
Kinetic Behavior of Glutathione Transferases: Understanding Cellular Protection from Reactive Intermediates


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Kinetic Mechanism Studies
3. Binding Function
4. Discussion
4.1. Random Sequential Kinetic Mechanism
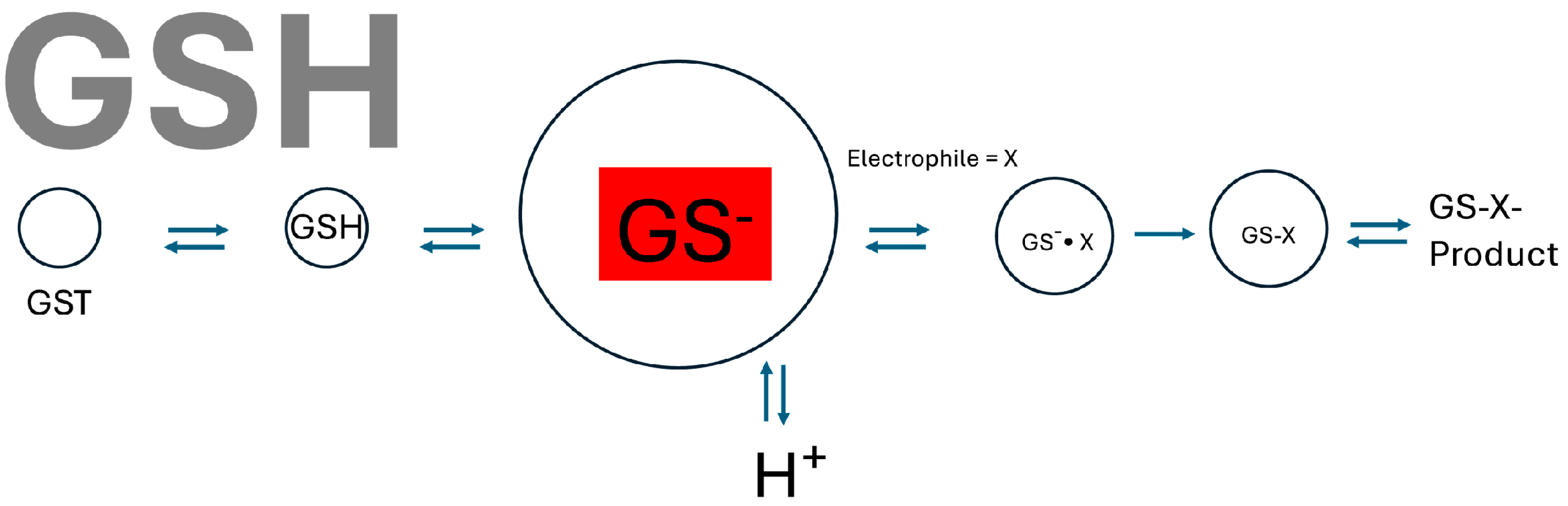
4.2. Co-Operativity
4.3. Partial Sites Reactivity
4.4. Covalent Binding
4.5. Spatial Aspects
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione Transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, B.; Danielson, U.H. Glutathione transferases--structure and catalytic activity. CRC Crit. Rev. Biochem. 1988, 23, 283–337. [Google Scholar] [CrossRef]
- Cheng, J.Z.; Sharma, R.; Yang, Y.; Singhal, S.S.; Sharma, A.; Saini, M.K.; Singh, S.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Accelerated metabolism and exclusion of 4-hydroxynonenal through induction of RLIP76 and hGST5.8 is an early adaptive response of cells to heat and oxidative stress. J. Biol. Chem. 2001, 276, 41213–41223. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Baez, S.; Widersten, M.; Welch, C.J.; Mannervik, B. Human Class Mu Glutathione Transferases, In Particular Isoenzyme M2-2, Catalyze Detoxication Of the Dopamine Metabolite Aminochrome. J. Biol. Chem. 1997, 272, 5727–5731. [Google Scholar] [CrossRef] [PubMed]
- Arias, I.M.; Ohmi, N.; Bhargava, M.; Listowsky, I. Ligandin: An adventure in liverland. Mol. Cell Biochem. 1980, 29, 71–80. [Google Scholar] [CrossRef] [PubMed]
- DePierre, J.W.; Morgenstern, R. Comparison of the distribution of microsomal and cytosolic glutathione S-transferase activities in different organs of the rat. Biochem. Pharmacol. 1983, 32, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, R.; Guthenberg, C.; Mannervik, B.; DePierre, J.W. The amount and nature of of glutathione transferases in rat liver microsomes determined by immunochemical methods. FEBS Lett. 1983, 160, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Askelöf, P.; Guthenberg, C.; Jakobson, I.; Mannervik, B. Purification and characterization of two glutathione S-aryltransferase activities from rat liver. Biochem. J. 1975, 147, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Chasseaud, L.F. The role of glutathione and glutathione S-transferases in the metabolism of chemical carcinogens and other electrophilic agents. Adv. Cancer Res. 1979, 29, 175–274. [Google Scholar]
- Jernström, B.; Gräslund, A. Covalent binding of benzo[a]pyrene 7,8-dihydrodiol 9,10-epoxides to DNA: Molecular structures, induced mutations and biological consequences. Biophys. Chem. 1994, 49, 185–199. [Google Scholar] [CrossRef]
- Tipping, E.; Ketterer, B.; Christodoulides, L.; Enderby, G. The non-convalent binding of small molecules by ligandin. Interactions with steroids and their conjugates, fatty acids, bromosulphophthalein, carcinogens, glutathione and related compounds. Eur. J. Biochem. 1976, 67, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.G.; Hayes, J.D. Mechanisms of induction of cytosolic and microsomal glutathione transferase (GST) genes by xenobiotics and pro-inflammatory agents. Drug Metab. Rev. 2011, 43, 92–137. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Strange, R.C. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology 2000, 61, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Mazari, A.M.A.; Zhang, L.; Ye, Z.W.; Zhang, J.; Tew, K.D.; Townsend, D.M. The Multifaceted Role of Glutathione S-Transferases in Health and Disease. Biomolecules 2023, 13, 688. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, J.R. The glutathione peroxidase activity of glutathione S-transferases. Biochim. Biophys. Acta 1980, 611, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Mosialou, E.; Piemonte, F.; Andersson, C.; Vos, R.; Van Bladeren, P.J.; Morgenstern, R. Microsomal glutathione transferase—Lipid-derived substrates and lipid dependence. Arch. Biochem. Biophys. 1995, 320, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Pabst, M.J.; Habig, W.H.; Jakoby, W.B. Glutathione S-transferase A. A novel kinetic mechanism in which the major reaction pathway depends on substrate concentration. J. Biol. Chem. 1974, 249, 7140–7147. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, B.; Askelöf, P. Absence of a ping-pong pathway in the kinetic mechanism of glutathione S-transferase A from rat liver. Evidence based on quantitative comparison of the asymptotic properties of experimental data and alternative rat equations. FEBS Lett. 1975, 56, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Arbildi, P.; Turell, L.; López, V.; Alvarez, B.; Fernández, V. Mechanistic insights into EgGST1, a Mu class glutathione S-transferase from the cestode parasite Echinococcus granulosus. Arch. Biochem. Biophys. 2017, 633, 15–22. [Google Scholar] [CrossRef]
- Chang, G.G.; Tsai, L.N.; Tang, S.S.; Wang, T.C. Purification and kinetic mechanism of the glutathione S-transferase from C6/36, an Aedes albopictus cell line. Arch. Biochem. Biophys. 1994, 310, 134–143. [Google Scholar] [CrossRef]
- Jakobson, I.; Askelöf, P.; Warholm, M.; Mannervik, B. A steady-state-kinetic random mechanism for glutathione S-transferase A from rat liver. A model involving kinetically significant enzyme-product complexes in the forward reaction. Eur. J. Biochem. 1977, 77, 253–262. [Google Scholar] [CrossRef]
- Jakobson, I.; Warholm, M.; Mannervik, B. The binding of substrates and a product of the enzymatic reaction to glutathione S-transferase A. J. Biol. Chem. 1979, 254, 7085–7089. [Google Scholar] [CrossRef]
- Jakobson, I.; Warholm, M.; Mannervik, B. Multiple inhibition of glutathione S-transferase A from rat liver by glutathione derivatives: Kinetic analysis supporting a steady-state random sequential mechanism. Biochem. J. 1979, 177, 861–868. [Google Scholar] [CrossRef]
- Nay, B.; Fournier, D.; Baudras, A.; Baudras, B. Mechanism of an insect glutathione S-transferase: Kinetic analysis supporting a rapid equilibrium random sequential mechanism with housefly I1 isoform. Insect Biochem. Mol. Biol. 1999, 29, 71–79. [Google Scholar] [CrossRef]
- Tang, S.S.; Chang, G.G. Steady-state kinetics and chemical mechanism of octopus hepatopancreatic glutathione transferase. Biochem. J. 1995, 309 Pt 1, 347–353. [Google Scholar] [CrossRef]
- Vorachek, W.R.; Pearson, W.R.; Rule, G.S. Cloning, expression, and characterization of a class-mu glutathione transferase from human muscle, the product of the GST4 locus. Proc. Natl. Acad. Sci. USA 1991, 88, 4443–4447. [Google Scholar] [CrossRef]
