
Genome-Wide Identification of Specific Genetic Loci Common to Sheep and Goat

 , and
, and
Abstract
:1. Introduction
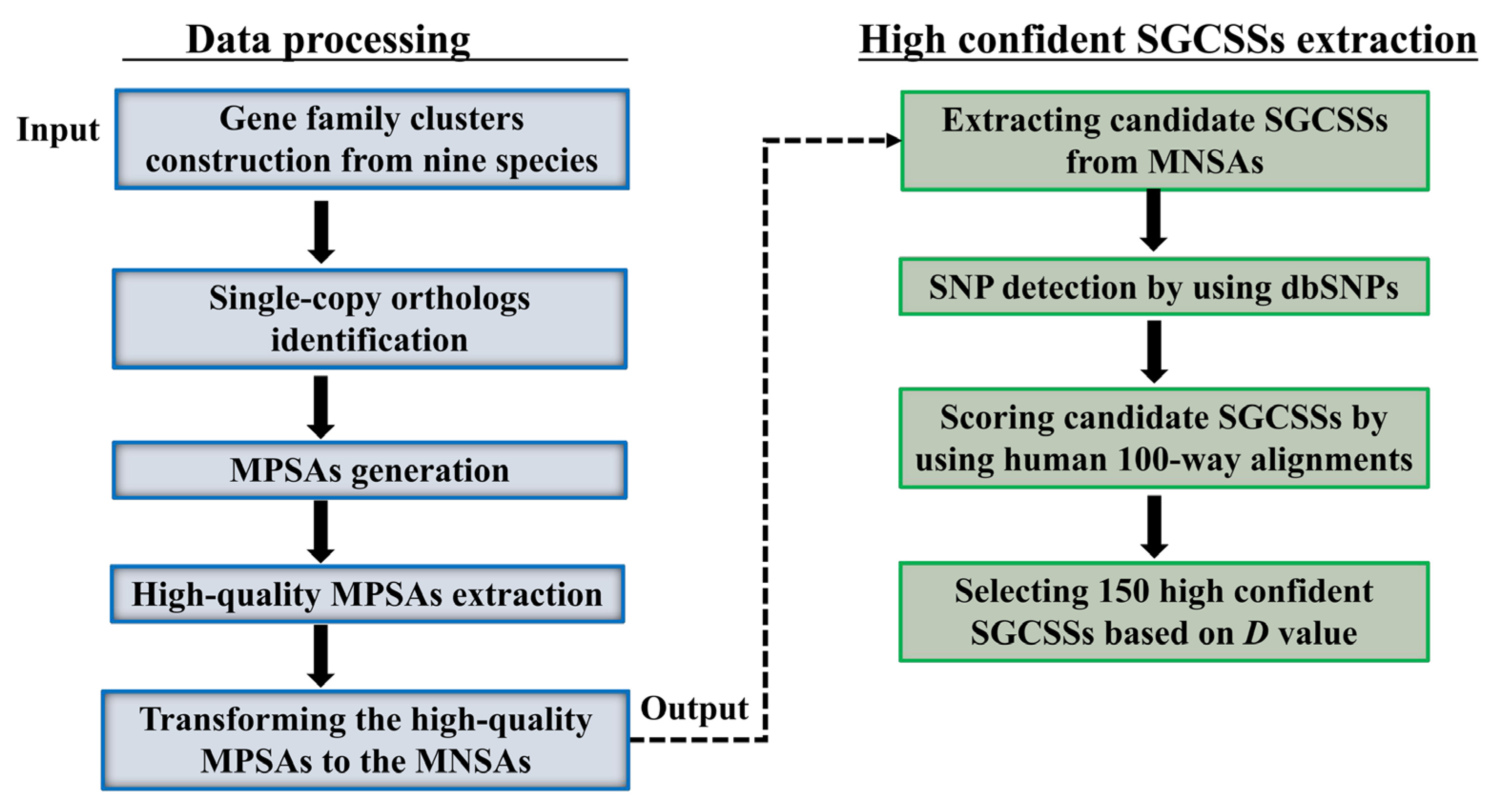
2. Materials and Methods
2.1. Data Collection
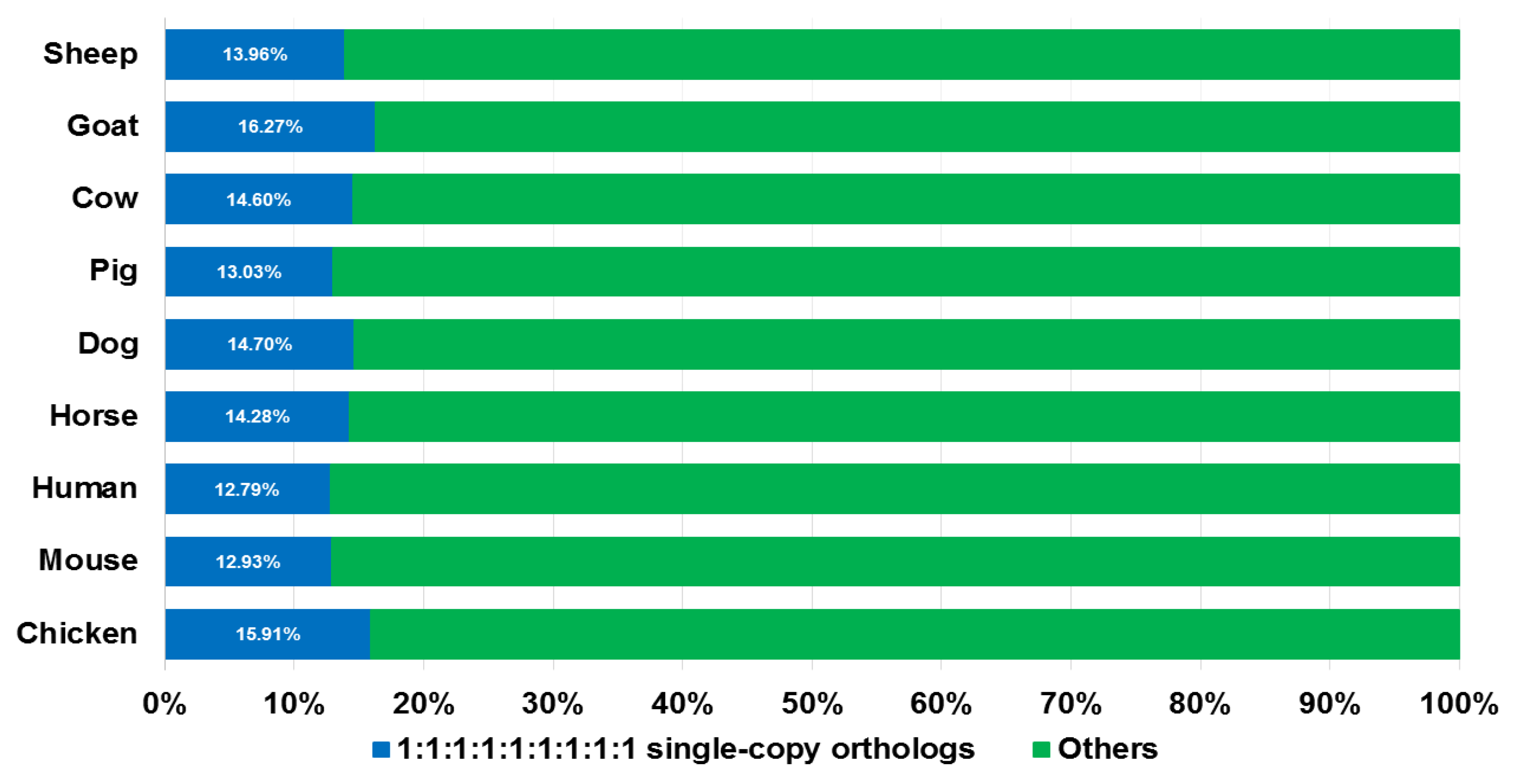
2.2. Gene Family Clusters Construction
2.3. Sequence Alignment Processing
2.4. Searching Candidate SGCSSs
2.5. Scoring Candidate SGCSSs
2.6. The Specificity of SGCSS Analysis
2.7. Tests for Selection Signals
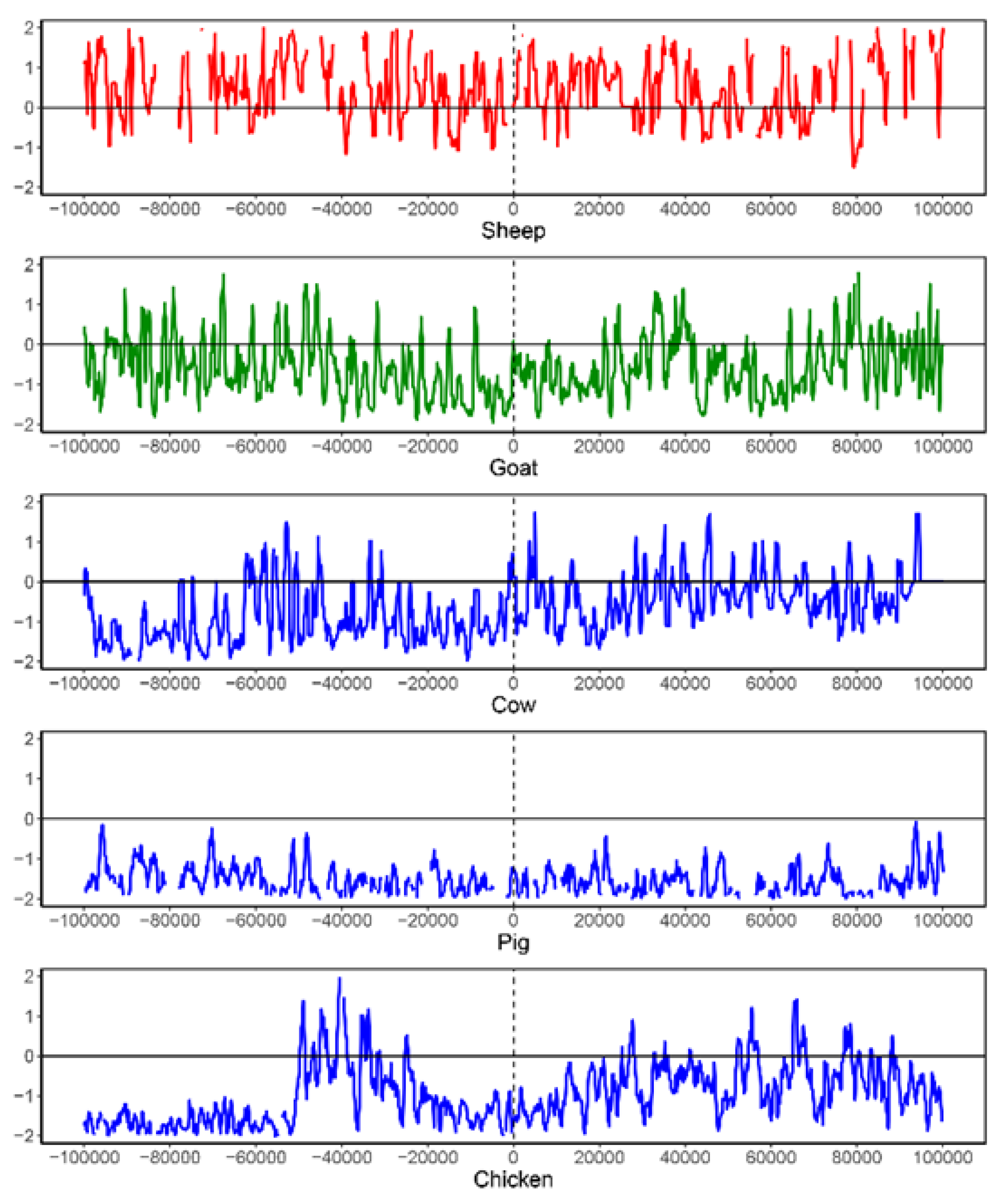
3. Results and Discussion
3.1. Identification of the Highly Confident SGCSSs
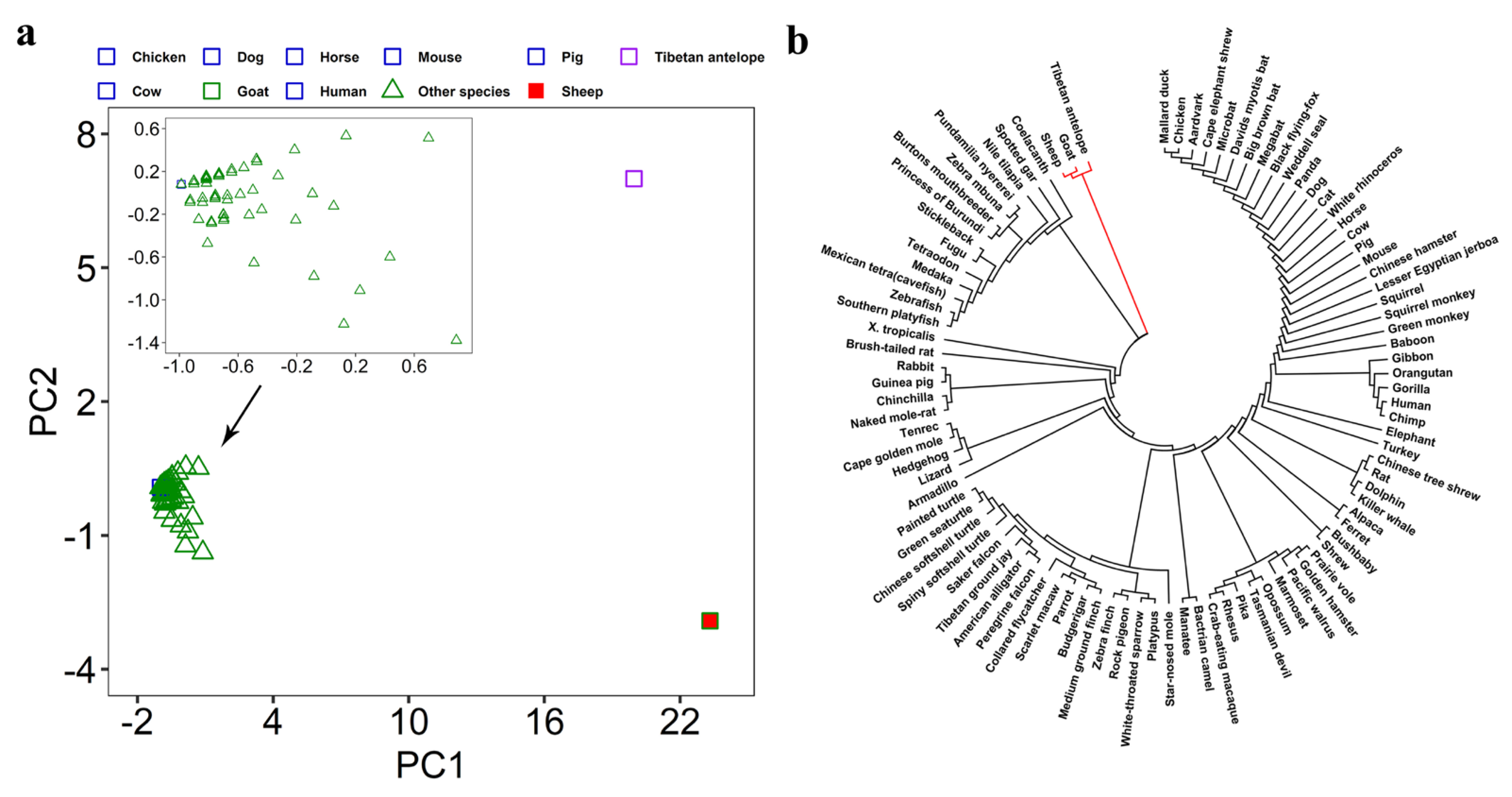
3.2. Evaluation of Intraspecific Heterogeneity
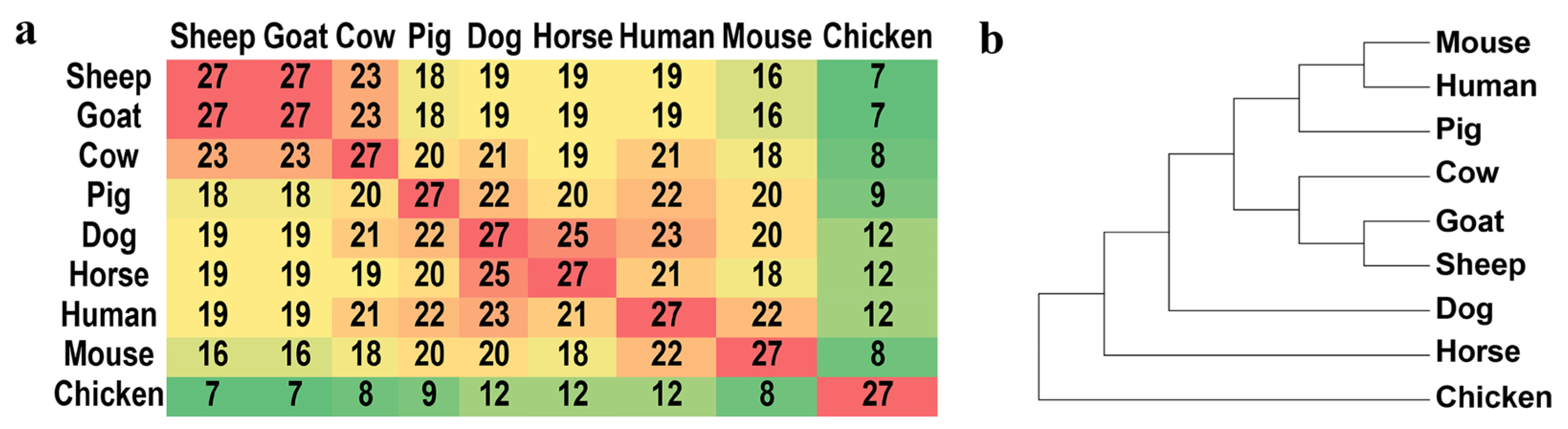
3.3. Evaluation of Interspecific Divergence
3.4. Characterization of Selected SGCSSs
3.5. Case Study for Functionality of SGCSSs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tedeschi, L.O.; Cannas, A.; Fox, D.G. A nutrition mathematical model to account for dietary supply and requirements of energy and other nutrients for domesticated small ruminants: The development and evaluation of the Small Ruminant Nutrition System. Small Rumin. Res. 2010, 89, 174–184. [Google Scholar] [CrossRef]
- Alberto, F.J.; Boyer, F.; Orozco-terWengel, P.; Streeter, I.; Servin, B.; de Villemereuil, P.; Benjelloun, B.; Librado, P.; Biscarini, F.; Colli, L.; et al. Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 2018, 9, 813. [Google Scholar] [CrossRef] [PubMed]
