Abstract
Manganese (Mn) is an essential heavy metal in the human body, while excess Mn leads to neurotoxicity, as observed in this study, where 100 µM of Mn was administered to the human neuroblastoma (SH-SY5Y) cell model of dopaminergic neurons in neurodegenerative diseases. We quantitated pathway and gene changes in homeostatic cell-based adaptations to Mn exposure. Utilizing the Gene Expression Omnibus, we accessed the GSE70845 dataset as a microarray of SH-SY5Y cells published by Gandhi et al. (2018) and applied statistical significance cutoffs at p < 0.05. We report 74 pathway and 10 gene changes with statistical significance. ReactomeGSA analyses demonstrated upregulation of histones (5 out of 10 induced genes) and histone deacetylases as a neuroprotective response to remodel/mitigate Mn-induced DNA/chromatin damage. Neurodegenerative-associated pathway changes occurred. NF-κB signaled protective responses via Sirtuin-1 to reduce neuroinflammation. Critically, Mn activated three pathways implicating deficits in purine metabolism. Therefore, we validated that urate, a purine and antioxidant, mitigated Mn-losses of viability in SH-SY5Y cells. We discuss Mn as a hypoxia mimetic and trans-activator of HIF-1α, the central trans-activator of vascular hypoxic mitochondrial dysfunction. Mn induced a 3-fold increase in mRNA levels for antioxidant metallothionein-III, which was induced 100-fold by hypoxia mimetics deferoxamine and zinc.
1. Introduction
The central nervous system is subject to acute, chronic, and latent perturbation by heavy metals, including mercury (Hg), lead (Pb), and manganese (Mn) [1]. The acute neurotoxicity of Hg and Pb is well known. Both of these metals are also implicated in Alzheimer’s disease (AD) [2,3,4]. Mn is associated with several neurodegenerative conditions, including movement disorders such as Parkinson’s disease (PD) [5,6]. There are multiple pleiotropic mechanisms of manganese toxicity within the nervous system [7,8]. At the cellular level, we previously reported pathways by which this divalent cationic metal disrupts translation of the L- and H-subunits of the iron storage multimer ferritin and the Alzheimer’s amyloid β (Aβ) precursor protein (APP) that activates iron efflux by ferroportin (FPN) [7,9,10,11,12]. The contribution of APP to iron homeostasis as a ferroportin-binding protein remains to be solved [9,13]. Nevertheless, perturbations of iron-dependent post-transcriptional regulatory events to both ferritin and APP are associated with damage to neurons by ferroptosis and AD [14,15,16]. Mn inhibited expression of APP at the level of translation via Iron-Responsive Elements (IREs) in 5′untranslated region (5′-UTR) sequences in its transcript, while IREs in other transcripts, such as transferrin receptor, are also potentially important regulatory determinants of AD [7,17]. The APP 5′-UTR IRE is part of a multi-target nexus that also responds to interleukin-1 and microRNA-346 [18]. This action of Mn can be modeled to embargo of iron export sufficient to cause ferroptosis by limiting the export of excess Mn and Fe by FPN [9,16]. Nevertheless, reduced levels of APP would also be predicted to limit Aβ accumulation after cleavage from the precursor, although monomer concentration is set by secretase cleavage and clearance [19,20,21]. Finally, the intracellular losses of both APP and ferritin significantly reduced the viability of neurons during Mn exposure [7].
Mn is an essential trace element at physiological concentrations, acting as a cofactor in many enzymatic reactions in humans, including the antioxidant enzyme superoxide dismutase [22]. Nevertheless, at elevated industrial concentrations, Mn damages the nervous system, causing a neurotoxic ailment that has been documented for about 150 years. Excess manganese (Mn) intake causes manganism, a disease of occupational Mn overexposures. Those with this chronic industrial occupational condition exhibit psychiatric and motor disturbances [23], resulting in symptoms of bradykinesia, rigidity, tremor, gait disturbance, postural instability, and dystonia and/or ataxia [24]. Elevated Mn in environmental water supplies also impairs childhood cognitive performances and IQ in less-developed countries that lack appropriate public water filtration capabilities [25,26], similar to better-known effects of Pb [27]. We reported on biomarkers of environmental manganese exposure and associations with childhood neurodevelopment [26], along with associations between APP and autism [28,29,30,31]. Mn excess is associated with rare cases of Parkinson’s disease (PD) in the field [23], and also results from mutations that prevent the Park-9 protein from detoxifying Mn in affected neurons and a-synuclein fibril formation [32,33].
Depending on the duration and amount of exposure, Mn2+ induces toxic reactive oxygen species in rats [34,35], while its exposure to cultured cells can facilitate toxic cellular Fe overload, since FPN and iron efflux are inhibited [36]. Excess toxic Mn exposure certainly causes motor deficits [37] as well as partial activation of GABAergic neurons in the Globus pallidus and Substantia nigra brain subregions [38]. In order to offset toxic Mn overload, the universal brain iron transporter FPN can function as a manganese exporter [39,40,41].
In seeking a therapy to offset Mn neurotoxicity, we tested the antioxidant purine urate, which exerts a currently undefined protective action towards neurons in cell-based models of Parkinson’s disease (PD) [42]. In this report, we addressed how increased neuronal urate might therapeutically oppose Mn toxicity, since our bioinformatic analysis demonstrated that, first, Mn disrupts purine metabolism via depurination and inhibition of HPRT [43]. In fact, purines are metabolically converted to uric acid by xanthine oxidase, an enzymatic activity that, in excess, causes gout. In the PD brain, however, at physiological pH, the ionized urate salts protect against 1-methyl-4-phenyl-pyridinium (MPP+) degeneration of dopaminergic neurons. In most mammals, urate is converted to allantoin by uricase (uroxidase; UOx), an enzyme primarily expressed in the liver [44,45,46,47]. Urate added to cultured neurons 24 h before and during treatment with MPP+ toxin attenuated the loss of enriched dopaminergic neurons and fully prevented their atrophy in neuron–astrocyte cultures [48]. Uric acid provides an antioxidant defense in humans that has a protective effect against oxidant and radical-caused aging and cancer [49,50]. Similar functions of uric acid and ascorbate were reported in humans [50]. UO knockout mice express brain urate at elevated levels and are less vulnerable than wild-type mice to Parkinsonian-specific neurodegenerative brain lesions [47,51].
In this communication, we provide a compelling rationale to further test urate’s capacity to mitigate manganese-induced liabilities to cell survival, as outlined in the bioinformatic and pilot cell-based data provided. Mn acts as a hypoxia mimetic in several cell types, including SH-SY5Y cells [52,53,54,55]. Therefore, in this report, we also highlighted the microarray-oriented bioinformatic findings, showing that metallothionein-III (MT-III) is a prominent Mn-induced transcript. We discuss MT-III may provide further cell-based antioxidant responses to mitigate Mn-induced ferroptosis and neurotoxicity [54,55,56,57,58].
2. Materials and Methods
2.1. Data Access
The data for the human neuroblastoma cell line (SH-SY5Y) exposed to manganese were accessed using accession GSE70845 in the Gene Expression Omnibus (GEO). This dataset was derived from SH-SY5Y cells that were chronically exposed to 100 µM (1/5 of LD50) of manganese as MnCl2 for 24 h; the experiment was performed in triplicate. Total RNA was harvested using TRIzol, and 100 ng was used to prepare biotinylated complementary RNA (cRNA). The cRNA was hybridized on Affymetrix Prime view Human arrays and scanned using the Affymetrix GeneChip scanner, uploaded to GEO [56].
2.2. Data Exploration
The normalized data were downloaded from GEO and analyzed using the GEO2R bioinformatics software (https://www.ncbi.nlm.nih.gov/geo/geo2r/, accessed on 6 October 2023) [59]. The three Mn exposure samples were assigned to the manganese treatment group, and the three control samples were assigned to the control group. These groups were analyzed using linear models for microarray analysis for statistical visualization [60].
2.3. Pathway and Gene Expression Statistical Analysis
ReactomGSA [61] was used to evaluate pathway and gene expression changes following manganese exposure. The Correlation-Adjusted Mean Rank gene set analysis (CAMERA) [62] was used to conduct a differential expression analysis to determine differentially expressed pathways and genes between two sample groups. The Binomial Test was used to evaluate the statistical significance for each hit pathway and gene analysis and a Benjamini–Hochberg p adjustment was subsequently utilized to manage the false discovery rate. Using the filter of adjusted p < 0.05 as a statistical significance cut-off, the top differentially expressed genes with adjusted p < 0.05 were plotted onto a heatmap and hierarchically clustered using the Morpheus matrix visualization and analysis software (https://software.broadinstitute.org/morpheus/, accessed on 11 October 2023).
2.4. Experimental Validation of Bioinformatic Model
Cell culture methods: The human neuroblastoma cell line SH-SY5Y was obtained from the American Type Culture Collection (ATCC) [63,64].
MTT viability assays: In six separate cotreatment assays, cells were cotreated for 72 h with 100 µM UA and separate increasing dosages of 25 µM Mn as MnCl2, 50 µM Mn as MnCl2, 75 µM Mn as MnCl2, and 100 µM MnCl2. In octuplicate pre-treatment assays, cells were pre-treated for 24 h with UA at 3 µM, 30 µM, 75 µM, 150 µM, and 300 µM. Then, the cells were washed and dosed with 100 µM Mn as MnCl2. After the Mn treatment period for both assay types, cell viability was measured by a colorimetric assay either by using the MTT (thiazolyl blue tetrazolium, Sigma-Aldrich (St. Louis, MO, USA)) viability assay or the MTS-Assay (CellTiter96 AQ assay, Promega [Madison, WI]) according to the protocol of the supplier, as reported previously [7]. In parallel, cells were grown in 12-well plates for cell growth analysis. Repetitive aliquots were taken and counted three times using a hemocytometer with trypan blue exclusion assay, as previously reported. For each treatment condition, triplicate wells were counted, and values were averaged.
Western blotting: SH-SY5Y cells (9 × 106), at a 60–70% confluency, were treated with the following conditions in duplicate, and all for 48 h. The APP Western blot had (i) Lanes 1–2: control; (ii) Lanes 3–4: 100 µM Mn as 100 µM MnCl2; (iii) Lanes 5–8: 2 sets of increasing UA at 75, 300 µM; and (iv) Lanes 9–12: 2 sets of 100 µM Mn and increasing UA at 75, 300 µM. The H-ferritin Western blot had (i) Lanes 1–2: control; (ii) Lanes 3–4: Fe as 100 µM ferric ammonium citrate (FAC); (iii) Lanes 5–6: 100 µM Mn as 100 µM MnCl2; (iv) Lanes 7–8: 100 µM UA; (v) Lanes 9–10: 100 µM FAC and 100 µM UA; and (vi) Lanes 11–12: 100 µM Mn and 100 µM UA. After subsequent washing in PBS, cells were scraped into RIPA lysis buffer. Total protein concentrations were analyzed using Bio-Rad protein assay. Cell extracts were immunoblotted as described elsewhere [7,63]. Western blots were repeated at least three times, and densitometric analysis was performed using ImageJ software (version 1.54 g) [65].
RT-PCR: Triplicate assays were performed in untreated versus deferoxamine (DFO)- and zinc-treated SH-SY5Y cells for 6 and 16 h timepoints with the following conditions (i) control; (ii) control; (iii) 100 µM DFO; (iv) 200 µM Zn as ZnSO4; (v) 100 µM Zn as ZnSO4; (vi) 200 µM Zn as ZnSO4 and 100 µM DFO; and (vii) 100 µM Zn as ZnSO4 100 µM DFO. Total RNA was isolated using TRI-reagent (Sigma-Aldrich) according to the manufacturer’s instructions. Assays were performed using an ABI Prism 7000 system (Applied Biosystems, Foster City, CA, USA). Metallothionein-III mRNA primers, Forward: 5′AGT GCG AGG GAT GCA AAT G 3′. Reverse: 5′ACA CAC AGT CCT TGG CAC ACT T3′. APP mRNA primers (forward, 5-GCCCTGCGGAATTGACAAG-3; reverse, 5-CCATCTGCATAGTCTGTGTCTG-3). transferrin receptor mRNA primers (forward, 5-GGCTACTTGGGCTATTGTAAAGG-3; reverse, 5-CAGTTTCTCCGACAACTTTCTCT-3); and beta-actin mRNA primers (forward, 5-CATGTACGTTGCTATCCAGGC-3; reverse, 5-CTCCTTAATGTCACGCACGAT-3) were purchased from Life Technology.