- Wang, B.; Peng, Y.; Zhang, T.; Ding, J. Crystal structures and kinetic studies of human Kappa class glutathione transferase provide insights into the catalytic mechanism. Biochem. J. 2011, 439, 215–225. [Google Scholar] [CrossRef]
- Kolawole, A.O. Catalysis of Silver catfish Major Hepatic Glutathione Transferase proceeds via rapid equilibrium sequential random Mechanism. Toxicol. Rep. 2016, 3, 598–607. [Google Scholar] [CrossRef]
- Thumser, A.E.; Ivanetich, K.M. Kinetic mechanism of human erythrocyte acidic isoenzyme rho. Biochim. Biophys. Acta 1993, 1203, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Labrou, N.E.; Karavangeli, M.; Tsaftaris, A.; Clonis, Y.D. Kinetic analysis of maize glutathione S-transferase I catalysing the detoxification from chloroacetanilide herbicides. Planta 2005, 222, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ivanetich, K.M.; Goold, R.D. A rapid equilibrium random sequential bi-bi mechanism for human placental glutathione S-transferase. Biochim. Biophys. Acta 1989, 998, 7–13. [Google Scholar] [CrossRef]
- Ivanetich, K.M.; Goold, R.D.; Sikakana, C.N. Explanation of the non-hyperbolic kinetics of the glutathione S-transferases by the simplest steady-state random sequential Bi Bi mechanism. Biochem. Pharmacol. 1990, 39, 1999–2004. [Google Scholar] [CrossRef]
- Schramm, V.L.; McCluskey, R.; Emig, F.A.; Litwack, G. Kinetic studies and active site-binding properties of glutathione S-transferase using spin-labeled glutathione, a product analogue. J. Biol. Chem. 1984, 259, 714–722. [Google Scholar] [CrossRef]
- Young, P.R.; Briedis, A.V. Purification and kinetic mechanism of the major glutathione S-transferase from bovine brain. Biochem. J. 1989, 257, 541–548. [Google Scholar] [CrossRef]
- Radika, K.; Northrop, D. A new kinetic diagnostic for enzymatic mechanisms using alternative substrates. Anal. Biochem. 1984, 141, 413–417. [Google Scholar] [CrossRef]
- Chen, W.J.; Graminski, G.F.; Armstrong, R.N. Dissection of the catalytic mechanism of isozyme 4-4 of glutathione S-transferase with alternative substrates. Biochemistry 1988, 27, 647–654. [Google Scholar] [CrossRef]
- Andersson, C.; Piemonte, F.; Mosialou, E.; Weinander, R.; Sun, T.H.; Lundqvist, G.; Adang, A.E.; Morgenstern, R. Kinetic studies on rat liver microsomal glutathione transferase: Consequences of activation. Biochim. Biophys. Acta 1995, 1247, 277–283. [Google Scholar] [CrossRef]
- Adang, A.E.P.; Brussee, J.; Meyer, D.J.; Coles, B.; Ketterer, B.; Van Der Gen, A.; Mulder, G.J. Substrate specificity of rat liver glutathione S-transferase isoenzymes for a series of glutathione analogues, modified at the g-glutamyl moiety. Biochem. J. 1988, 255, 721–724. [Google Scholar]
- Adang, A.E.P.; Brussee, J.; Vandergen, A.; Mulder, G.J. The Glutathione-Binding Site in Glutathione S-Transferases—Investigation of the Cysteinyl, Glycyl and Gamma-Glutamyl Domains. Biochem. J. 1990, 269, 47–54. [Google Scholar] [CrossRef]
- Caccuri, A.M.; Antonini, G.; Board, P.G.; Flanagan, J.; Parker, M.W.; Paolesse, R.; Turella, P.; Chelvanayagam, G.; Ricci, G. Human glutathione transferase T2-2 discloses some evolutionary strategies for optimization of the catalytic activity of glutathione transferases. J. Biol. Chem. 2001, 276, 5432–5437. [Google Scholar] [CrossRef]
