative Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Nyati, S. Molecular targets that sensitize cancer to radiation killing: From the bench to the bedside. Biomed. Pharmacother. 2022, 158, 114126. [Google Scholar] [CrossRef]
- Kong, A.; Good, J.; Kirkham, A.; Savage, J.; Mant, R.; Llewellyn, L.; Parish, J.; Spruce, R.; Forster, M.; Schipani, S.; et al. Phase I trial of WEE1 inhibition with chemotherapy and radiotherapy as adjuvant treatment, and a window of opportunity trial with cisplatin in patients with head and neck cancer: The WISTERIA trial protocol. BMJ Open 2020, 10, e033009. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Tung, N.; Garber, J.E. PARP inhibition in breast cancer: Progress made and future hopes. NPJ Breast Cancer 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, M.; Fujita, M.; Karasawa, K. Combining Carbon-Ion Irradiation and PARP Inhibitor, Olaparib Efficiently Kills BRCA1-Mutated Triple-Negative Breast Cancer Cells. Breast Cancer 2022, 16, 11782234221080553. [Google Scholar] [CrossRef]
- Feng, F.Y.; Speers, C.; Liu, M.; Jackson, W.C.; Moon, D.; Rinkinen, J.; Wilder-Romans, K.; Jagsi, R.; Pierce, L.J. Targeted radiosensitization with PARP1 inhibition: Optimization of therapy and identification of biomarkers of response in breast cancer. Breast Cancer Res. Treat. 2014, 147, 81–94. [Google Scholar] [CrossRef]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef]
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in PARP Inhibitors for the Treatment of Ovarian Cancer: Mechanisms of Action, Pharmacology, Safety, and Efficacy. Int. J. Mol. Sci. 2021, 22, 4203. [Google Scholar] [CrossRef]
- Xia, Q.; Cai, Y.; Peng, R.; Wu, G.; Shi, Y.; Jiang, W. The CDK1 inhibitor RO3306 improves the response of BRCA-pro fi cient breast cancer cells to PARP inhibition. Int. J. Oncol. 2014, 44, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Jang, H.; Kim, S.; Lee, K.H.; Kim, D.K.; Suh, K.J.; Yang, Y.; Elvin, P.; O’Connor, M.J.; Im, S.A. Androgen Receptor Inhibitor Enhances the Antitumor Effect of PARP Inhibitor in Breast Cancer Cells by Modulating DNA Damage Response. Mol. Cancer Ther. 2018, 17, 2507–2518. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Weng, X.; Xu, D.; Cai, S.; Lou, H.; Ding, L. Non-small cell lung cancer cells with deficiencies in homologous recombination genes are sensitive to PARP inhibitors. Biochem. Biophys. Res. Commun. 2020, 522, 121–126. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Hamilton, T.C.; O’Dwyer, P.J.; Ozols, R.F. Cisplatin and its analogues. In Cancer: Principles and Practice of Oncology; DeVita, V.T., Rosenberg, S.A., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1997; pp. 467–483. [Google Scholar]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef]
- Horwitz, S.B. Taxol (paclitaxel): Mechanisms of action. Ann. Oncol. 1994, 5 (Suppl. S6), S3–S6. [Google Scholar]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef]
- Dibra, D.; Moyer, S.M.; El-Naggar, A.K.; Qi, Y.; Su, X.; Lozano, G. Triple-negative breast tumors are dependent on mutant p53 for growth and survival. Proc. Natl. Acad. Sci. USA 2023, 120, e2308807120. [Google Scholar] [CrossRef]
- Lee, H.; Trainer, A.H.; Friedman, L.S.; Thistlethwaite, F.C.; Evans, M.J.; Ponder, B.A.; Venkitaraman, A.R. Mitotic checkpoint inactivation fosters transformation in cells lacking the breast cancer susceptibility gene, Brca2. Mol. Cell 1999, 4, 1–10. [Google Scholar] [CrossRef]
- Liu, J.; Adhav, R.; Miao, K.; Su, S.M.; Mo, L.; Chan, U.I.; Zhang, X.; Xu, J.; Li, J.; Shu, X.; et al. Characterization of BRCA1-deficient premalignant tissues and cancers identifies Plekha5 as a tumor metastasis suppressor. Nat. Commun. 2020, 11, 4875. [Google Scholar] [CrossRef]
- Wang, R.H.; Yu, H.; Deng, C.X. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proc. Natl. Acad. Sci. USA 2004, 101, 17108–17113. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sriramulu, S.; Thoidingjam, S.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Walker, E.; Nyati, S. BUB1 Inhibition Sensitizes TNBC Cell Lines to Chemotherapy and Radiotherapy. Biomolecules 2024, 14, 625. https://doi.org/10.3390/biom14060625
Sriramulu S, Thoidingjam S, Siddiqui F, Brown SL, Movsas B, Walker E, Nyati S. BUB1 Inhibition Sensitizes TNBC Cell Lines to Chemotherapy and Radiotherapy. Biomolecules. 2024; 14(6):625. https://doi.org/10.3390/biom14060625
Chicago/Turabian StyleSriramulu, Sushmitha, Shivani Thoidingjam, Farzan Siddiqui, Stephen L. Brown, Benjamin Movsas, Eleanor Walker, and Shyam Nyati. 2024. "BUB1 Inhibition Sensitizes TNBC Cell Lines to Chemotherapy and Radiotherapy" Biomolecules 14, no. 6: 625. https://doi.org/10.3390/biom14060625