2.5. Experimental Statistics
Following the MTT co-treatment assays, one-tailed Student’s t-tests were conducted between Mn and Mn + UA treatment groups. Statistical significance was deemed to be p < 0.05, indicating that UA recovery of cells did not occur by chance. Pre-treatment assays were evaluated for statistical significance via the use of one-tailed Student’s t-tests for control versus treatments. Using densitometric data, unpaired two-tailed Student’s t-tests were performed for control versus treatment lanes, as indicated by the Western blot treatment conditions.
3. Results
3.1. Data Exploration
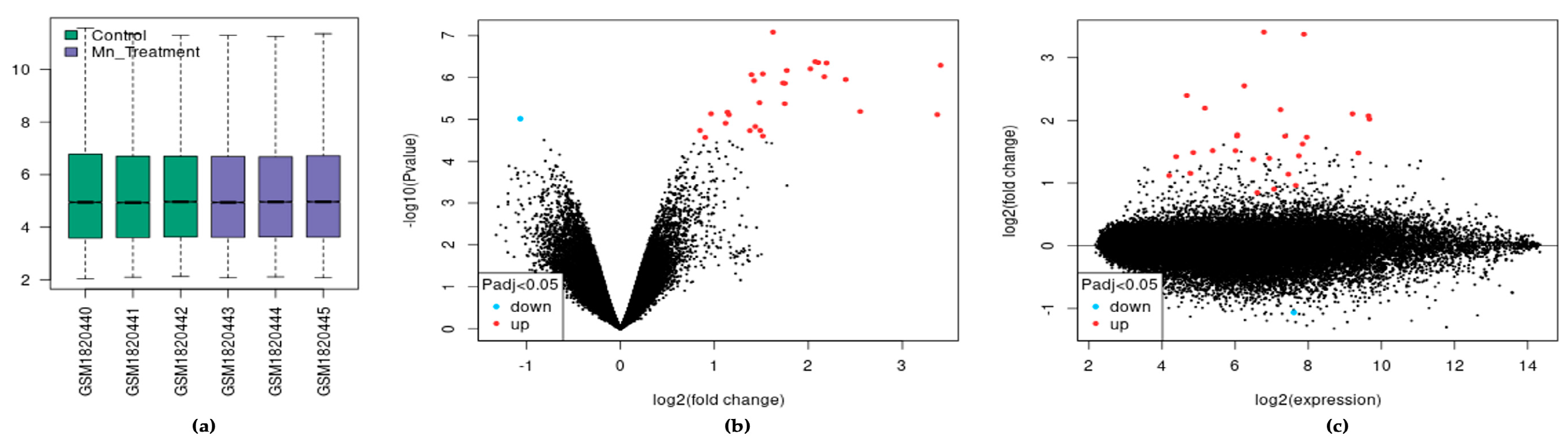
In the dataset analyzed by GEO2R, each sample was log2 normalized with the median-centering values being present, indicating cross-comparability between samples in the dataset (Figure 1a). We visualized 27,770 transcripts by volcano plot and found that 29 of them were significantly differentially expressed (adjusted p < 0.05). These transcripts mapped to 22 differentially expressed genes (DEGs) (Figure 1b). Of the 22 DEGs, 21 were upregulated and one was downregulated following chronic Mn exposure. Similarly, the mean difference (MD) plot represents the differential expression showing the most differentially expressed genes on the right region of the plot (Figure 1c).
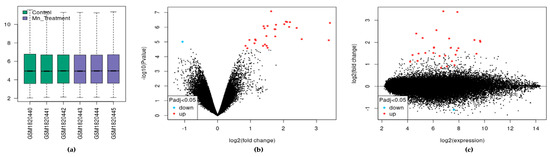
Figure 1.
The effect of exposure of SH-SY5Y cells to Mn. (a) Expression boxplot of the three treatments and three control samples. (b) Volcano plot of differential expression and significance by adjusted p-value. (c) MD plot of differential expression.
3.2. Manganese Pathway Modulation
We found significant changes via the ReactomeGSA pathway analysis tool utilizing the efficient CAMERA algorithm, which further filters DEGs from GEO2R analyses. Table 1 shows six pathways to be upregulated in Mn exposure; these pathways are participants in histone deacetylase (HDAC) expression as a neuroprotective response to DNA damage, depurination, and amyloidosis. Two pathways were downregulated, resulting from mitochondrial dysfunction and a protective oxidative stress pathway.

Table 1.
Critical pathways modulated by Mn exposure.
3.3. Manganese Gene Modulation
Table 2 represents the top gene changes following 24 h of manganese treatment of SH-SY5Y cells. After ReactomeGSA CAMERA analysis, the 22 DEGs identified in GEO2R were narrowed down to 10 DEGs; these genes were involved in and indicate neuroprotection, histone expression, mitochondrial stress response, and detrimental expression of proinflammatory cytokines.
Table 2.
Top differentially expressed genes (DEGs) modulated by manganese exposure.
3.4. DEG Similarity and Expression Analysis
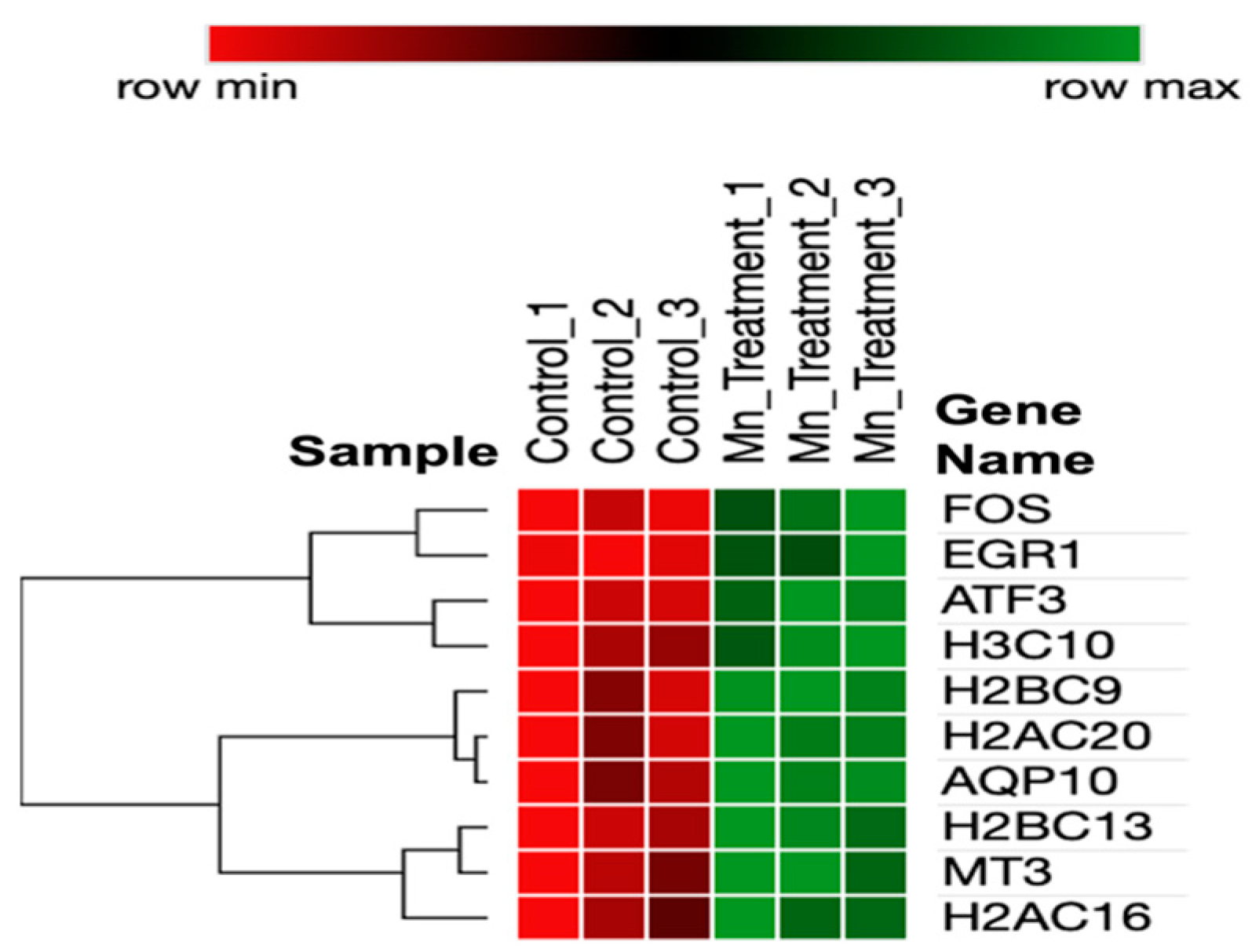
A heatmap of the DEGs (Figure 2) represents interrelationships via a dendrogram created using one minus the Pearson correlation and complete linkage. Clustering identified that genes have potential co-expression with histone-related and cellular stress-response genes.
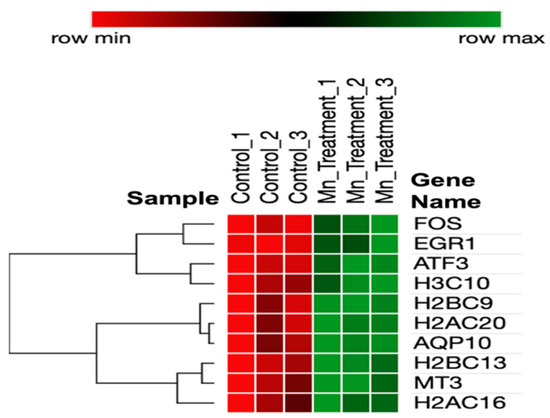
Figure 2.
Heatmap of DEGs in chronic manganese exposure.