- Codreanu, S.G.; Ladner, J.E.; Xiao, G.; Stourman, N.V.; Hachey, D.L.; Gilliland, G.L.; Armstrong, R.N. Local protein dynamics and catalysis: Detection of segmental motion associated with rate-limiting product release by a glutathione transferase. Biochemistry 2002, 41, 15161–15172. [Google Scholar] [CrossRef]
- Wolkoff, A.W.; Ketley, J.N.; Waggoner, J.G.; Berk, P.D.; Jakoby, W.B. Hepatic accumulation and intracellular binding of conjugated bilirubin. J. Clin. Investig. 1978, 61, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Alander, J.; Lengqvist, J.; Holm, P.J.; Svensson, R.; Gerbaux, P.; Heuvel, R.H.; Hebert, H.; Griffiths, W.J.; Armstrong, R.N.; Morgenstern, R. Microsomal glutathione transferase 1 exhibits one-third-of-the-sites-reactivity towards glutathione. Arch. Biochem. Biophys. 2009, 487, 42–48. [Google Scholar] [CrossRef]
- Mosialou, E.; Morgenstern, R. Inhibition studies on rat liver microsomal glutathione transferase. Chem. Biol. Interact. 1990, 74, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Bocedi, A.; Fabrini, R.; Lo Bello, M.; Caccuri, A.M.; Federici, G.; Mannervik, B.; Cornish-Bowden, A.; Ricci, G. Evolution of Negative Cooperativity in Glutathione Transferase Enabled Preservation of Enzyme Function. J. Biol. Chem. 2016, 291, 26739–26749. [Google Scholar] [CrossRef]
- Martos-Maldonado, M.C.; Casas-Solvas, J.M.; Téllez-Sanz, R.; Mesa-Valle, C.; Quesada-Soriano, I.; García-Maroto, F.; Vargas-Berenguel, A.; García-Fuentes, L. Binding properties of ferrocene-glutathione conjugates as inhibitors and sensors for glutathione S-transferases. Biochimie 2012, 94, 541–550. [Google Scholar] [CrossRef]
- McManus, G.; Costa, M.; Canals, A.; Coll, M.; Mantle, T.J. Site-directed mutagenesis of mouse glutathione transferase P1-1 unlocks masked cooperativity, introduces a novel mechanism for ‘ping pong’ kinetic behaviour, and provides further structural evidence for participation of a water molecule in proton abstraction from glutathione. FEBS J. 2011, 278, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Niegowski, D.; Wetterholm, A.; Haeggström, J.Z.; Morgenstern, R.; Rinaldo-Matthis, A. Catalytic characterization of human microsomal glutathione S-transferase 2: Identification of rate-limiting steps. Biochemistry 2013, 52, 1755–1764. [Google Scholar] [CrossRef]
- Ahmad, S.; Thulasingam, M.; Palombo, I.; Daley, D.O.; Johnson, K.A.; Morgenstern, R.; Haeggström, J.Z.; Rinaldo-Matthis, A. Trimeric microsomal glutathione transferase 2 displays one third of the sites reactivity. Biochim. Biophys. Acta 2015, 1854, 1365–1371. [Google Scholar] [CrossRef]
- Morgenstern, R.; Svensson, R.; Bernat, B.A.; Armstrong, R.N. Kinetic analysis of the slow ionization of glutathione by microsomal glutathione transferase MGST1. Biochemistry 2001, 40, 3378–3384. [Google Scholar] [CrossRef]
- Brown, A.P.; Gandolfi, A.J. Glutathione-S-transferase is a target for covalent modification by a halothane reactive intermediate in the guinea pig liver. Toxicology 1994, 89, 35–47. [Google Scholar] [CrossRef]