- Hammer, R.E.; Pursel, V.G.; Rexroad, C.E., Jr.; Wall, R.J.; Bolt, D.J.; Ebert, K.M.; Palmiter, R.D.; Brinster, R.L. Production of transgenic rabbits, sheep and pigs by microinjection. Nature 1985, 315, 680–683. [Google Scholar] [CrossRef]
- Shumbusho, F.; Raoul, J.; Astruc, J.M.; Palhiere, I.; Lemarie, S.; Fugeray-Scarbel, A.; Elsen, J.M. Economic evaluation of genomic selection in small ruminants: A sheep meat breeding program. Animal 2016, 10, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, D.; Li, G.; Angeles, C.F.; Patterson, J.T.; Huang, L.Y. A large animal neuropathic pain model in sheep: A strategy for improving the predictability of preclinical models for therapeutic development. J. Pain Res. 2012, 5, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Ganter, M. Zoonotic risks from small ruminants. Vet. Microbiol. 2015, 181, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.R.; Will, R.; Ironside, J.; Nguyen, H.O.; Tremblay, P.; DeArmond, S.J.; Prusiner, S.B. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc. Natl. Acad. Sci. USA 1999, 96, 15137–15142. [Google Scholar] [CrossRef] [PubMed]
- Padilla, D.; Beringue, V.; Espinosa, J.C.; Andreoletti, O.; Jaumain, E.; Reine, F.; Herzog, L.; Gutierrez-Adan, A.; Pintado, B.; Laude, H.; et al. Sheep and Goat BSE Propagate More Efficiently than Cattle BSE in Human PrP Transgenic Mice. PLoS Pathog. 2011, 7, e1001319. [Google Scholar] [CrossRef] [PubMed]