3.5. Experimental Validation
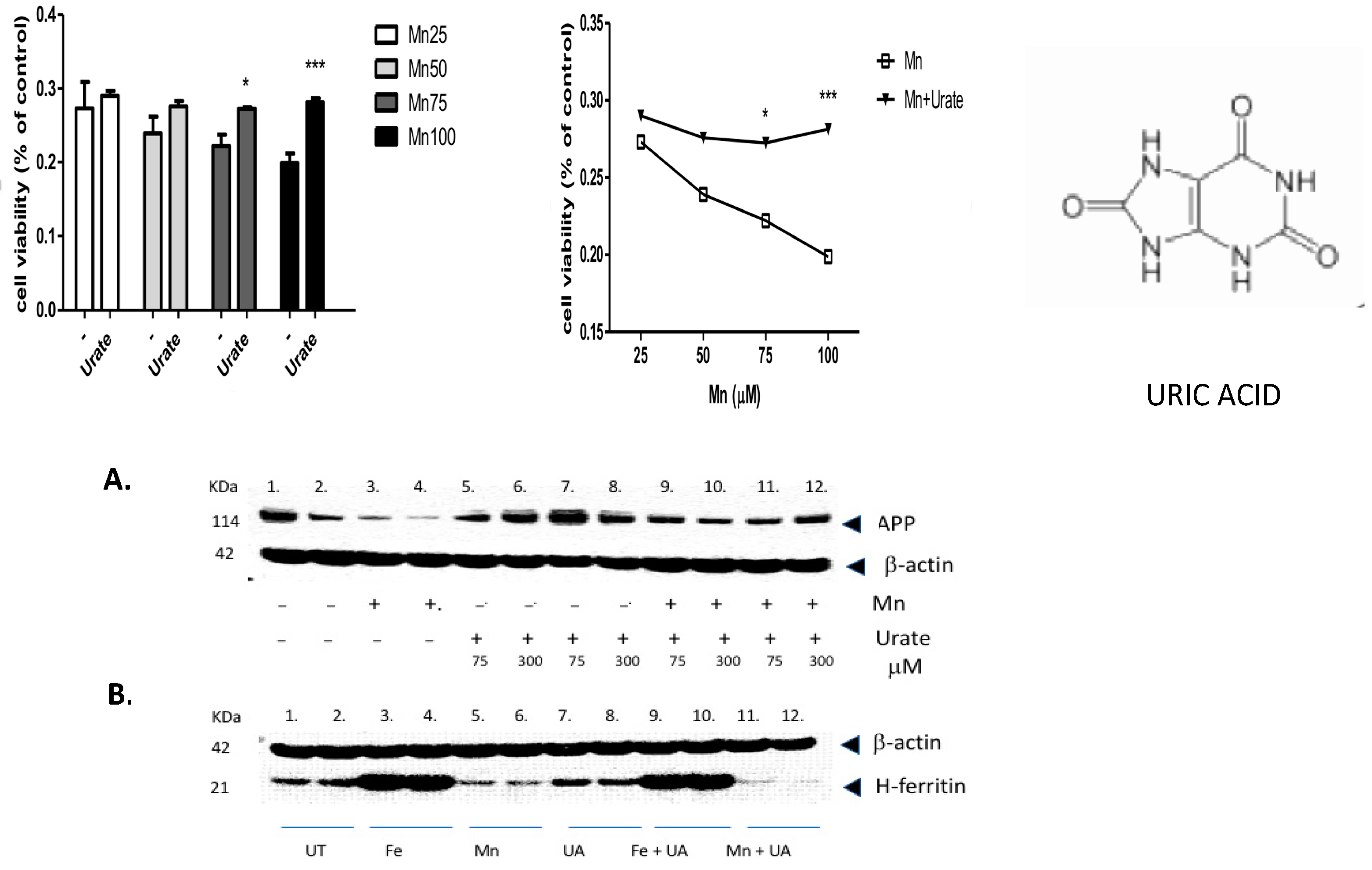
We found that multiple pathways were activated after Mn exposure, including sirtuin-dependent rRNA translation and inhibition of NF-κB, as well as the capacity to inhibit pro-oxidant pathways via NRF-2 (Table 1). However, the most significant effect of manganese exposure was to perturb three major pathways involving purine metabolism (nucleotide salvage defects were highly expressed, as well as pathways involving cleavage and damage to purines and also depurination steps of DNA and RNA). Therefore, we further tested whether pre- and co-incubation of the antioxidant purine urate might mitigate manganese toxicity (Figure 3, Figure 4 and Figure 5). The top panel in Figure 3 shows MTT assay results indicating that co-treatment of cells with uric acid (100 mM UA) mitigated manganese toxicity to SH-SY5Y neuroblastoma cells relative to controls (i.e., UA reduced the toxic effects of treatments with 25 mM, 50 mM, 75 mM, and 100 mM MnCl2 to cells for 72 h).
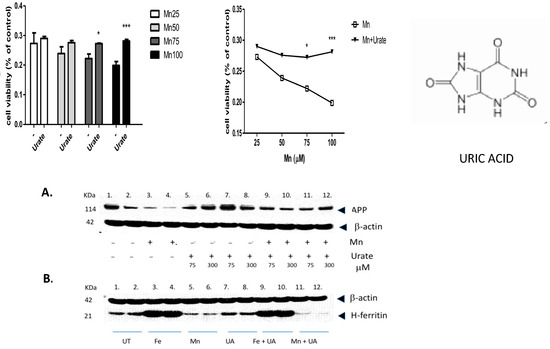
Figure 3.
The purine and antioxidant urate offsets manganese-dependent pathways of cell-based toxicity to SH-SY5Y neuroblastoma cells. Top Panels: MTT assay of cell viability: The effects of co-treatment of cells with uric acid (100 mM) on dose-responsive Mn toxicity to SH-SY5Y neuroblastoma cells relative to controls (i.e., Plus/minus urate to mitigate toxicity of 25 mM MnCl2, 50 mM MnCl2, 75 mM MnCl2 and 100 mM MnCl2 to cells for 72 h, N = 6. Bottom Panels: A: Representative Western blot depicting the effects of urate and manganese to modulate APP (FPN-APP complexes export excess iron from cells) in SH-SY5Y neuroblastoma cells. Western blot shows the effect of urate on Mn-dependent repression of APP by Mn in SH-SY5Y cells (9 × 106 cells) when treated for 48 h, (A) (i) Lanes 1–2: control; (ii) Lanes 3–4: 100 µM Mn as 100 µM MnCl2; (iii) Lanes 5–8: 2 sets of increasing UA at 75, 300 µM; and (iv) Lanes 9–12: 2 sets of 100 µM Mn and increasing UA at 75, 300 µM. B: Effect of urate towards Fe- and Mn-dependent modulation of H-ferritin translation; (B) (i) Lanes 1–2: control; (ii) Lanes 3–4: Fe as 100µM ferric ammonium citrate (FAC); (iii) Lanes 5–6: 100 µM Mn as 100 µM MnCl2; (iv) Lanes 7–8: 100 µM UA; (v) Lanes 9–10: 100 µM Mn and 100 µM UA; and (vi) Lanes 11–12: 100 µM Mn and 100 µM UA. Full-length Western blot Gels #95 and #97 are provided in the Supplemental Materials section of this paper. * p < 0.05 for Student’s t-test conducted between Mn and Mn + UA groups. *** p = 0.000165; Student’s t-test conducted between Mn and Mn + UA groups.
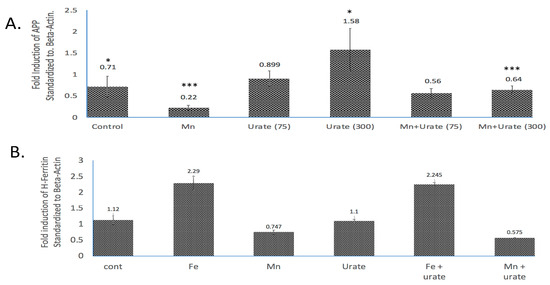
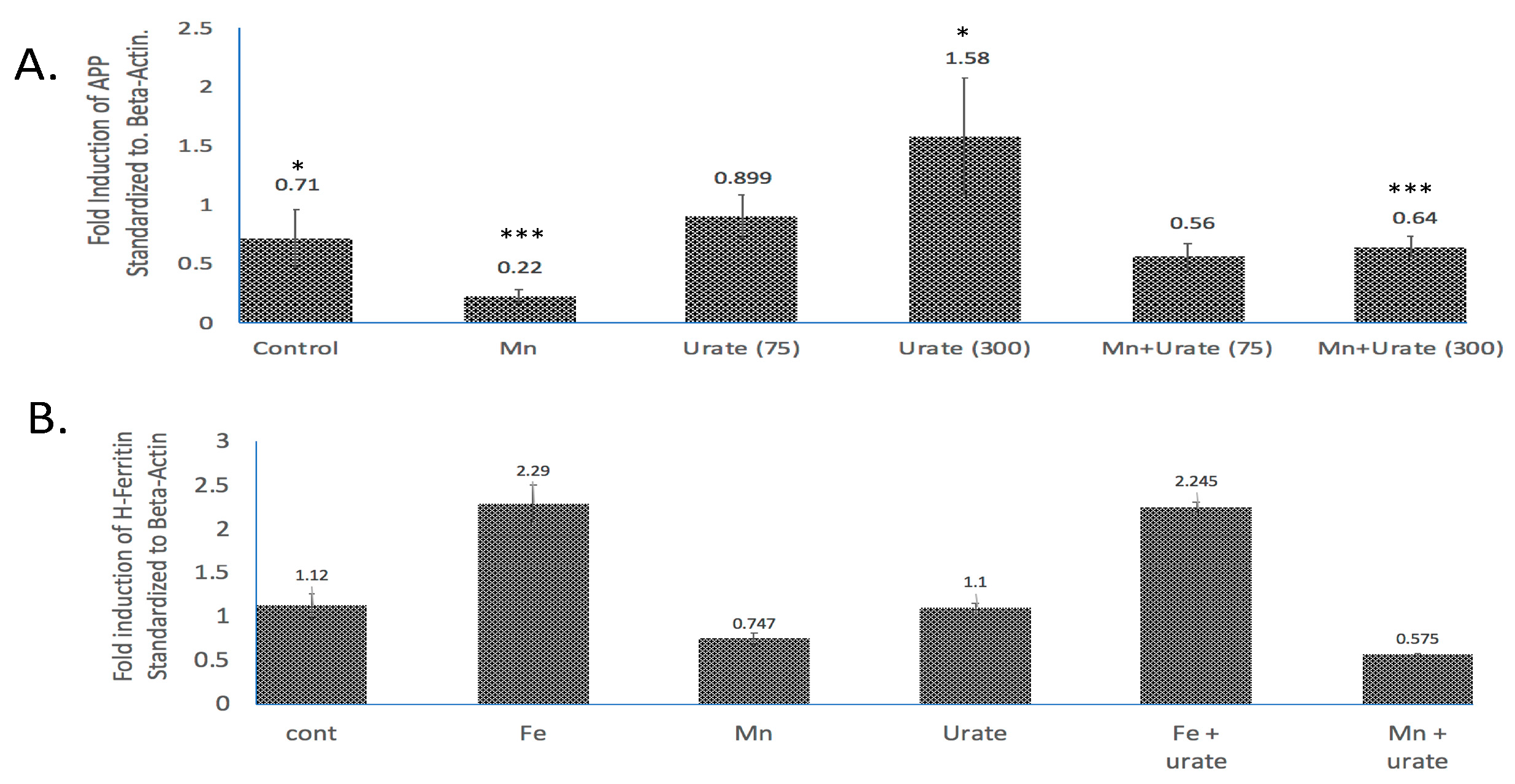
Figure 4.
Densitometric quantization of the Western blots shown in Figure 3 after normalization with β-actin. (A) Standardized fold modulations (1 = 100%) of the APP/b-actin levels in response to Mn dose (100 mM, 48 h) as compared to control and Mn treatment in the presence of urate (75 mM and 300 mM urate; see Western blots in Supplemental Materials). Prior to normalization, the data showed β-actin was unchanged by all treatments. (B) Standardized fold modulations of the H-ferritin/b-actin levels in response to Fe (100 mM FAC, 48 h) and Mn (100 mM MnCl2, 48 h) as compared to controls; Fe and Mn treatments in the co-presence of 100 mM urate (see Supplemental Materials). Data without normalization with β-actin were unchanged by treatments. To test statistical significance for changes in APP expression, unpaired two-tailed t-tests were performed for controls vs. treated lanes and are indicated on the graphs. * & *** p ≤ 0.05, i.e., for Cont. vs. UA and for Mn vs. Mn + UA treatments. Statistical P values reflect the capacity of urate to restore APP expression after its inhibition by Mn treatments, n = 3 independent WB experiments, each in duplicate.