- Weis, M.; Morgenstern, R.; Cotgreave, I.A.; Nelson, S.D.; Moldeus, P. N-acetyl-p-benzoquinone imine-induced protein thiol modification in isolated rat hepatocytes. Biochem. Pharmacol. 1992, 43, 1493–1505. [Google Scholar] [CrossRef]
- Morgenstern, R.; DePierre, J.W.; Ernster, L. Activation of microsomal glutathione transferase activity by sulfhydryl reagents. Biochem. Biophys. Res. Commun. 1979, 87, 657–663. [Google Scholar] [CrossRef]
- van Ommen, B.; Bogaards, J.J.; Peters, W.H.; Blaauboer, B.; van Bladeren, P.J. Quantification of human hepatic glutathione S-transferases. Biochem. J. 1990, 269, 609–613. [Google Scholar] [CrossRef]
- Seutter-Berlage, F.; van Dorp, H.L.; Kosse, H.G.; Henderson, P.T. Urinary mercapturic acid excretion as a biological parameter of exposure to alkylating agents. Int. Arch. Occup. Environ. Health 1977, 39, 45–51. [Google Scholar] [CrossRef]
- Pirie, N.W.; Godwin Pinhey, K. The titration curve of glutathione. J. Biol. Chem. 1929, 84, 321–333. [Google Scholar] [CrossRef]
- Coles, B.; Wilson, I.; Wardman, P.; Hinson, J.A.; Nelson, S.D.; Ketterer, B. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: A stopped-flow kinetic study. Arch. Biochem. Biophys. 1988, 264, 253–260. [Google Scholar] [CrossRef]
- Rinaldi, R.; Eliasson, E.; Swedmark, S.; Morgenstern, R. Reactive intermediates and the dynamics of glutathione transferases. Drug Metab. Dispos. 2002, 30, 1053–1058. [Google Scholar] [CrossRef]
- Deponte, M. The Incomplete Glutathione Puzzle: Just Guessing at Numbers and Figures? Antioxid. Redox Signal. 2017, 27, 1130–1161. [Google Scholar] [CrossRef]
- Geenen, S.; du Preez, F.B.; Snoep, J.L.; Foster, A.J.; Sarda, S.; Kenna, J.G.; Wilson, I.D.; Westerhoff, H.V. Glutathione metabolism modeling: A mechanism for liver drug-robustness and a new biomarker strategy. Biochim. Biophys. Acta 2013, 1830, 4943–4959. [Google Scholar] [CrossRef]
- Reed, M.C.; Thomas, R.L.; Pavisic, J.; James, S.J.; Ulrich, C.M.; Nijhout, H.F. A mathematical model of glutathione metabolism. Theor. Biol. Med. Model. 2008, 5, 8. [Google Scholar] [CrossRef]
- Ben-Shachar, R.; Chen, Y.; Luo, S.; Hartman, C.; Reed, M.; Nijhout, H.F. The biochemistry of acetaminophen hepatotoxicity and rescue: A mathematical model. Theor. Biol. Med. Model. 2012, 9, 55. [Google Scholar] [CrossRef]
- Lee, H.C.; Toung, Y.P.; Tu, Y.S.; Tu, C.P. A molecular genetic approach for the identification of essential residues in human glutathione S-transferase function in Escherichia coli. J. Biol. Chem. 1995, 270, 99–109. [Google Scholar] [CrossRef]
- McLellan, R.A.; Oscarson, M.; Alexandrie, A.K.; Seidegård, J.; Evans, D.A.; Rannug, A.; Ingelman-Sundberg, M. Characterization of a human glutathione S-transferase mu cluster containing a duplicated GSTM1 gene that causes ultrarapid enzyme activity. Mol. Pharmacol. 1997, 52, 958–965. [Google Scholar] [CrossRef]
- Caccuri, A.M.; Antonini, G.; Board, P.G.; Parker, M.W.; Nicotra, M.; Lo Bello, M.; Federici, G.; Ricci, G. Proton release on binding of glutathione to alpha, Mu and Delta class glutathione transferases. Biochem. J. 1999, 344 Pt 2, 419–425. [Google Scholar] [CrossRef]
- Svensson, R.; Alander, J.; Armstrong, R.N.; Morgenstern, R. Kinetic Characterization of Thiolate Anion Formation and Chemical Catalysis of Activated Microsomal Glutathione Transferase 1. Biochemistry 2004, 43, 8869–8877. [Google Scholar] [CrossRef]
