
Identification of Prospective Ebola Virus VP35 and VP40 Protein Inhibitors from Myxobacterial Natural Products

Abstract
:1. Introduction
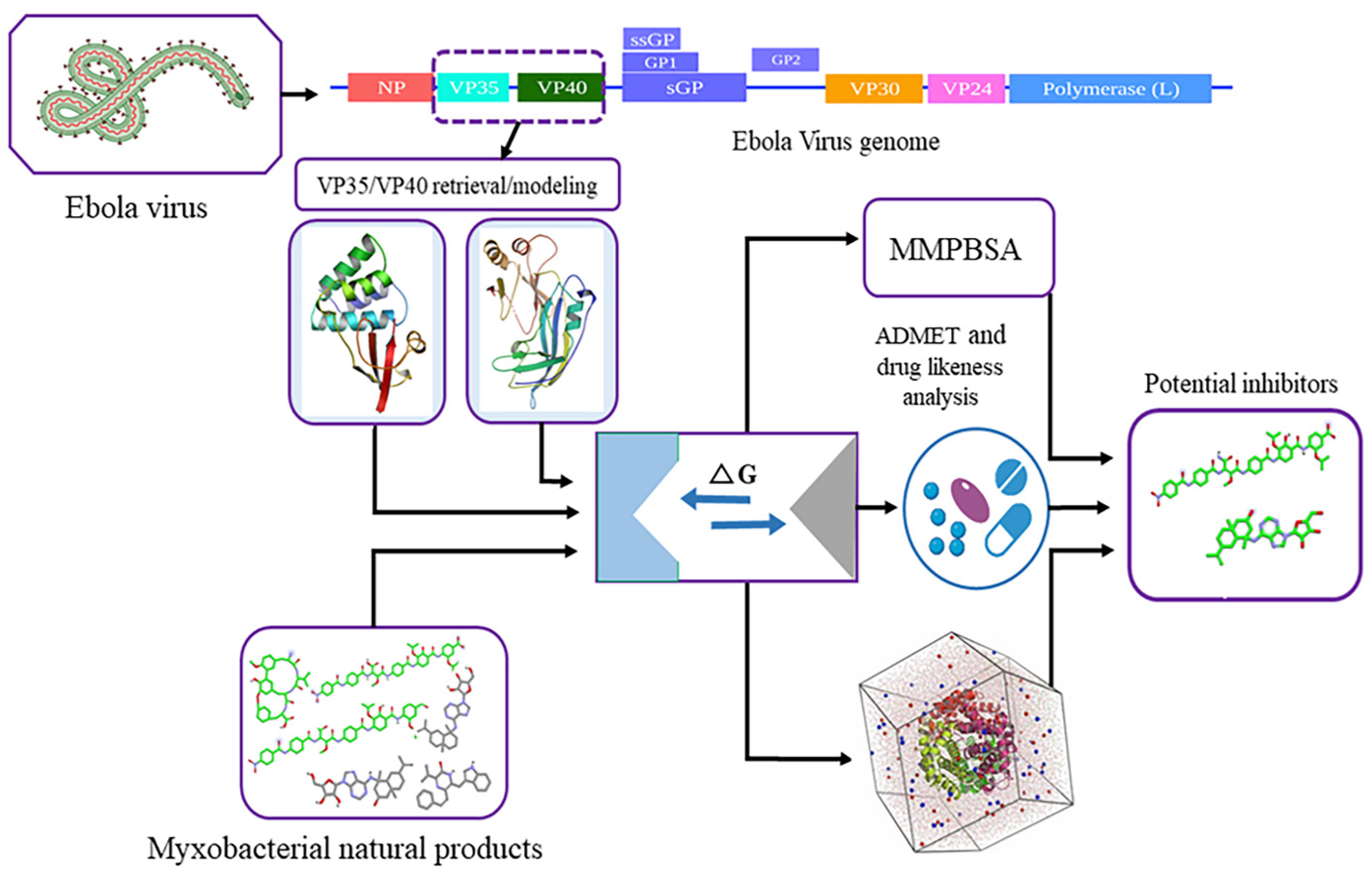
2. Materials and Methods
2.1. Retrieval and Structural Evaluation of Proteins
2.2. Remodeling of VP40 Protein
2.3. Myxobacterial Natural Product Dataset Preparation
2.4. Binding Site Evaluation
2.5. Virtual Library Screening
2.6. Molecular Dynamic Simulation Analysis
2.7. Binding Energy Calculations
2.8. Computational Pharmacokinetics
3. Results
3.1. Protein Structure and Binding Sites Prediction of EBOV VP35 and VP40
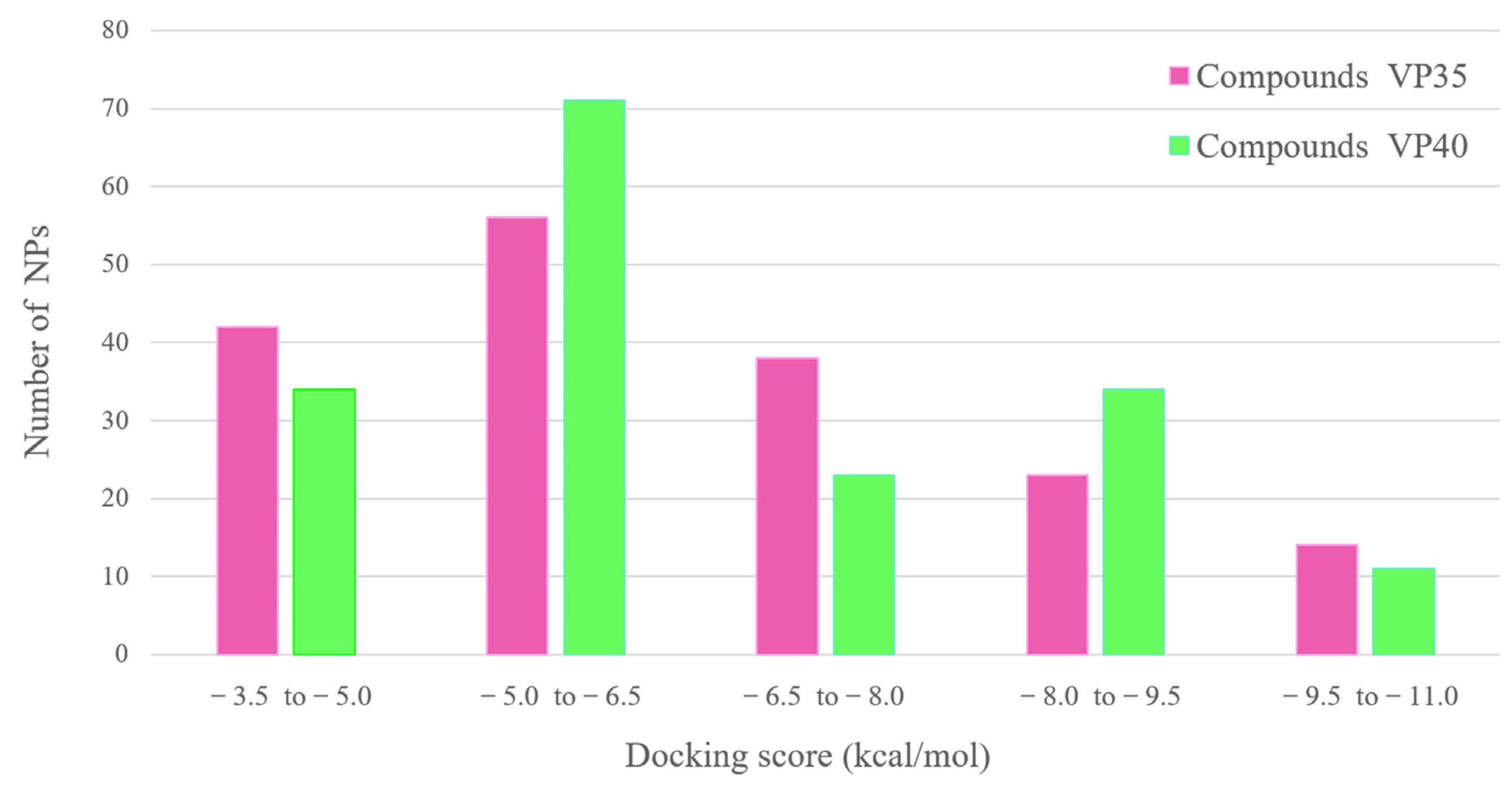
3.2. Molecular Docking Analysis of Myxobacterial Natural Products against EBOV VP35 or VP40
3.3. Molecular Dynamic Simulations of the Main Compounds
3.4. Protein–Ligand Contacts and MMPBSA Binding Energy Calculations of the Main Compounds
3.5. Drug-Likeness and Pharmacokinetics of Bioactive Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geisbert, T.W.; Hensley, L.E. Ebola Virus: New Insights into Disease Aetiopathology and Possible Therapeutic Interventions. Expert Rev. Mol. Med. 2004, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, H.; Sprecher, A.; Geisbert, T.W. Ebola. N. Engl. J. Med. 2020, 382, 1832–1842. [Google Scholar] [CrossRef]
- Jacob, S.T.; Crozier, I.; Fischer, W.A.; Hewlett, A.; Kraft, C.S.; de L. Vega, M.-A.; Soka, M.J.; Wahl, V.; Griffiths, A.; Bollinger, L.; et al. Ebola Virus Disease. Nat. Rev. Dis. Primer 2020, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- WHO Ebola Response Team; Aylward, B.; Barboza, P.; Bawo, L.; Bertherat, E.; Bilivogui, P.; Blake, I.; Brennan, R.; Briand, S.; Chakauya, J.M.; et al. Ebola Virus Disease in West Africa--the First 9 Months of the Epidemic and Forward Projections. N. Engl. J. Med. 2014, 371, 1481–1495. [Google Scholar] [CrossRef] [PubMed]
- Billioux, B.J.; Smith, B.; Nath, A. Neurological Complications of Ebola Virus Infection. Neurotherapeutics 2016, 13, 461–470. [Google Scholar] [CrossRef] [PubMed]
- MacDermott, N.; Herberg, J.A. Ebola: Lessons Learned. Paediatr. Child Health 2017, 27, 128–134. [Google Scholar] [CrossRef]
- Redding, D.W.; Atkinson, P.M.; Cunningham, A.A.; Lo Iacono, G.; Moses, L.M.; Wood, J.L.N.; Jones, K.E. Impacts of Environmental and Socio-Economic Factors on Emergence and Epidemic Potential of Ebola in Africa. Nat. Commun. 2019, 10, 4531. [Google Scholar] [CrossRef]