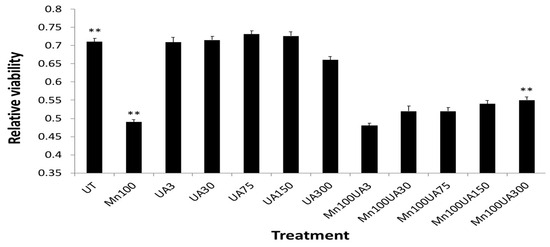
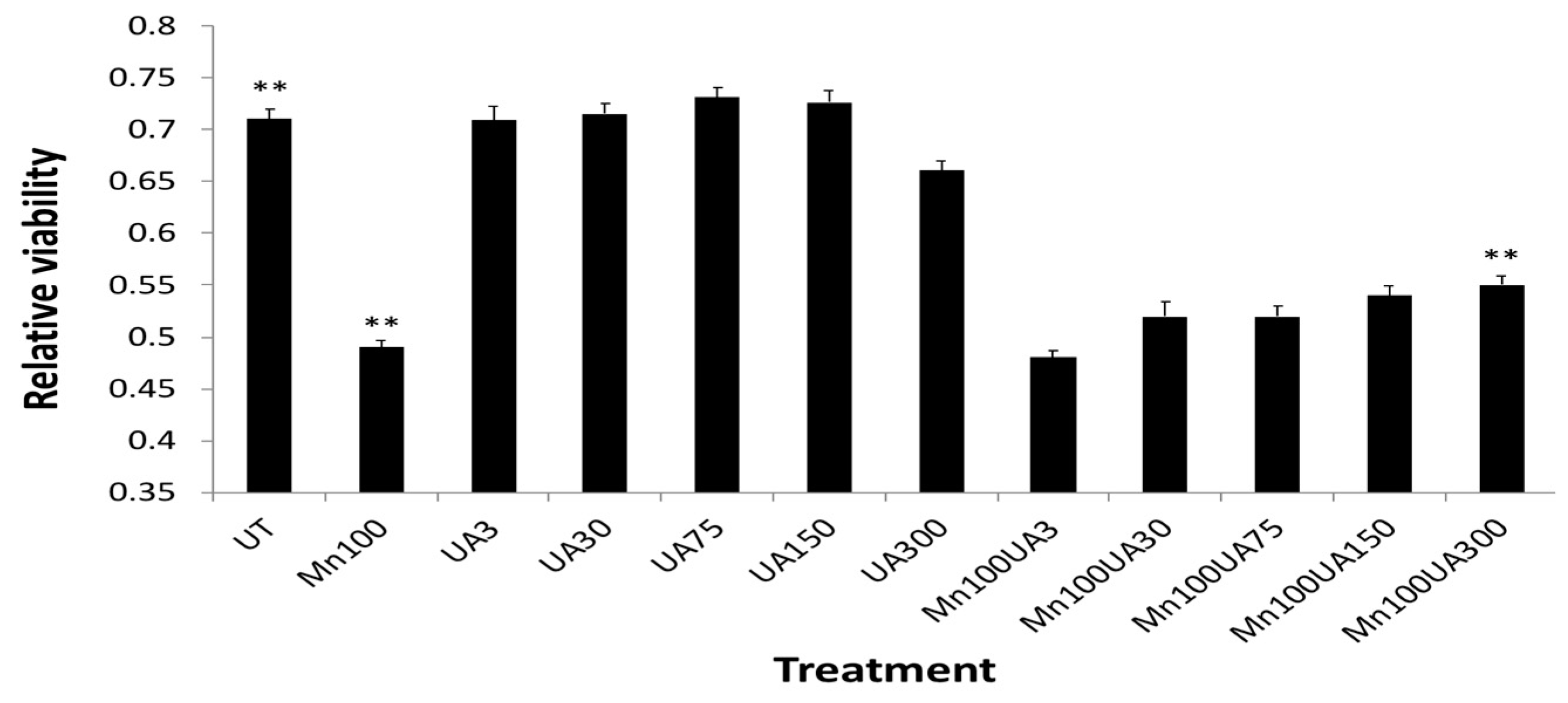
Figure 5.
Averaged Urate (@ 3, 30 75, 150 300 mM) pre-treatments afforded a dose graded up to 14%, which increased neuroprotection to SH-SY5Y cells after washout when exposed to Mn (100 mM 24 h) (n = 8). ** One-tailed t-tests were performed for controls vs. Mn-treated, p = 4.00 × 10−8; Mn-treated vs. Mn + UA treated, p = 3.70 × 10−2; control vs. Mn + UA treated, p = 6.689 × 10−5 (N = 8).
The top right-hand panel in Figure 3 exhibits a representative experiment demonstrating that the presence of urate completely mitigated a 33% loss of SHY5Y cell viability over the three-day period of exposure. This representative experiment was conducted to high statistical significance (n = 6), p < 0.05) as indicated in the histogram on the left-hand side (Figure 3, next page).
In terms of statistical significance, we observed a 33% loss of viability at the max dose of 100 mM manganese relative to untreated controls, while the presence of UA (100 mM) mitigated 33% of the loss of viability (MTT @ 72 h *** p = 0.000165; Student’s t-test conducted between Mn and Mn + UA groups). We noted that at 72 h, cotreatment of the cells with 100 mM urate completely rescued the cells from such toxicity. Losses of cellular viability were dose-responsive, and the degree of cell death and recovery with urate co-treatment was statistically significant (p < 0.05, n = 6) over several separate assays.
As we observed in a previous publication [7], Mn reduced the expression of neuroprotective APP and ferritin. Western blots are shown in Figure 3 and densitometric quantitation is provided in the histogram in Figure 4 (See Supplemental Materials for full-length gels). In three separate experiments performed in duplicate, we observed that both APP and ferritin were reduced by >50% after 48 h Mn exposure. During these studies, we noted that the presence of urate co-treatment restored the steady-state levels of APP, although this was not the case for steady-state levels of H-ferritin during urate + Mn co-exposures of SH-SY5Y cells. Densitometric quantitation of APP and H-ferritin expression relative to that of b-actin is graphically represented in Figure 4. The results of treatments with Mn alone and Mn in the presence of urate are also depicted. Duplicate assays in Figure 4A showed that Mn reduced APP levels by 3-fold, while the presence of urate enhanced APP protein levels 2–3-fold, such that the presence of co-added urate mitigated Mn inhibition of APP levels to 90 percent of control levels. We quantitated that 100 mM iron as ferric ammonium citrate (FAC) treatment increased ferritin expression by 2.2-fold, as standardized to b-actin in the representative densitometry of the averaged duplicate lanes. Contrary to APP expression, the presence of urate did not change the Mn inhibition or Fe induction of H-ferritin (n = 3) (Figure 4B).
In addition to urate co-treatment with Mn exposures (Figure 3 and Figure 4), we also tested whether pre-treatment with urate might offset Mn neurotoxicity (Figure 5). Indeed, 24 h pre-treatment with urate dose afforded 14% neuroprotection to SH-SY5Y cells exposed to Mn (100 mM 24 h) [54,66].
Our bioinformatics findings showed a three-fold increase in MT-III mRNA (Table 2) when characterizing the transcriptomic profile of manganese-exposed SH-SY5Y cells. Manganese is a divalent cation and may well exhibit biology typical of other divalent cations, whereas Mn, cobalt, zinc, and DFO are known hypoxia mimetics [52,53]. Wang et al. demonstrated that hypoxia is an active inducer of MT-3 mRNA via RT-PCR analyses in human adipocytes [53].
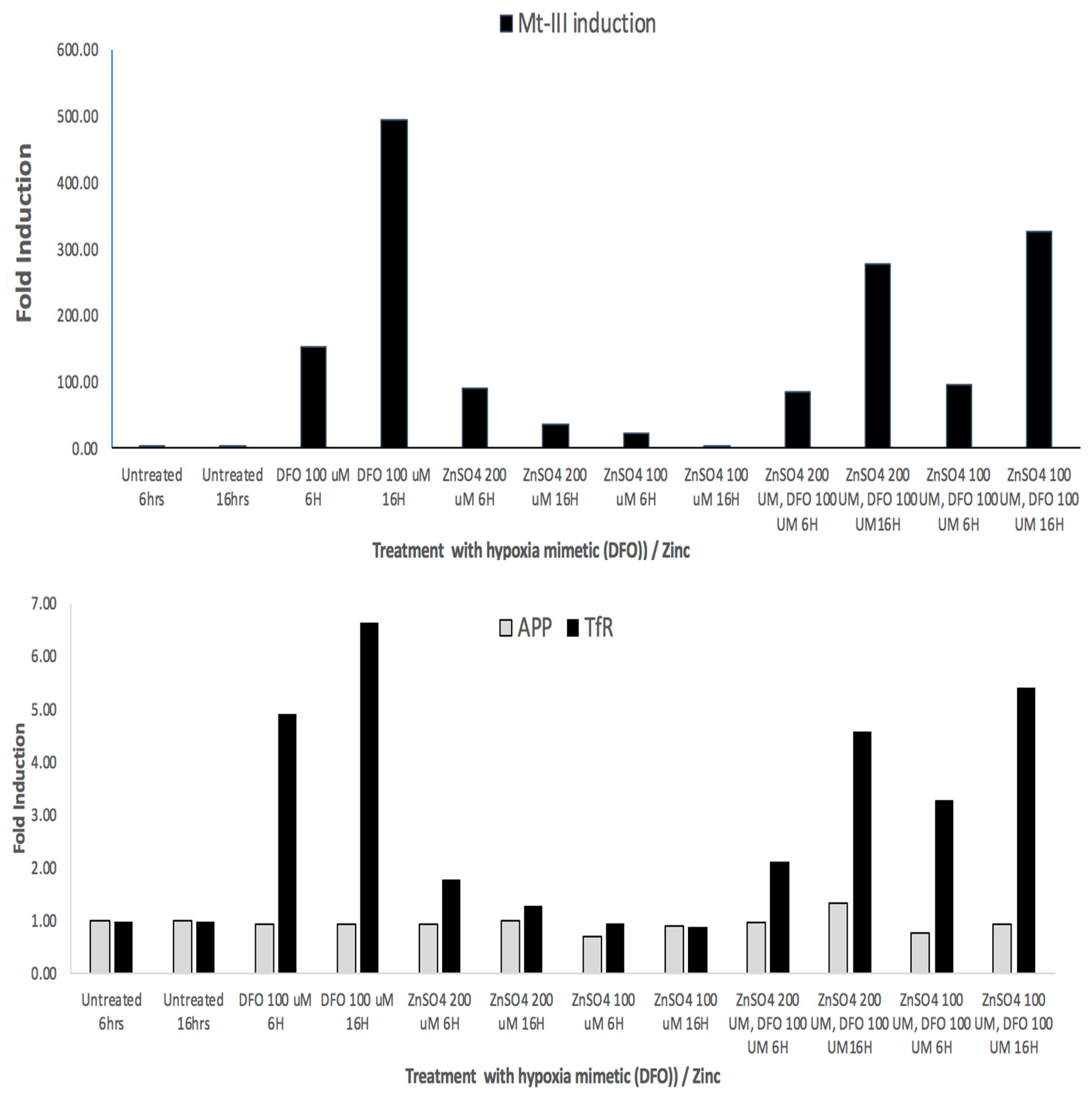
To follow up, we demonstrated RT-PCR data in Figure 6 to show that MT-3 mRNA, transferrin receptor (TfR) TfR-mRNA and APP mRNAs were all induced in response to DFO and Zn treatments (hypoxia mimetics). We obtained three independent RT-PCR measurements from SH-SY5Y cells to quantify inductions of MT-III mRNA in order to determine whether they occurred as a result of treatment of cells with the hypoxia mimetics DFO and Zn. RT-PCR data (N = 3) showed that MT-3 mRNA levels were induced by 500-fold by DFO > 3–30-fold inductions of MT-3 mRNA by ZnCl2 (Figure 6).
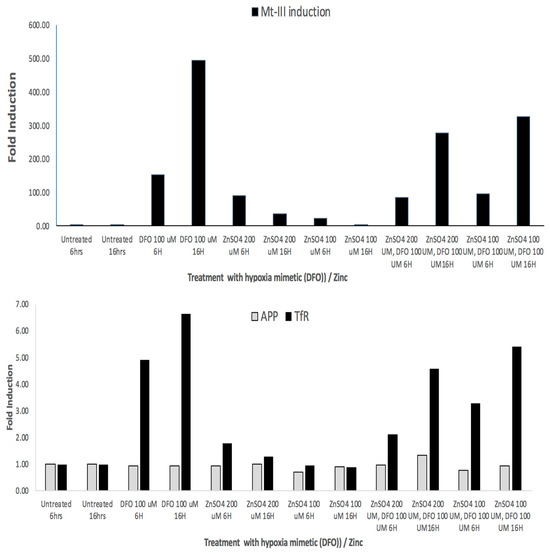
Figure 6.