- Tipping, E.; Ketterer, B. The influence of soluble binding proteins on lipophile transport and metabolism in hepatocytes. Biochem. J. 1981, 195, 441–452. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. Quantitative assessment of the multiple processes responsible for bilirubin homeostasis in health and disease. Clin. Exp. Gastroenterol. 2014, 7, 307–328. [Google Scholar] [CrossRef]
- Setchell, K.D.; Rodrigues, C.M.; Clerici, C.; Solinas, A.; Morelli, A.; Gartung, C.; Boyer, J. Bile acid concentrations in human and rat liver tissue and in hepatocyte nuclei. Gastroenterology 1997, 112, 226–235. [Google Scholar] [CrossRef]
- Roda, A.; Minutello, A.; Angellotti, M.A.; Fini, A. Bile acid structure-activity relationship: Evaluation of bile acid lipophilicity using 1-octanol/water partition coefficient and reverse phase HPLC. J. Lipid Res. 1990, 31, 1433–1443. [Google Scholar] [CrossRef]
- Dreij, K.; Chaudhry, Q.A.; Jernstrom, B.; Morgenstern, R.; Hanke, M. A method for efficient calculation of diffusion and reactions of lipophilic compounds in complex cell geometry. PLoS ONE 2011, 6, e23128. [Google Scholar] [CrossRef]
- Turella, P.; Pedersen, J.Z.; Caccuri, A.M.; De Maria, F.; Mastroberardino, P.; Lo Bello, M.; Federici, G.; Ricci, G. Glutathione transferase superfamily behaves like storage proteins for dinitrosyl-diglutathionyl-iron complex in heterogeneous systems. J. Biol. Chem. 2003, 278, 42294–42299. [Google Scholar] [CrossRef]
- Caccuri, A.M.; Antonini, G.; Ascenzi, P.; Nicotra, M.; Nuccetelli, M.; Mazzetti, A.P.; Federici, G.; Lo Bello, M.; Ricci, G. Temperature adaptation of glutathione S-transferase P1-1. A case for homotropic regulation of substrate binding. J. Biol. Chem. 1999, 274, 19276–19280. [Google Scholar] [CrossRef]
- Lien, S.; Gustafsson, A.; Andersson, A.K.; Mannervik, B. Human glutathione transferase A1-1 demonstrates both half-of-the-sites and all-of-the-sites reactivity. J. Biol. Chem. 2001, 276, 35599–35605. [Google Scholar] [CrossRef]
- Thulasingam, M.; Orellana, L.; Nji, E.; Ahmad, S.; Rinaldo-Matthis, A.; Haeggstrom, J.Z. Crystal structures of human MGST2 reveal synchronized conformational changes regulating catalysis. Nat. Commun. 2021, 12, 1728. [Google Scholar] [CrossRef]
- Rinaldo-Matthis, A.; Wetterholm, A.; Martinez Molina, D.; Holm, J.; Niegowski, D.; Ohlson, E.; Nordlund, P.; Morgenstern, R.; Haeggstrom, J.Z. Arginine 104 is a key catalytic residue in leukotriene C4 synthase. J. Biol. Chem. 2010, 285, 40771–40776. [Google Scholar] [CrossRef]
- Ketterer, B. Amino-azo-dye-binding protein in the soluble cytoplasm of the rat liver. Biochem. J. 1971, 122, 53–54. [Google Scholar] [CrossRef]
- Ricci, G.; Caccuri, A.M.; Lo Bello, M.; Parker, M.W.; Nuccetelli, M.; Turella, P.; Stella, L.; Di Iorio, E.E.; Federici, G. Glutathione transferase P1-1: Self-preservation of an anti-cancer enzyme. Biochem. J. 2003, 376, 71–76. [Google Scholar] [CrossRef]
- Shimoji, M.; Aniya, Y.; Morgenstern, R. Activation of Microsomal Glutathione Transferase 1 in Toxicology. In Toxicology of Glutathione Transferases; Awasthi, Y.C., Ed.; CRC Press/Taylor & Francis Group: Boca Raton, FL, USA, 2007; pp. 293–319. [Google Scholar]