- Takamatsu, Y.; Kolesnikova, L.; Becker, S. Ebola Virus Proteins NP, VP35, and VP24 Are Essential and Sufficient to Mediate Nucleocapsid Transport. Proc. Natl. Acad. Sci. USA 2018, 115, 1075–1080. [Google Scholar] [CrossRef]
- Balmith, M.; Soliman, M.E.S. Potential Ebola Drug Targets—Filling the Gap: A Critical Step Forward towards the Design and Discovery of Potential Drugs. Biologia 2017, 72, 1–13. [Google Scholar] [CrossRef]
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.-J.; Feldmann, H. A New Ebola Virus Nonstructural Glycoprotein Expressed through RNA Editing. J. Virol. 2011, 85, 5406–5414. [Google Scholar] [CrossRef]
- Takada, A.; Kawaoka, Y. The Pathogenesis of Ebola Hemorrhagic Fever. Trends Microbiol. 2001, 9, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Olukitibi, T.A.; Ao, Z.; Mahmoudi, M.; Kobinger, G.A.; Yao, X. Dendritic Cells/Macrophages-Targeting Feature of Ebola Glycoprotein and Its Potential as Immunological Facilitator for Antiviral Vaccine Approach. Microorganisms 2019, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Mühlberger, E.; Lötfering, B.; Klenk, H.D.; Becker, S. Three of the Four Nucleocapsid Proteins of Marburg Virus, NP, VP35, and L, Are Sufficient to Mediate Replication and Transcription of Marburg Virus-Specific Monocistronic Minigenomes. J. Virol. 1998, 72, 8756–8764. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Wang, X.; Mühlberger, E.; Volchkov, V.; Paragas, J.; Klenk, H.D.; García-Sastre, A.; Palese, P. The Ebola Virus VP35 Protein Functions as a Type I IFN Antagonist. Proc. Natl. Acad. Sci. USA 2000, 97, 12289–12294. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; García-Sastre, A. Viruses and the Type I Interferon Antiviral System: Induction and Evasion. Int. Rev. Immunol. 2002, 21, 305–337. [Google Scholar] [CrossRef] [PubMed]
- Haasnoot, J.; de Vries, W.; Geutjes, E.-J.; Prins, M.; de Haan, P.; Berkhout, B. The Ebola Virus VP35 Protein Is a Suppressor of RNA Silencing. PLoS Pathog. 2007, 3, e86. [Google Scholar] [CrossRef] [PubMed]
- Jasenosky, L.D.; Cadena, C.; Mire, C.E.; Borisevich, V.; Haridas, V.; Ranjbar, S.; Nambu, A.; Bavari, S.; Soloveva, V.; Sadukhan, S.; et al. The FDA-Approved Oral Drug Nitazoxanide Amplifies Host Antiviral Responses and Inhibits Ebola Virus. iScience 2019, 19, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Rüth, F.X.; Dessen, A.; Timmins, J.; Bracher, A.; Kolesnikowa, L.; Becker, S.; Klenk, H.D.; Weissenhorn, W. The Matrix Protein VP40 from Ebola Virus Octamerizes into Pore-like Structures with Specific RNA Binding Properties. Struct. Lond. Engl. 1993 2003, 11, 423–433. [Google Scholar] [CrossRef]