Top Panel: RT-PCR: Fold inductions of MT-3 mRNA in response to the hypoxia mimetics DFO and zinc in SH-SY5Y cells. Second Panel: RT-PCR showing fold inductions of SH-SY5Y-specific APP mRNA and Transferrin receptor mRNA in response to the hypoxia mimetics DFO and Zn (negative and positive controls, respectively).
We noted from the RTPCR experiments in Figure 6 that DFO increased levels of mRNA for neuroprotectant MT-III by 500-fold (N = 3). The divalent cation zinc, another hypoxia mimetic, increased MT-3 mRNA by up to 30-fold in SH-SY5Y cells. Transferrin receptor mRNA and APP mRNA are known to be abundant transcripts. In these experiments, DFO and zinc were found to increase TfR mRNAs to a less inductive, although similar, extent relative to MT-3 mRNA when responding to hypoxia mimetic (DFO and Zn). APP mRNA is regulated via translational control circuits, and thus its mRNA was unchanged by DFO and Mn and Zn, as predicted.
Our bioinformatic and pre- and post-published experimental data are shown in Table 3 to quantitate, list, and rank the cellular biomarkers of Mn toxicity. We reported a notable increase in the transferrin receptor (TfR) mRNA in reference [7] in MnCl2-exposed SHSY5Y cells. However, in this study, we statistically reevaluated the source q-RT-PCR data to demonstrate a precise 7.8-fold increase in TfR mRNA levels (N = 3 standardized to b-actin controls). Iron regulatory protein 1 (IRP1) was also noted to be slightly increased at the basal level with enhanced IRE binding after Mn exposures (pre-published data). Iron-regulatory Protein 2 (IRP2) decreased to 9.05% (St dev (SD), ±1.47%) of controls by the same Mn exposures [7]. Table 3 presents our reanalysis of densitometry from Western blots to confirm our previous report [7] that H-ferritin deceased to 5% of that exhibited by the controls in SHSY5Y cells treated with 100 mM MnCl2. By contrast, a study of welders exposed to airborne Mn showed serum ferritin, which is rich in the L-subunit and was slightly increased by chronic Mn exposures while also providing an existing biomarker for iron overload [67]. Metallothionein 3 (MT-3) also implicated its role as a potential manganese chelator, elevating its expression as a neuroprotectant 3.36-fold, as listed in Table 3. Alpha-synuclein protein (60 KDa tetramer) levels decreased 2-fold in Mn-exposed SH-SY5Y cells.
Table 3.
Potential cellular biomarkers of manganese toxicity in SH-SY5Y cells (100 mM, 24 h).
We propose that the ratio of APP protein/APP mRNA constitutes a top candidate cellular biomarker for Mn toxicity. In Table 3, APP mRNA increased by 4.02-fold (10 mM, 24 h) and 3.68-fold (100 mM, 24 h) in response to Mn treatments of SH-SY5Y cells while APP protein was greatly inhibited, i.e., by 15% (10 mM MnCl2, 24 h), 50% (50 mM MnCl2, 24 h), 85% (100 mM MnCl2, 24 h) in the same SH-SY5Y cell lineage. Therefore, the ratio of APP protein/APP mRNA (index ratio of APP gene expression) provides a clear cellular biomarker signal unique to Mn relative to iron exposure (i.e., Mn causes APP translational repression, while APP mRNA levels are increased by Mn exposures).
4. Discussion
We conducted bioinformatic analyses based on microarray data in triplicate to identify the metabolic pathways modulated by Mn exposures in SH-SY5Y cells [56]. Here, we report which gene transcripts are indeed activated by Mn exposure in SH-SY5Y cells. The study pertains to inductions in human cells in SHSY5Y, while comparative findings for mouse cell lines, including mouse N2a neuroblastoma cells, will be investigated. Although we have reported our work here using SH SY5Y cells, our lab has used other neuroblastoma lines, such as SK-N-SH, derived from the same stock as well as human primary neuronal enriched cultures (18). Based on our experience, the effect we report here most likely would work in other neuroblastoma cells; however, it needs additional future work to confirm the effect in other neuro-derivative cultures.
This is particularly relevant, since Mn influences APP expression via the human IRE-type-II sequences in the 5′UTR of its transcript, which is relevant to this study. Our findings in Figure 3 and Figure 4 are consistent with the model that the human APP 5′UTR-specific iron-responsive element-Type-II drives APP translation as the major conduit though which urate operates, providing therapeutic value to offset Mn toxicity (Table 3). Our current findings support future tests to establish species-specific differences by which Mn exposures might induce APP translation as a biomarker for predicting toxicity in human SHSY5Y cells compared to mouse cell N2A types and ultimately in vivo. There may be differences for manganese toxic actions acting through the APP 5′UTR sequences in humans compared to mouse models (Supplementary Figure S3).
Our bioinformatic findings were experimentally validated via MTT viability assays and RT-PCR analyses. We found, as a first example, that Mn activated histone deacetylases, which act to prevent chromatin condensations from oxidative stresses induced by Mn. Consistent with this finding, Mn exposures induced five histone gene variants H3C10, H2BC13, H2AC16, H2AC20, and H2BC9 of the best ten differentially expressed genes (DEGs).
The transcriptomics microarray analyses demonstrated that the antioxidant, low molecular weight, neuroprotectant metallothionein-III (MT-3) was among the top Mn induced mRNAs in SH-SY5Y cells. MT-3 is a neuroprotectant capable of offsetting AD [69] and PD, as well as biochemically limiting Mn-induced damage [58,70]. Like cobalt, zinc, and DFO, the divalent metal Mn has been experimentally employed in lung epithelial and adipocytes as a chemical mimetic of hypoxia [52,53]. The data in Figure 6 further validated Mn as a hypoxia mimic by the use of RT-PCR, as is consistent with a model for MT-3 providing neuroprotection to SH-SY5Y cells.
Overall, using the SH-SY5Y model of dopaminergic-transformed neurons, the results from our bioinformatic analysis indicated that Mn exposures primarily modulated pathways involved in depurination, HDAC-related neuroprotection, histone modulation, and amyloid fiber formation. Manganese also specifically upregulated specific target genes listed in Table 2, such as the histone genes (as stated above), stress response, transport genes (MT3, AQP10), and immediate early genes (ATF3, c-Fos, EGR1). Neuronal damage resulting from Mn exposure elevated reactive oxygen species (ROS) concentrations, which we reported to be mediated via IRE/IRP control of ferritin (iron storage) and APP (iron efflux by APP/FPN complexes) translational control circuits [7]. Consequentially, ROS surplus by Mn can activate NF-κB, which may exacerbate neuronal damage by further elevating concentrations of reactive oxygen species to cause pro-inflammatory reactions [71].
As an adaptive cellular response, we note that SIRT1 deacetylases the p65 subunit of NF-κB at its Lys310 residue, an event that could protect cells from apoptosis by reducing transcriptional activity [72]. Overall, the adaptive upregulation of SIRT1 (Table 1) and downregulation of NF-kB pathways is a cellular marker indicating accelerated activation of an ameliorative SIRT1/NF-κB signaling pathway to reduce neuroinflammation [73]. Furthermore, increases in the HDAC expression pathway are specifically involved in this functional pathway, i.e., the Class I HDACs (HDAC1, HDAC3, HDAC8) and HDAC10. Notably in neuronal cells, HDAC1 binds to SIRT1 and provides neuroprotective effects [74]. HDAC expression was increased and involved in neurodegeneration through inflammation [75].
Following exposure of manganese to SH-SY5Y cells, there was increased expression of three depurination-related pathways from the eight top Mn-induced pathways listed in Table 1. These adaptations are likely in response to damage to adenine and guanine structures. This prompted our report that aspects of Mn neurotoxicity could be quelled by urate (Figure 3, Figure 4 and Figure 5). We reasoned that the purine damage can be ameliorated by supplementing urate to repair the transversion mutation; this theory was also supported by our prior publications [45,46]. The purine metabolism pathway we observed after Mn perturbations controls the rates of mutation load to affected cells [43,76]. It is noteworthy that increased intracellular urate is the metabolic product of the oral purine supplement inosine and has long been shown to be a neuro-protective antioxidant [42,44,46,47,77]. The Mn-induced loss of APP and ferritin we observed here was consistent with our previously reported losses of IRE/IRP-dependent translation of both H-ferritin and APP mRNAs [7].
In addition to our own findings, our colleagues have shown that the APP 5′-UTR-directed drugs posiphen and phenserine inhibit APP translation to generate anti-amyloid efficacies [78,79] for the purpose of treating AD as effectively as current anti-inflammatory pathways [80]. Antioxidant and APP-processing pathways also proved to be highly relevant as key therapies for AD [81,82]. However, in younger adults and children, amyloidosis is not a risk factor for developing AD. Therefore, as a model for mitigating metal toxicity and oxidative neurotoxicity in younger age groups, we suggest that antioxidants such as urate might favor APP translation, possibly via the same 5′-UTR, and thus increases of APP/FPN complexes, sufficient to efflux excess embargoed Mn and Fe in SH-SY5Y cells, as outlined in our previously reported models [7,9]. Here, urate treatments left ferritin expression unchanged, as shown in Figure 3. Thus, the canonical IRE was unresponsive to the intracellular therapeutic presence of urate, while the 5-’UTR specific IRE-type-II of APP mRNA may be tested as being responsive to urate (iron efflux-associated event [9,83]). These events are consistent with the theory that urate could induce neuroprotective APP(s), an event that facilitates FPN-dependent Fe efflux and thus counteracted ferroptosis resulting from prolonged cellular exposures to Mn [16].
Mn inhibition of APP translation requires further testing to detect decreases in levels of APP(s) as well as c-terminal fragments of APP (APP-CTF-b) which are produced after BACE cleavage. Certainly, APPs and APP-CTF-b fragments of APP have differing consequences to influence cellular iron homeostasis [84,85]. Here, we employed conditions of Mn exposure where APP was limited at the translational level so as to limit the appearance of APP(s) and APP CTF [86]. We previously stated that APP 5′UTR translation blockers reduced both fragments in parallel, as detected by 22C11 and A8717 antibodies on Western blots [86]. Thus, Mn does not appear to separately induce CTF-b via compensatory activation of BACE in SHSY5Y cells although this remains to be compared in human and mouse cell lines [86]. The biological action of APP CTF fragments might change intracellular iron levels via perturbations to the mitochondrial ATPases that induce lysosomal driven pH changes and inclusion bodies [84,85,87]. Here, lysosomal dysfunction was shown to occur when APP is overexpressed in Down syndrome (triple APP) and in Alzheimer mouse models so as to proportionately also increase APP CTF (see citations [85,88]. By contrast, Mn might not promote the appearance of Tyr(682)-phosphorylated APP beta-CTF if shown to induce parallel losses of both of APP and APP CTF.
Metallothionein-III is a known key neuroprotectant that was noticeably induced in SH-SY5Y cells exposed to manganese. Mn is a divalent cation and may well exhibit biology typical of other divalent cations such as cobalt and zinc, which are known hypoxia mimetics [54,66]. Our bioinformatics findings showed that Mn induced MT-III mRNA three-fold, while the data in Figure 6 confirmed hypoxic mimetics are active inducers of MT-3 mRNA, which is consistent with published RT-PCR analyses in human adipocytes [53]. In this context, our data in Figure 6 confirm MT-3 mRNAs were 100-fold induced in response to the hypoxia mimetics DFO and zinc.