- Cebula, M.; Morgenstern, R. Enzymology of reactive intermediate protection: Kinetic analysis and temperature dependence of the mesophilic membrane protein catalyst MGST1. FEBS J. 2023, 290, 3448–3460. [Google Scholar] [CrossRef]
- Morgenstern, R.; DePierre, J.W.; Ernster, L. Reversible activation of microsomal glutathione S-transferase activity by 5,5′-dithiobis(2-nitrobenzoic acid) and 2,2′-dipyridyl disulfide. Acta Chem. Scand. B 1980, 34, 229–230. [Google Scholar] [CrossRef]
- Armstrong, R.N.; Morgenstern, R.; Board, P.G. Glutathione Transferases. In Comprehensive Toxicology, 3rd ed.; McQueen, C.A., Ed.; Elsevier Ltd.: Oxford, UK, 2018; Volume 10, pp. 326–362. [Google Scholar]
- Bennett, C.F.; Yeoman, L.C. Microinjected glutathione S-transferase Yb subunits translocate to the cell nucleus. Biochem. J. 1987, 247, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, R.; Lundqvist, G.; Andersson, G.; Balk, L.; DePierre, J.W. The distribution of microsomal glutathione transferase among different organelles, different organs and different organisms. Biochem. Pharmacol. 1984, 33, 3609–3614. [Google Scholar] [CrossRef] [PubMed]
- Fabrini, R.; Bocedi, A.; Pallottini, V.; Canuti, L.; De Canio, M.; Urbani, A.; Marzano, V.; Cornetta, T.; Stano, P.; Giovanetti, A.; et al. Nuclear shield: A multi-enzyme task-force for nucleus protection. PLoS ONE 2010, 5, e14125. [Google Scholar] [CrossRef] [PubMed]
- Busenlehner, L.S.; Alander, J.; Jegerscohld, C.; Holm, P.J.; Bhakat, P.; Hebert, H.; Morgenstern, R.; Armstrong, R.N. Location of substrate binding sites within the integral membrane protein microsomal glutathione transferase-1. Biochemistry 2007, 46, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Q.; Purhonen, P.; Alander, J.; Svensson, R.; Hoogland, V.; Winerdal, J.; Spahiu, L.; Ottosson-Wadlund, A.; Jegerschold, C.; Morgenstern, R.; et al. Dead-end complex, lipid interactions and catalytic mechanism of microsomal glutathione transferase 1, an electron crystallography and mutagenesis investigation. Sci. Rep. 2017, 7, 7897. [Google Scholar] [CrossRef]
- Hargus, S.J.; Fitzsimmons, M.E.; Aniya, Y.; Anders, M.W. Stereochemistry of the Microsomal Glutathione S-Transferase Catalyzed Addition of Glutathione to Chlorotrifluoroethene. Biochemistry 1991, 30, 717–721. [Google Scholar] [CrossRef]
- Cebula, M.; Turan, I.S.; Sjodin, B.; Thulasingam, M.; Brock, J.; Chmyrov, V.; Widengren, J.; Abe, H.; Mannervik, B.; Haeggstrom, J.Z.; et al. Catalytic Conversion of Lipophilic Substrates by Phase constrained Enzymes in the Aqueous or in the Membrane Phase. Sci. Rep. 2016, 6, 38316. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgenstern, R. Kinetic Behavior of Glutathione Transferases: Understanding Cellular Protection from Reactive Intermediates. Biomolecules 2024, 14, 641. https://doi.org/10.3390/biom14060641
Morgenstern R. Kinetic Behavior of Glutathione Transferases: Understanding Cellular Protection from Reactive Intermediates. Biomolecules. 2024; 14(6):641. https://doi.org/10.3390/biom14060641
Chicago/Turabian StyleMorgenstern, Ralf. 2024. "Kinetic Behavior of Glutathione Transferases: Understanding Cellular Protection from Reactive Intermediates" Biomolecules 14, no. 6: 641. https://doi.org/10.3390/biom14060641