- Harty, R.N.; Brown, M.E.; Wang, G.; Huibregtse, J.; Hayes, F.P. A PPxY Motif within the VP40 Protein of Ebola Virus Interacts Physically and Functionally with a Ubiquitin Ligase: Implications for Filovirus Budding. Proc. Natl. Acad. Sci. USA 2000, 97, 13871–13876. [Google Scholar] [CrossRef]
- Noda, T.; Sagara, H.; Suzuki, E.; Takada, A.; Kida, H.; Kawaoka, Y. Ebola Virus VP40 Drives the Formation of Virus-like Filamentous Particles along with GP. J. Virol. 2002, 76, 4855–4865. [Google Scholar] [CrossRef]
- Stahelin, R.V. Membrane Binding and Bending in Ebola VP40 Assembly and Egress. Front. Microbiol. 2014, 5, 300. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.; Scianimanico, S.; Schoehn, G.; Weissenhorn, W. Vesicular Release of Ebola Virus Matrix Protein VP40. Virology 2001, 283, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dessen, A.; Volchkov, V.; Dolnik, O.; Klenk, H.D.; Weissenhorn, W. Crystal Structure of the Matrix Protein VP40 from Ebola Virus. EMBO J. 2000, 19, 4228–4236. [Google Scholar] [CrossRef] [PubMed]
- Adu-Gyamfi, E.; Soni, S.P.; Xue, Y.; Digman, M.A.; Gratton, E.; Stahelin, R.V. The Ebola Virus Matrix Protein Penetrates into the Plasma Membrane: A Key Step in Viral Protein 40 (VP40) Oligomerization and Viral Egress. J. Biol. Chem. 2013, 288, 5779–5789. [Google Scholar] [CrossRef]
- McCarthy, S.E.; Johnson, R.F.; Zhang, Y.-A.; Sunyer, J.O.; Harty, R.N. Role for Amino Acids 212KLR214 of Ebola Virus VP40 in Assembly and Budding. J. Virol. 2007, 81, 11452–11460. [Google Scholar] [CrossRef] [PubMed]
- Fabozzi, G.; Nabel, C.S.; Dolan, M.A.; Sullivan, N.J. Ebolavirus Proteins Suppress the Effects of Small Interfering RNA by Direct Interaction with the Mammalian RNA Interference Pathway. J. Virol. 2011, 85, 2512–2523. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kuo, C.-J.; Shin, J.S.; Jung, E.; Liang, P.-H.; Jung, Y.-S. Identification of Non-Covalent 3C-like Protease Inhibitors against Severe Acute Respiratory Syndrome Coronavirus-2 via Virtual Screening of a Korean Compound Library. Bioorg. Med. Chem. Lett. 2021, 42, 128067. [Google Scholar] [CrossRef]