The bioinformatic findings in this communication therefore provide rationale to reintroduce urate as a neuroprotective agent to treat Mn neurotoxicity. In this context, depurination is a chemical reaction of purine deoxyribonucleosides, deoxyadenosine, and deoxyguanosine, in which the beta-n-glycosidic bond is hydrolytically cleaved, releasing the nucleic base, adenine and guanine, respectively [76]. The removal of purine structures elicits a transversion mutation that needs a new purine to repair the DNA [43].
Metallothioneins (MTs) are cysteine-rich metal-binding proteins that chelate metals and inhibit oxidative stress, inflammation, and mitochondrial dysfunction induced by metals, while MT-III is reduced in Alzheimer’s disease [69]. Furthermore, MT proteins scavenge free radicals and exhibit anti-inflammatory effects through the suppression of microglial activation; MTs are also critical, as they can act as targets for reducing metal-induced α-synuclein aggregation [58]. Our study showed that MT-3 was increased three-fold following manganese exposure in SH-SY5Y cells. MT-3 is also transcriptionally induced several hundred-fold by our hypoxia mimetics and in [55]. Our current findings in Figure 6 confirm that deferoxamine induced MT-3 mRNA in SH-SY5Y cells by several multiples of 10 while leaving APP mRNA unchanged; also, we determined that there is a putative IRE in the 3′UTR of MT-3 mRNA [89,90]. We therefore conducted validating RT-PCR studies of SH-SY5Y cells dosed with zinc and deferoxamine, agents known to induce hypoxia, and we found these agents, like Mn, exhibited similar effects to significantly induce MT-3 mRNA. Overall, like deferoxamine and zinc, manganese is a hypoxia mimetic that significantly upregulates the expression of MT-3 mRNA levels in SH-SY5Y cells.
In the literature, both DFO and manganese can function to induce hypoxia, while DFO is known to upregulate HIF1α in rat brains after injury [91]. In lung cells, manganese induces chemical hypoxia by inhibiting HIF-prolyl hydroxylase. This action interferes with the hydroxylation of HIF-1α, a key post-translational modification for the Von Hippel–Lindau dependent degradation of HIF-1α [52]. Manganese was also shown to mediate the upregulation of HIF-1α protein in Hep2 human laryngeal epithelial cells by activating the MAPK family of enzymes [92]. These manganese-mediated hypoxic conditions appear potentially to play a role in the induction of MT-3, a highly inducible gene in hypoxia [55]. Based on these considerations, Metallothionein-III, like urate, may present new therapeutic angles for treating neurotoxic assaults from manganese overexposures, such as those that occur in children suffering from the effects of industrial pollution or for those suffering from neurodegenerative Manganism [7,26].
At a more global level, Mn exposures increase caspase-3 promoter activity through the Sp1-binding regions, increasing the caspase-3 mRNA and triggering the apoptotic chromatin condensation [70], which may be similar to the latent disruption of Sp1 induced by early-life exposure to Pb that we have previously reported [3,93,94]. Our results indicate an increase in the expression of core histones; specifically, H2BC9, H2AC20, H2AC16, H2BC13, and H3C10 in SH-SY5Y cells. We note that the increase in histones in response to DNA damage is essential for protecting cells against hydroxyl radical-induced DNA damage [95]. Histone mRNA is the only replication-dependent mRNA in eukaryotes, and is typically expressed during the S-phase of the cell cycle to coincide with DNA replication [96]. It has been shown that cadmium, nickel, and chromium, divalent cations similar to manganese, deplete the stem–loop binding protein (SLBP). This SLBP structure is essential for histone pre-mRNA pre-processing, histone mRNA stabilization and migration, and proper histone expression. This occurs because the histone mRNAs have a stem–loop structure at their 3′ ends, meaning they are not polyadenylated; these stem–loop structures are also highly critical to the histone mRNA regulation. Depletion of the SLBP will increase histone mRNAs outside the S-phase of the cell cycle through elevated polyadenylation. This depletion is also implicated in altered chromatin assembly, supporting the findings that manganese increases chromatin condensation, [97] while compensatory histone expression was observed in Table 2. Furthermore, the polyadenylated genes have been shown to be expressed in response to DNA damage or senescence, which is also present in manganese-induced toxicity, functionally acting as a biomarker of DNA damage [98].
Increases occur not only for MT-3 mRNA (Table 2) but also for TfR mRNA levels and these data are included in Table 3 to represent top potential cellular biomarkers for Mn toxicity. We re-quantitated our q-RT-PCR data that we had previously reported [7]. In Table 3, TfR mRNA levels are listed to be increased by >7-fold in SH-SY5Y cells when exposed to acute MnCl2 bolus (i.e., 7.6-fold, 10 mM MnCl2 (SD = 0.2219), and 7.8-fold, 100 mM. (SD = 2); each result was obtained after 24 h treatments [7]. Under the same conditions, iron (as ferric ammonium citrate) did not alter TfR mRNA levels in SH-SY5Y cells, while iron chelation with DFO caused a 9.7-fold increase in the steady state of TfR-mRNA levels. We note that TfR protein is readily detected in the serum as a blood-based biomarker and elevated in conditions of anemia. The reference range of serum TfR varies by sex in adults. Normal findings are as follows: Men: 2–5 mg/L, Women: 1.9–4.4 mg/L [99]. Certainly, increases in TfR mRNA expression in certain blood cell types, including lymphocytes, might be tested as a biomarker for Mn exposure while, conversely, levels of serum TfR were already reported to be reduced in welding factory workers who were chronically exposed to Mn in airborne fumes [67]. RNA stability factors may confound this direct correlation between TfR mRNA and TfR protein levels in the blood of Mn-exposed patients [16].
Several immediate early genes (IEGs), ATF3, FOS, and EGR1, were significantly upregulated by manganese exposures. First, ATF3 encodes the CREB protein family of transcription factors. ATF3 expression also mediates the mitochondrial stress response [100]. Secondly, c-Fos and the associated c-Jun potentially form heterodimer AP-1, expressing subsequent survival genes [101]. c-Fos, a proto-oncogene, has been determined to show an immediate increase in response to brain injury, and it increases to protect neurons [102]. Thirdly, EGR1 has been associated with the control of neuronal cell death and inflammation [103,104,105]. Fourth, AQP10, a member of the aquaporin family, was significantly upregulated in response to manganese. Fifth, this aquaporin potentially plays a potential role in the transport of glycerol [106]. We provide a highly useful dendrogram that represents the relationships between Mn-induced genes based on their expression patterns (as shown in the heatmap in Figure 2). The dendrogram branches indicate the presence of co-expression. MT3, H2AC16, and H2BC13 express similar patterns that potentially function through similar neuroprotective pathways. FOS, EGR1, and ATF3 are also biologically related due to the inferred dendrogram relatedness functioning through an immediate early gene pathway.
Our demonstration of key acute expression effects for Mn treatment can be used to interlock with other pathways vital to understanding neurodegeneration. PD is not usually associated with acute toxic responses any more than AD. However, chronic elevation levels of Mn have been reported for the movement disturbances of manganism which is distinct from, although often compared with, Parkinson’s disease. Should Mn have a place as an important factor in neurodegenerative disorders, it may be through latent pathways, such as changing epigenetic markers, as has been explored for Pb and dementia [27,107,108]. For example, Mn overexposure of SH-SY5Y cells alters the methylation of PD-associated genes [109] and induces histone acetylation changes [110]. Thus, we consider this work to be an important foundation in establishing not only acute but potentially long-term effects of Mn overexposure in neurological health.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biom14060647/s1, Figure S1: Original Western Blot; Figure S2: Original Western Blot; Figure S3: Homology between APP IRE-Type II in the human and mouse genomes.
Author Contributions
Conceptualization by C.M.C., S.S.S., D.K.L. and J.T.R.; methodology by C.M.C., R.B., S.S.S. and N.X.; software, S.S.S.; validation, R.B. and N.X.; formal analysis, D.K.L., J.T.R. and S.S.S.; investigation, C.M.C., R.B., S.S.S. and N.X.; resources, J.T.R.; data curation, S.S.S.; writing—original draft preparation, S.S.S. and J.T.R.; writing—review and editing, D.K.L. and J.T.R.; visualization, J.T.R. and C.M.C. supervised; J.T.R. and C.M.C. administered the project, C.M.C. and J.T.R. acquired funding. D.K.L. read the manuscript, revised the manuscript, and participated in the early design of the experiment. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Michael J. Fox Foundation for Parkinson’s Research, grant number 007382 (J.T.R. and C.M.C), JTR thanks the NIH for R21NS077079 and R01AG021081). D.K.L. thanks NIH grants (R21AG076202, R56AG072810, P01AG014449, P30AG072976).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Acknowledgments
J.T.R. and C.M.C. thank Maurizio Fava for his support. JTR thanks Yanyan Liu for technical input and D.K.L. thanks Bryan Maloney for his help. We acknowledge the advice of Satinder Sarang, Roderick Jensen and Steven Gullans and technical input of Lee Jae Morse for RT-PCR experiments.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Neal, A.P.; Guilarte, T.R. Mechanisms of lead and manganese neurotoxicity. Toxicol. Res. 2013, 2, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Paduraru, E.; Iacob, D.; Rarinca, V.; Rusu, A.; Jijie, R.; Ilie, O.D.; Ciobica, A.; Nicoara, M.; Doroftei, B. Comprehensive review regarding mercury poisoning and its complex involvement in alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 1992. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Basha, M.R.; Brock, B.; Cox, D.P.; Cardozo-Pelaez, F.; McPherson, C.A.; Harry, J.; Rice, D.C.; Maloney, B.; Chen, D.; et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal Lead (Pb): Evidence for a developmental origin and environmental link for ad. J. Neurosci. 2008, 28, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Bolin, C.M.; Basha, R.; Cox, D.; Zawia, N.H.; Maloney, B.; Lahiri, D.K.; Cardozo-Pelaez, F. Exposure to Lead and the developmental origin of oxidative DNA damage in the aging brain. FASEB J. 2006, 20, 788–790. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.C., Jr.; Gubert, P.; Villas Boas, G.R.; Meirelles Paes, M.; Santamaria, A.; Lee, E.; Tinkov, A.A.; Bowman, A.B.; Aschner, M. Manganese-induced neurodegenerative diseases and possible therapeutic approaches. Expert. Rev. Neurother. 2020, 20, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, D.; Ganguly, J.; Jog, M. Manganese and movement disorders: A review. J. Mov. Disord. 2021, 14, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Doeppner, T.R.; Willkommen, D.; Cahill, C.M.; Xin, Y.; Ye, G.; Liu, Y.; Southon, A.; Aron, A.; Au-Yeung, H.Y.; et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-ferritin. J. Neurochem. 2018, 147, 831–848. [Google Scholar] [CrossRef]