- Reviriego, F.J.; Moreno, M.A.; Dominguez, L. Risk factors for brucellosis seroprevalence of sheep and goat flocks in Spain. Prev. Vet. Med. 2000, 44, 167–173. [Google Scholar] [CrossRef]
- Seleem, M.N.; Boyle, S.M.; Sriranganathan, N. Brucellosis: A re-emerging zoonosis. Vet. Microbiol. 2010, 140, 392–398. [Google Scholar] [CrossRef]
- Wareth, G.; Melzer, F.; Tomaso, H.; Roesler, U.; Neubauer, H. Detection of Brucella abortus DNA in aborted goats and sheep in Egypt by real-time PCR. BMC Res. Notes 2015, 8, 212. [Google Scholar] [CrossRef]
- Atig, R.K.; Hsouna, S.; Beraud-Colomb, E.; Abdelhak, S. Mitochondrial DNA: Properties and applications. Arch. De L’Institut Pasteur De Tunis 2009, 86, 3–14. [Google Scholar]
- Hebert, P.D.N.; Ratnasingham, S.; deWaard, J.R. Barcoding animal life: Cytochrome oxidase subunit 1 divergences among closely related species. Proc. R. Soc. B Boil. Sci. 2003, 270, S96–S99. [Google Scholar] [CrossRef]
- Haider, N.; Nabulsi, I.; Al-Safadi, B. Identification of meat species by PCR-RFLP of the mitochondrial COI gene. Meat Sci. 2012, 90, 490–493. [Google Scholar] [CrossRef]
- Jahura, F.; Munira, S.; Bhuiyan, A.; Hoque, M.; Bhuiyan, M. Molecular detection of goat and sheep meat origin using mitochondrial cytochrome b gene. Bangladesh J. Anim. Sci. 2016, 45, 41–45. [Google Scholar] [CrossRef]
- Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; Gabriel, S.B.; et al. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Abatepaulo, A.R.R.; Abbey, C.A.; Adelson, D.L.; Aerts, J.; Ahola, V.; Alexander, L.; et al. The Genome Sequence of Taurine Cattle: A Window to Ruminant Biology and Evolution. Science 2009, 324, 522–528. [Google Scholar]
- Groenen, M.A.M.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fan, W.; Tian, G.; Zhu, H.; He, L.; Cai, J.; Huang, Q.; Cai, Q.; Li, B.; Bai, Y.; et al. The sequence and de novo assembly of the giant panda genome. Nature 2010, 463, 311–317. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Li, R.; Li, Y.; Tian, G.; Goodman, L.; Fan, W.; Zhang, J.; Li, J.; Zhang, J.; et al. The diploid genome sequence of an Asian individual. Nature 2008, 456, 60–65. [Google Scholar] [CrossRef]
- Green, R.E.; Braun, E.L.; Armstrong, J.; Earl, D.; Nguyen, N.; Hickey, G.; Vandewege, M.W.; St John, J.A.; Capella-Gutierrez, S.; Castoe, T.A.; et al. Three crocodilian genomes reveal ancestral patterns of evolution among archosaurs. Science 2014, 346, 1254449. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Kosiol, C.; Vinar, T.; da Fonseca, R.R.; Hubisz, M.J.; Bustamante, C.D.; Nielsen, R.; Siepel, A. Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008, 4, e1000144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, Q.; Wang, Y.; Luo, L.; Yang, J.; Yang, L.; Liu, M.; Li, Y.; Qian, T.; Zheng, Y.; et al. Gekko japonicus genome reveals evolution of adhesive toe pads and tail regeneration. Nat. Commun. 2015, 6, 10033. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Guang, X.; Al-Fageeh, M.B.; Cao, J.; Pan, S.; Zhou, H.; Zhang, L.; Abutarboush, M.H.; Xing, Y.; Xie, Z.; et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 2014, 5, 5188. [Google Scholar] [CrossRef] [PubMed]
- Köppel, R.; Daniels, M.; Felderer, N.; Brünen-Nieweler, C. Multiplex real-time PCR for the detection and quantification of DNA from duck, goose, chicken, turkey and pork. Eur. Food Res. Technol. 2013, 236, 1093–1098. [Google Scholar] [CrossRef]
- Laube, I.; Zagon, J.; Broll, H. Quantitative determination of commercially relevant species in foods by real-time PCR. Int. J. Food Sci. Technol. 2007, 42, 336–341. [Google Scholar] [CrossRef]
- Li, H.; Coghlan, A.; Ruan, J.; Coin, L.J.; Hériché, J.K.; Osmotherly, L.; Li, R.Q.; Liu, T.; Zhang, Z.; Bolund, L.; et al. TreeFam: A curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 2006, 34, D572–D580. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xie, M.; Chen, W.B.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.H.; Muzny, D.M.; Li, Y.X.; Zhang, W.G.; et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef]
- Stajich, J.E.; Block, D.; Boulez, K.; Brenner, S.E.; Chervitz, S.A.; Dagdigian, C.; Fuellen, G.; Gilbert, J.G.R.; Korf, I.; Lapp, H.; et al. The bioperl toolkit:: Perl modules for the life sciences. Genome Res. 2002, 12, 1611–1618. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Liu, K.; Raghavan, S.; Nelesen, S.; Linder, C.R.; Warnow, T. Rapid and Accurate Large-Scale Coestimation of Sequence Alignments and Phylogenetic Trees. Science 2009, 324, 1561–1564. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Olsen, H.G.; Knutsen, T.M.; Kohler, A.; Svendsen, M.; Gidskehaug, L.; Grove, H.; Nome, T.; Sodeland, M.; Sundsaasen, K.K.; Kent, M.P.; et al. Genome-wide association mapping for milk fat composition and fine mapping of a QTL for de novo synthesis of milk fatty acids on bovine chromosome 13. Genet. Sel. Evol. 2017, 49, 20. [Google Scholar] [CrossRef] [PubMed]
- Puig-Oliveras, A.; Revilla, M.; Castelló, A.; Fernández, A.I.; Folch, J.M.; Ballester, M. Expression-based GWAS identifies variants, gene interactions and key regulators affecting intramuscular fatty acid content and composition in porcine meat. Sci. Rep. 2016, 6, 31803. [Google Scholar] [CrossRef]
- Singh, K.; Davis, S.R.; Dobson, J.M.; Molenaar, A.J.; Wheeler, T.T.; Prosser, C.G.; Farr, V.C.; Oden, K.; Swanson, K.M.; Phyn, C.V.C.; et al. cDNA microarray analysis reveals that antioxidant and immune genes are upregulated during involution of the bovine mammary gland. J. Dairy Sci. 2008, 91, 2236–2246. [Google Scholar] [CrossRef]
- Sturm, R.A.; Duffy, D.L. Human pigmentation genes under environmental selection. Genome Biol. 2012, 13, 248. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Before | After | Filter Factors |
|---|---|---|
| - | 3245 | Non-candidate SGCSSs |