- Hansen, F.; Feldmann, H.; Jarvis, M.A. Targeting Ebola Virus Replication through Pharmaceutical Intervention. Expert Opin. Investig. Drugs 2021, 30, 201–226. [Google Scholar] [CrossRef]
- Markham, A. REGN-EB3: First Approval. Drugs 2021, 81, 175–178. [Google Scholar] [CrossRef]
- Saxena, D.; Kaul, G.; Dasgupta, A.; Chopra, S. Atoltivimab/Maftivimab/Odesivimab (Inmazeb) Combination to Treat Infection Caused by Zaire Ebolavirus. Drugs Today Barc. Spain 1998 2021, 57, 483–490. [Google Scholar] [CrossRef]
- Lane, T.; Anantpadma, M.; Freundlich, J.S.; Davey, R.A.; Madrid, P.B.; Ekins, S. The Natural Product Eugenol Is an Inhibitor of the Ebola Virus In Vitro. Pharm. Res. 2019, 36, 104. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, H. Myxobacteria, Producers of Novel Bioactive Substances. J. Ind. Microbiol. Biotechnol. 2001, 27, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Gerth, K.; Pradella, S.; Perlova, O.; Beyer, S.; Müller, R. Myxobacteria: Proficient Producers of Novel Natural Products with Various Biological Activities--Past and Future Biotechnological Aspects with the Focus on the Genus Sorangium. J. Biotechnol. 2003, 106, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Mohr, K.I.; Moradi, A.; Glaeser, S.P.; Kämpfer, P.; Gemperlein, K.; Nübel, U.; Schumann, P.; Müller, R.; Wink, J. Nannocystis Konarekensis Sp. Nov., a Novel Myxobacterium from an Iranian Desert. Int. J. Syst. Evol. Microbiol. 2018, 68, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Fayad, A.A.; Müller, R. Natural Products from Myxobacteria: Novel Metabolites and Bioactivities. Nat. Prod. Rep. 2017, 34, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J.; Müller, R. Myxobacterial Secondary Metabolites: Bioactivities and Modes-of-Action. Nat. Prod. Rep. 2010, 27, 1276–1295. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Müller, R. The Impact of Genomics on the Exploitation of the Myxobacterial Secondary Metabolome. Nat. Prod. Rep. 2009, 26, 1385. [Google Scholar] [CrossRef] [PubMed]
- Hüttel, S.; Testolin, G.; Herrmann, J.; Planke, T.; Gille, F.; Moreno, M.; Stadler, M.; Brönstrup, M.; Kirschning, A.; Müller, R. Discovery and Total Synthesis of Natural Cystobactamid Derivatives with Superior Activity against Gram-Negative Pathogens. Angew. Chem. Int. Ed Engl. 2017, 56, 12760–12764. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Romero-Brey, I.; Berger, C.; Pérez-Vilaró, G.; Monteiro Perin, P.; Vondran, F.W.R.; Kalesse, M.; Harmrolfs, K.; Müller, R.; Martinez, J.P.; et al. Soraphen A: A Broad-Spectrum Antiviral Natural Product with Potent Anti-Hepatitis C Virus Activity. J. Hepatol. 2015, 63, 813–821. [Google Scholar] [CrossRef]