- Peres, T.V.; Schettinger, M.R.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef]
- Cho, H.H.; Cahill, C.M.; Vanderburg, C.R.; Scherzer, C.R.; Wang, B.; Huang, X.; Rogers, J.T. Selective translational control of the Alzheimer’s amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 2010, 285, 31217–31232. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type-II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef]
- Lumsden, A.L.; Rogers, J.T.; Majd, S.; Newman, M.; Sutherland, G.T.; Verdile, G.; Lardelli, M. Dysregulation of neuronal iron homeostasis as an alternative unifying effect of mutations causing familial alzheimer’s disease. Front. Neurosci. 2018, 12, 533. [Google Scholar] [CrossRef]
- Dlouhy, A.C.; Bailey, D.K.; Steimle, B.L.; Parker, H.V.; Kosman, D.J. Fluorescence resonance energy transfer links membrane ferroportin, hephaestin but not ferroportin, amyloid precursor protein complex with iron efflux. J. Biol. Chem. 2019, 294, 4202–4214. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.M.; Rogers, J.T.; Leedman, P.J. Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation. Int. J. Biochem. Cell Biol. 1999, 31, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.M.; Cahill, C.M.; Cho, H.H.; Kassachau, K.D.; Epis, M.R.; Bridges, K.R.; Leedman, P.J.; Rogers, J.T. The acute box cis-element in human heavy ferritin mRNA 5′-untranslated region is a unique translation enhancer that binds poly(c)-binding proteins. J. Biol. Chem. 2005, 280, 30032–30045. [Google Scholar] [CrossRef]
- Rogers, J.T.; Cahill, C.M. Iron-responsive-like Elements and neurodegenerative ferroptosis. Learn. Mem. 2020, 27, 395–413. [Google Scholar] [CrossRef]
- Hin, N.; Newman, M.; Pederson, S.; Lardelli, M. Iron-responsive Element-mediated responses to iron dyshomeostasis in Alzheimer’s disease. J. Alzheimers Dis. 2021, 84, 1597–1630. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Maloney, B.; Rogers, J.T.; Lahiri, D.K. Novel upregulation of amyloid-beta precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5′-untranslated region: Implications in Alzheimer’s disease. Mol. Psychiatry 2019, 24, 345–363. [Google Scholar] [CrossRef]
- Moussavi Nik, S.H.; Wilson, L.; Newman, M.; Croft, K.; Mori, T.A.; Musgrave, I.; Lardelli, M. The bace1-psen-abetapp regulatory axis has an ancient role in response to low oxygen/oxidative stress. J. Alzheimers Dis. 2012, 28, 515–530. [Google Scholar] [CrossRef]
- Newman, M.; Nornes, S.; Martins, R.N.; Lardelli, M.T. Robust homeostasis of presenilin1 protein levels by transcript regulation. Neurosci. Lett. 2012, 519, 14–19. [Google Scholar] [CrossRef]
- Wang, Z.; Jin, M.; Hong, W.; Liu, W.; Reczek, D.; Lagomarsino, V.N.; Hu, Y.; Weeden, T.; Frosch, M.P.; Young-Pearse, T.L.; et al. Learnings about Abeta from human brain recommend the use of a live-neuron bioassay for the discovery of next generation Alzheimer’s disease immunotherapeutics. Acta Neuropathol. Commun. 2023, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Keen, C.L.; Hurley, L.S. Effects of zinc deficiency on prenatal and postnatal development. Neurotoxicology 1987, 8, 379–387. [Google Scholar] [PubMed]
- Chia, S.E.; Foo, S.C.; Gan, S.L.; Jeyaratnam, J.; Tian, C.S. Neurobehavioral functions among workers exposed to manganese ore. Scand. J. Work. Environ. Health 1993, 19, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.K.; Samii, A.; Calne, D.B. Manganese neurotoxicity: A review of clinical features, imaging and pathology. Neurotoxicology 1999, 20, 227–238. [Google Scholar] [PubMed]
- O’Neal, S.L.; Zheng, W. Manganese toxicity upon overexposure: A decade in review. Curr. Environ. Health Rep. 2015, 2, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xin, Y.; Li, Q.; Shang, Y.; Ping, Z.; Min, J.; Cahill, C.M.; Rogers, J.T.; Wang, F. Biomarkers of environmental manganese exposure and associations with childhood neurodevelopment: A systematic review and meta-analysis. Environ. Health 2020, 19, 104. [Google Scholar] [CrossRef] [PubMed]
- Maloney, B.; Bayon, B.L.; Zawia, N.H.; Lahiri, D.K. Latent consequences of early-life Lead (Pb) exposure and the future: Addressing the Pb crisis. Neurotoxicology 2018, 68, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Maloney, B.; Song, W.; Sokol, D.K. Crossing the “birth border” for epigenetic effects. Biol. Psychiatry 2022, 92, e21–e23. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Maloney, B.; Wang, R.; Sokol, D.K.; Rogers, J.T.; Westmark, C.J. How autism and alzheimer’s disease are trapped. Mol. Psychiatry 2021, 26, 26–29. [Google Scholar] [CrossRef]
- Sokol, D.K.; Chen, D.; Farlow, M.R.; Dunn, D.W.; Maloney, B.; Zimmer, J.A.; Lahiri, D.K. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J. Child. Neurol. 2006, 21, 444–449. [Google Scholar] [CrossRef]
- Westmark, C.J.; Sokol, D.K.; Maloney, B.; Lahiri, D.K. Novel roles of amyloid-beta precursor protein metabolites in fragile x syndrome and autism. Mol. Psychiatry 2016, 21, 1333–1341. [Google Scholar] [CrossRef]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes Park9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef]
- Medici, S.; Peana, M.; Delogu, L.G.; Zoroddu, M.A. Mn(ii) and Zn(ii) interactions with peptide fragments from Parkinson’s disease genes. Dalton Trans. 2012, 41, 4378–4388. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.F.; Duhart, H.M.; Newport, G.D.; Lipe, G.W.; Slikker, W., Jr. Manganese-induced reactive oxygen species: Comparison between Mn+2 and Mn+3. Neurodegeneration 1995, 4, 329–334. [Google Scholar] [CrossRef]
- Roels, H.; Lauwerys, R.; Buchet, J.P.; Genet, P.; Sarhan, M.J.; Hanotiau, I.; de Fays, M.; Bernard, A.; Stanescu, D. Epidemiological survey among workers exposed to manganese: Effects on lung, central nervous system, and some biological indices. Am. J. Ind. Med. 1987, 11, 307–327. [Google Scholar] [CrossRef] [PubMed]
- Malecki, E.A.; Devenyi, A.G.; Beard, J.L.; Connor, J.R. Existing and emerging mechanisms for transport of iron and manganese to the brain. J. Neurosci. Res. 1999, 56, 113–122. [Google Scholar] [CrossRef]
- Cordova, F.M.; Aguiar, A.S., Jr.; Peres, T.V.; Lopes, M.W.; Goncalves, F.M.; Pedro, D.Z.; Lopes, S.C.; Pilati, C.; Prediger, R.D.; Farina, M.; et al. Manganese-exposed developing rats display motor deficits and striatal oxidative stress that are reversed by trolox. Arch. Toxicol. 2013, 87, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; An, J.; Wang, Y.; Luo, W.; Wang, W.; Mei, X.; Wu, S.; Chen, J. Intrastriatal manganese chloride exposure causes acute locomotor impairment as well as partial activation of substantia nigra gabaergic neurons. Environ. Toxicol. Pharmacol. 2011, 31, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Madejczyk, M.S.; Ballatori, N. The iron transporter ferroportin can also function as a manganese exporter. Biochim. Biophys. Acta 2012, 1818, 651–657. [Google Scholar] [CrossRef]
- Seo, Y.A.; Elkhader, J.A.; Wessling-Resnick, M. Distribution of manganese and other biometals in flatiron mice. Biometals 2016, 29, 147–155. [Google Scholar] [CrossRef]
- Seo, Y.A.; Wessling-Resnick, M. Ferroportin deficiency impairs manganese metabolism in flatiron mice. FASEB J. 2015, 29, 2726–2733. [Google Scholar] [CrossRef]
- Cipriani, S.; Bakshi, R.; Schwarzschild, M.A. Protection by inosine in a cellular model of Parkinson’s disease. Neuroscience 2014, 274, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Sagher, D.; Strauss, B. Insertion of nucleotides opposite apurinic/apyrimidinic sites in deoxyribonucleic acid during in vitro synthesis: Uniqueness of adenine nucleotides. Biochemistry 1983, 22, 4518–4526. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Bakshi, R.; Logan, R.; Ascherio, A.; Macklin, E.A.; Schwarzschild, M.A. Oral inosine persistently elevates plasma antioxidant capacity in Parkinson’s disease. Mov. Disord. 2016, 31, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, R.; Xu, Y.; Mueller, K.A.; Chen, X.; Granucci, E.; Paganoni, S.; Sadri-Vakili, G.; Schwarzschild, M.A. Urate mitigates oxidative stress and motor neuron toxicity of astrocytes derived from ALS-linked SOD-1(G93A) mutant mice. Mol. Cell Neurosci. 2018, 92, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, R.; Macklin, E.A.; Logan, R.; Zorlu, M.M.; Xia, N.; Crotty, G.F.; Zhang, E.; Chen, X.; Ascherio, A.; Schwarzschild, M.A. Higher urate in LRRK2 mutation carriers resistant to Parkinson disease. Ann. Neurol. 2019, 85, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Burdett, T.C.; Desjardins, C.A.; Logan, R.; Cipriani, S.; Xu, Y.; Schwarzschild, M.A. Disrupted and transgenic urate oxidase alter urate and dopaminergic neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Desjardins, C.A.; Burdett, T.C.; Xu, Y.; Xu, K.; Schwarzschild, M.A. Urate and its transgenic depletion modulate neuronal vulnerability in a cellular model of Parkinson’s disease. PLoS ONE 2012, 7, e37331. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef]
- Proctor, P. Similar functions of uric acid and ascorbate in man? Nature 1970, 228, 868. [Google Scholar] [CrossRef]
- Li, R.C.; Saleem, S.; Zhen, G.; Cao, W.; Zhuang, H.; Lee, J.; Smith, A.; Altruda, F.; Tolosano, E.; Dore, S. Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J. Cereb. Blood Flow. Metab. 2009, 29, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, J.S.; Choi, D.; Lee, Y.; Hong, S.; Choi, J.; Han, S.; Ko, Y.; Kim, J.A.; Kim, Y.M.; et al. Manganese (ii) induces chemical hypoxia by inhibiting hif-prolyl hydroxylase: Implication in manganese-induced pulmonary inflammation. Toxicol. Appl. Pharmacol. 2009, 235, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wood, I.S.; Trayhurn, P. Pcr arrays identify metallothionein-3 as a highly hypoxia-inducible gene in human adipocytes. Biochem. Biophys. Res. Commun. 2008, 368, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.F.; Wilkins, S.E.; Hopkinson, R.J.; Sekirnik, R.; Flashman, E.; Kawamura, A.; McCullagh, J.S.O.; Walport, L.J.; Schofield, C.J. Hypoxia and hypoxia mimetics differentially modulate histone post-translational modifications. Epigenetics 2021, 16, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Bousleiman, J.; Pinsky, A.; Ki, S.; Su, A.; Morozova, I.; Kalachikov, S.; Wiqas, A.; Silver, R.; Sever, M.; Austin, R.N. Function of metallothionein-3 in neuronal cells: Do metal ions alter expression levels of 3? Int. J. Mol. Sci. 2017, 18, 1133. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, D.; Sivanesan, S.; Kannan, K. Manganese-induced neurotoxicity and alterations in gene expression in human neuroblastoma sh-sy5y cells. Biol. Trace Elem. Res. 2018, 183, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Meloni, G.; Vasak, M. Redox activity of alpha-synuclein-Cu is silenced by Zn7-metallothionein-3. Free Radic. Biol. Med. 2011, 50, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M. Multifunctional metallothioneins as a target for neuroprotection in Parkinson’s disease. Antioxidants 2023, 12, 894. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBIgeo: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models for microarray data. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. In Statistics for Biology and Health; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. Reactomegsa-efficient multi-omics comparative pathway analysis. Mol. Cell Proteom. 2020, 19, 2115–2125. [Google Scholar] [CrossRef]