| 3245 | 3230 | No genomic coordinates |
| 3230 | 2136 | SNPs observed in dpSNP |
| 2136 | 1158 | No records or quality control (human 100-way alignments) |
| 1158 | 150 | D value |
| Chr | Position | Genotype1 | Genotype2 | Sheep | Goat | Cow | Pig | Chicken |
|---|---|---|---|---|---|---|---|---|
| 5 | 47181590 | GG, CC | AA, TT | 1 | 1 | 1 | 0.984 | 1 |
| 12 | 10283861 | TT | GG, CC | 1 | 1 | 1 | 1 | 0.993 |
| 12 | 40543138 | AA | GG, CC | 1 | 1 | 1 | 1 | 0.993 |
| 21 | 39262113 | CC | AA, TT | 0.95 | 1 | 1 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Z.; Yue, X.; Liu, Y.; Ye, M.; Zhong, L.; Luan, Y.; Wang, Q. Genome-Wide Identification of Specific Genetic Loci Common to Sheep and Goat. Biomolecules 2024, 14, 638. https://doi.org/10.3390/biom14060638
Liang Z, Yue X, Liu Y, Ye M, Zhong L, Luan Y, Wang Q. Genome-Wide Identification of Specific Genetic Loci Common to Sheep and Goat. Biomolecules. 2024; 14(6):638. https://doi.org/10.3390/biom14060638
Chicago/Turabian StyleLiang, Zuoxiang, Xiaoyu Yue, Yangxiu Liu, Mengyan Ye, Ling Zhong, Yue Luan, and Qin Wang. 2024. "Genome-Wide Identification of Specific Genetic Loci Common to Sheep and Goat" Biomolecules 14, no. 6: 638. https://doi.org/10.3390/biom14060638
APA StyleLiang, Z., Yue, X., Liu, Y., Ye, M., Zhong, L., Luan, Y., & Wang, Q. (2024). Genome-Wide Identification of Specific Genetic Loci Common to Sheep and Goat. Biomolecules, 14(6), 638. https://doi.org/10.3390/biom14060638