- Beck, S.; Henß, L.; Weidner, T.; Herrmann, J.; Müller, R.; Chao, Y.-K.; Grimm, C.; Weber, C.; Sliva, K.; Schnierle, B.S. Identification of Entry Inhibitors of Ebola Virus Pseudotyped Vectors from a Myxobacterial Compound Library. Antiviral Res. 2016, 132, 85–91. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB Protein Data Bank: Integrative View of Protein, Gene and 3D Structural Information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Willard, L. VADAR: A Web Server for Quantitative Evaluation of Protein Structure Quality. Nucleic Acids Res. 2003, 31, 3316–3319. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Cousins, K.R. ChemDraw Ultra 9.0. CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. Www. Cambridgesoft.Com. See Web Site for Pricing Options. J. Am. Chem. Soc. 2005, 127, 4115–4116. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; IEEE: Piscataway Township, NJ, USA, 2006; p. 43. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Shaw, D.E. A Fast, Scalable Method for the Parallel Evaluation of Distance-Limited Pairwise Particle Interactions. J. Comput. Chem. 2005, 26, 1318–1328. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Tsai, H.-H.G.; Tsai, C.-J.; Ma, B.; Nussinov, R. In Silico Protein Design by Combinatorial Assembly of Protein Building Blocks. Protein Sci. 2009, 13, 2753–2765. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Nasution, M.A.F.; Alkaff, A.H.; Tambunan, U.S.F. Discovery of Indonesian Natural Products as Potential Inhibitor of Ebola Virus VP40 through Molecular Docking Simulation; AIP. Conference Proceedings: Bali, Indonesia, 2018; p. 020055. [Google Scholar]
- Yu, W.; MacKerell, A.D. Computer-Aided Drug Design Methods. Methods Mol. Biol. Clifton NJ 2017, 1520, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K.S. Bringing Molecular Dynamics Simulation Data into View. Trends Biochem. Sci. 2019, 44, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of Lead Compounds against Scm (Fms10) in Enterococcus Faecium Using Computer Aided Drug Designing. Life 2021, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Karlgren, M.; Bergström, C.A.S. How Physicochemical Properties of Drugs Affect Their Metabolism and Clearance. In New Horizons in Predictive Drug Metabolism