- Wu, D.; Smyth, G.K. Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012, 40, e133. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Rossner, C.; Iffland, L.; Schweyer, S.; Tamboli, I.Y.; Walter, J.; Wirths, O.; Bayer, T.A. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the alzheimer amyloid precursor protein. J. Biol. Chem. 2010, 285, 10678–10689. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Venkataramani, V.; Washburn, C.; Liu, Y.; Tummala, V.; Jiang, H.; Smith, A.; Cahill, C.M. A role for amyloid precursor protein translation to restore iron homeostasis and ameliorate lead (Pb) neurotoxicity. J. Neurochem. 2016, 138, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, Y.S.; Kim, J.M.; Kim, K.L.; Lee, J.S.; Jang, H.S.; Shin, I.S.; Suh, W.; Jeon, E.S.; Byun, J.; et al. A novel chimeric promoter that is highly responsive to hypoxia and metals. Gene Ther. 2006, 13, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, L.L.; Li, G.J.; Guo, W.; Liang, W.; Zheng, W. Alteration of serum concentrations of manganese, iron, ferritin, and transferrin receptor following exposure to welding fumes among career welders. Neurotoxicology 2005, 26, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Harischandra, D.S.; Rokad, D.; Neal, M.L.; Ghaisas, S.; Manne, S.; Sarkar, S.; Panicker, N.; Zenitsky, G.; Jin, H.; Lewis, M.; et al. Manganese promotes the aggregation and prion-like cell-to-cell exosomal transmission of alpha-synuclein. Sci. Signal 2019, 12, eaau4543. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Lukiw, W.J.; Bergeron, C.; Niznik, H.B.; Fraser, P.E. Metallothionein-III is reduced in Alzheimer’s disease. Brain Res. 2001, 894, 37–45. [Google Scholar] [CrossRef]
- Smith, M.R.; Fernandes, J.; Go, Y.M.; Jones, D.P. Redox dynamics of manganese as a mitochondrial life-death switch. Biochem. Biophys. Res. Commun. 2017, 482, 388–398. [Google Scholar] [CrossRef]
- Cahill, C.M.; Rogers, J.T. Interleukin (il) 1beta induction of il-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent akt/ikappab kinase alpha pathway targeting activator protein-1. J. Biol. Chem. 2008, 283, 25900–25912. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, S.; Gan, L.; Vosler, P.S.; Gao, Y.; Zigmond, M.J.; Chen, J. Protective effects and mechanisms of sirtuins in the nervous system. Prog. Neurobiol. 2011, 95, 373–395. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Mittal, A.K.; Kalonia, H.; Madan, S.; Ghosh, S.; Sinha, J.K.; Rajput, S.K. Sirt1 promotes neuronal fortification in neurodegenerative diseases through attenuation of pathological hallmarks and enhancement of cellular lifespan. Curr. Neuropharmacol. 2021, 19, 1019–1037. [Google Scholar] [PubMed]
- Morrison, B.E.; Majdzadeh, N.; Zhang, X.; Lyles, A.; Bassel-Duby, R.; Olson, E.N.; D’Mello, S.R. Neuroprotection by histone deacetylase-related protein. Mol. Cell Biol. 2006, 26, 3550–3564. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the role of histone deacetylase and their inhibitors in neurodegenerative disorders: Current targets and future perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Bakshi, R.; Zhang, H.; Logan, R.; Joshi, I.; Xu, Y.; Chen, X.; Schwarzschild, M.A. Neuroprotective effects of urate are mediated by augmenting astrocytic glutathione synthesis and release. Neurobiol. Dis. 2015, 82, 574–579. [Google Scholar] [CrossRef]
- Shaw, K.T.; Utsuki, T.; Rogers, J.; Yu, Q.S.; Sambamurti, K.; Brossi, A.; Ge, Y.W.; Lahiri, D.K.; Greig, N.H. Phenserine regulates translation of beta -amyloid precursor protein mrna by a putative interleukin-1 responsive element, a target for drug development. Proc. Natl. Acad. Sci. USA 2001, 98, 7605–7610. [Google Scholar] [CrossRef]
- Yu, Q.S.; Reale, M.; Kamal, M.A.; Holloway, H.W.; Luo, W.; Sambamurti, K.; Ray, B.; Lahiri, D.K.; Rogers, J.T.; Greig, N.H. Synthesis of the alzheimer drug posiphen into its primary metabolic products (+)-n1-norposiphen, (+)-n8-norposiphen and (+)-n1, n8-bisnorposiphen, their inhibition of amyloid precursor protein, alpha-synuclein synthesis, interleukin-1beta release, and cholinergic action. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2013, 12, 117–128. [Google Scholar]
- Kopp, K.O.; Greer, M.E.; Glotfelty, E.J.; Hsueh, S.C.; Tweedie, D.; Kim, D.S.; Reale, M.; Vargesson, N.; Greig, N.H. A new generation of imids as treatments for neuroinflammatory and neurodegenerative disorders. Biomolecules 2023, 13, 747. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Perry, G.; Fang, X.; Zagorski, M.; Sambamurti, K.; Poeggeler, B. Indoles as essential mediators in the gut-brain axis. Their role in alzheimer’s disease. Neurobiol. Dis. 2021, 156, 105403. [Google Scholar] [CrossRef]
- Sambamurti, K.; Greig, N.H.; Utsuki, T.; Barnwell, E.L.; Sharma, E.; Mazell, C.; Bhat, N.R.; Kindy, M.S.; Lahiri, D.K.; Pappolla, M.A. Targets for ad treatment: Conflicting messages from gamma-secretase inhibitors. J. Neurochem. 2011, 117, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Lei, P.; Hare, D.J.; Duce, J.A.; George, J.L.; Adlard, P.A.; McLean, C.; Rogers, J.T.; Cherny, R.A.; Finkelstein, D.I.; et al. Parkinson’s disease iron deposition caused by nitric oxide-induced loss of beta-amyloid precursor protein. J. Neurosci. 2015, 35, 3591–3597. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Cahill, C.M. Iron responsiveness to lysosomal disruption: A novel pathway to Alzheimer’s disease. J. Alzheimers Dis. 2023, 96, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Lardelli, M. An alternative view of familial Alzheimer’s disease genetics. J. Alzheimers Dis. 2023, 96, 13–39. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Cahill, C.; Balleidier, A.; Huang, C.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Novel 5′ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: Implications for down syndrome and alzheimer’s disease. PLoS ONE 2013, 8, e65978. [Google Scholar] [CrossRef] [PubMed]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. eLife 2019, 8, e51031. [Google Scholar] [CrossRef] [PubMed]
- Im, E.; Jiang, Y.; Stavrides, P.H.; Darji, S.; Erdjument-Bromage, H.; Neubert, T.A.; Choi, J.Y.; Wegiel, J.; Lee, J.H.; Nixon, R.A. Lysosomal dysfunction in Down syndrome and Alzheimer mouse models is caused by v-atpase inhibition by tyr682-phosphorylated app betactf. Sci. Adv. 2023, 9, eadg1925. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Morse, L.; Lombardi, M.; Devaney, P.; Jensen, R.V. Metallothionein 3 gene expression by microarray profiling is greater than 100 fold up-regulated during intracellular iron chelation, the role of a putative iron-responsive element in the 3′ untranslated region. Biomark. Res. 2004, 12, 38. [Google Scholar]
- Morse, L.J.; Payton, S.M.; Cuny, G.D.; Rogers, J.T. Fda-preapproved drugs targeted to the translational regulation and processing of the amyloid precursor protein. J. Mol. Neurosci. 2004, 24, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yin, X.; Ma, N.; Lin, F.; Kong, X.; Chi, J.; Feng, Z. Desferrioxamine regulates Hif-1 alpha expression in neonatal rat brain after hypoxia-ischemia. Am. J. Transl. Res. 2014, 6, 377–383. [Google Scholar]
- Shin, H.J.; Choi, M.S.; Ryoo, N.H.; Nam, K.Y.; Park, G.Y.; Bae, J.H.; Suh, S.I.; Baek, W.K.; Park, J.W.; Jang, B.C. Manganese-mediated up-regulation of hif-1alpha protein in hep2 human laryngeal epithelial cells via activation of the family of mapks. Toxicol Vitr. 2010, 24, 1208–1214. [Google Scholar] [CrossRef]
- Dosunmu, R.; Wu, J.; Adwan, L.; Maloney, B.; Basha, M.R.; McPherson, C.A.; Harry, G.J.; Rice, D.C.; Zawia, N.H.; Lahiri, D.K. Lifespan profiles of alzheimer’s disease-associated genes and products in monkeys and mice. J. Alzheimers Dis. 2009, 18, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Maloney, B.; Basha, M.R.; Ge, Y.W.; Zawia, N.H. How and when environmental agents and dietary factors affect the course of alzheimer’s disease: The “learn” model (latent early-life associated regulation) may explain the triggering of ad. Curr. Alzheimer Res. 2007, 4, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M.; Hanawalt, P.C. Efficient protection against oxidative DNA damage in chromatin. Mol. Carcinog. 1992, 5, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(a) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.R.; Jin, C. Stem-loop binding protein and metal carcinogenesis. Semin. Cancer Biol. 2021, 76, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.M.; Cunningham, C.H.; Welch, J.D.; Groh, B.; Guo, A.Y.; Wei, B.; Whitfield, M.L.; Xiong, Y.; Marzluff, W.F. A subset of replication-dependent histone mRNAs are expressed as polyadenylated RNAs in terminally differentiated tissues. Nucleic Acids Res. 2016, 44, 9190–9205. [Google Scholar] [CrossRef] [PubMed]
- Pagana, K.; Pagana, T.; Pagana, T. Emedicine blood transferrin receptor measurements. In Mosby’s Diagnostic and Laboratory Test Reference, 14th ed.; Mosby Inc.: St. Louis, MO, USA, 2019; p. 906. [Google Scholar]
- Zhang, S.; Chen, L.; Chen, T.; Zhang, Y.; Ma, J.; Ji, H.; Guo, C.; Xiao, Z.; Li, J.; Niu, P. Transcriptome evidence reveals mitochondrial unfolded protein response participate in sh-sy5y cells exposed to manganese. J. Integr. Neurosci. 2022, 21, 127. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Niu, L.; Zhang, C.; Zhu, C.; Xie, F.; Cao, C.; Li, G. Brain edema and protein expression of c-fos and c-jun in the brain after diffused brain injury. Int. J. Clin. Exp. Pathol. 2014, 7, 2809–2817. [Google Scholar] [PubMed]
- Tanaka, K.; Nagata, E.; Suzuki, S.; Dembo, T.; Nogawa, S.; Fukuuchi, Y. Immunohistochemical analysis of cyclic amp response element binding protein phosphorylation in focal cerebral ischemia in rats. Brain Res. 1999, 818, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Rossler, O.G.; Thiel, G. Specificity of stress-responsive transcription factors nrf2, atf4, and ap-1. J. Cell Biochem. 2017, 118, 127–140. [Google Scholar] [CrossRef]
- Thiel, G.; Rossler, O.G. Hyperforin activates gene transcription involving transient receptor potential c6 channels. Biochem. Pharmacol. 2017, 129, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rossler, O.G. Resveratrol regulates gene transcription via activation of stimulus-responsive transcription factors. Pharmacol. Res. 2017, 117, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Amro, Z.; Ryan, M.; Collins-Praino, L.E.; Yool, A.J. Unexpected classes of aquaporin channels detected by transcriptomic analysis in human brain are associated with both patient age and alzheimer’s disease status. Biomedicines 2023, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W.; Schumacher, A.; Maloney, B.; Lahiri, D.K.; Zawia, N.H. Do epigenetic pathways initiate late onset alzheimer disease (load): Towards a new paradigm. Curr. Alzheimer Res. 2012, 9, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Maloney, B.; Lahiri, D.K. Epigenetics of dementia: Understanding the disease as a transformation rather than a state. Lancet Neurol. 2016, 15, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Tarale, P.; Sivanesan, S.; Daiwile, A.P.; Stoger, R.; Bafana, A.; Naoghare, P.K.; Parmar, D.; Chakrabarti, T.; Kannan, K. Global DNA methylation profiling of manganese-exposed human neuroblastoma sh-sy5y cells reveals epigenetic alterations in Parkinson’s disease-associated genes. Arch. Toxicol. 2017, 91, 2629–2641. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, Z.; Wang, Q.; Zhang, J.; Wang, L.; Zhang, Q.; Li, H.; Wu, S. Manganese chloride induces histone acetylation changes in neuronal cells: Its role in manganese-induced damage. Neurotoxicology 2018, 65, 255–263. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).