and Pharmacokinetics; Wilson, A.G.E., Ed.; The Royal Society of Chemistry: London, UK, 2015; pp. 1–26. ISBN 978-1-84973-828-6. [Google Scholar]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. G_mmpbsa —A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lohit, N.; Singh, A.K.; Kumar, A.; Singh, H.; Yadav, J.P.; Singh, K.; Kumar, P. Description and In Silico ADME Studies of US-FDA Approved Drugs orDrugs under Clinical Trial Which Violate the Lipinski’s Rule of 5. Lett. Drug Des. Discov. 2024, 21, 1334–1358. [Google Scholar] [CrossRef]
- Testolin, G.; Cirnski, K.; Rox, K.; Prochnow, H.; Fetz, V.; Grandclaudon, C.; Mollner, T.; Baiyoumy, A.; Ritter, A.; Leitner, C.; et al. Synthetic Studies of Cystobactamids as Antibiotics and Bacterial Imaging Carriers Lead to Compounds with High in Vivo Efficacy. Chem. Sci. 2020, 11, 1316–1334. [Google Scholar] [CrossRef] [PubMed]
- Elston, J.W.T.; Cartwright, C.; Ndumbi, P.; Wright, J. The Health Impact of the 2014-15 Ebola Outbreak. Public Health 2017, 143, 60–70. [Google Scholar] [CrossRef]
- Broni, E.; Ashley, C.; Adams, J.; Manu, H.; Aikins, E.; Okom, M.; Miller, W.A.; Wilson, M.D.; Kwofie, S.K. Cheminformatics-Based Study Identifies Potential Ebola VP40 Inhibitors. Int. J. Mol. Sci. 2023, 24, 6298. [Google Scholar] [CrossRef]
- Mirza, M.; Ikram, N. Integrated Computational Approach for Virtual Hit Identification against Ebola Viral Proteins VP35 and VP40. Int. J. Mol. Sci. 2016, 17, 1748. [Google Scholar] [CrossRef] [PubMed]
- Pleško, S. In Silico Study of Plant Polyphenols’ Interactions with VP24–Ebola Virus Matrix Protein. Acta Chim. Slov. 2015, 62, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Raj, U.; Varadwaj, P.K. Flavonoids as Multi-Target Inhibitors for Proteins Associated with Ebola Virus: In Silico Discovery Using Virtual Screening and Molecular Docking Studies. Interdiscip. Sci. Comput. Life Sci. 2016, 8, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, S. Comparative insillico studies on phytochemicals of ocimum as natural inhibitors of ebola vp-35 protein. Indo Am. J. Pharm. Sci. 2019, 10, 489–511. [Google Scholar] [CrossRef]
- Ekins, S.; Freundlich, J.S.; Coffee, M. A Common Feature Pharmacophore for FDA-Approved Drugs Inhibiting the Ebola Virus. F1000Research 2014, 3, 277. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Borek, D.; Luthra, P.; Binning, J.M.; Anantpadma, M.; Liu, G.; Harvey, I.B.; Su, Z.; Endlich-Frazier, A.; Pan, J.; et al. An Intrinsically Disordered Peptide from Ebola Virus VP35 Controls Viral RNA Synthesis by Modulating Nucleoprotein-RNA Interactions. Cell Rep. 2015, 11, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Shu, T.; Gan, T.; Bai, P.; Wang, X.; Qian, Q.; Zhou, H.; Cheng, Q.; Qiu, Y.; Yin, L.; Zhong, J.; et al. Ebola Virus VP35 Has Novel NTPase and Helicase-like Activities. Nucleic Acids Res. 2019, 47, 5837–5851. [Google Scholar] [CrossRef]
- Banerjee, A.; Mitra, P. Ebola Virus VP35 Protein: Modeling of the Tetrameric Structure and an Analysis of Its Interaction with Human PKR. J. Proteome Res. 2020, 19, 4533–4542. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.S.; Lee, M.S.; Leung, D.W.; Wang, T.; Xu, W.; Luthra, P.; Anantpadma, M.; Shabman, R.S.; Melito, L.M.; MacMillan, K.S.; et al. In Silico Derived Small Molecules Bind the Filovirus VP35 Protein and Inhibit Its Polymerase Cofactor Activity. J. Mol. Biol. 2014, 426, 2045–2058. [Google Scholar] [CrossRef]
- Dilley, K.A.; Voorhies, A.A.; Luthra, P.; Puri, V.; Stockwell, T.B.; Lorenzi, H.; Basler, C.F.; Shabman, R.S. The Ebola Virus VP35 Protein Binds Viral Immunostimulatory and Host RNAs Identified through Deep Sequencing. PLoS ONE 2017, 12, e0178717. [Google Scholar] [CrossRef]
- Prins, K.C.; Binning, J.M.; Shabman, R.S.; Leung, D.W.; Amarasinghe, G.K.; Basler, C.F. Basic Residues within the Ebolavirus VP35 Protein Are Required for Its Viral Polymerase Cofactor Function. J. Virol. 2010, 84, 10581–10591. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, C.R.; Bornholdt, Z.A.; Li, S.; Woods, V.L.; MacRae, I.J.; Saphire, E.O. Ebolavirus VP35 Uses a Bimodal Strategy to Bind dsRNA for Innate Immune Suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Ginder, N.D.; Fulton, D.B.; Nix, J.; Basler, C.F.; Honzatko, R.B.; Amarasinghe, G.K. Structure of the Ebola VP35 Interferon Inhibitory Domain. Proc. Natl. Acad. Sci. USA 2009, 106, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Prins, K.C.; Borek, D.M.; Farahbakhsh, M.; Tufariello, J.M.; Ramanan, P.; Nix, J.C.; Helgeson, L.A.; Otwinowski, Z.; Honzatko, R.B.; et al. Structural Basis for dsRNA Recognition and Interferon Antagonism by Ebola VP35. Nat. Struct. Mol. Biol. 2010, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Darko, L.K.S.; Broni, E.; Amuzu, D.S.Y.; Wilson, M.D.; Parry, C.S.; Kwofie, S.K. Computational Study on Potential Novel Anti-Ebola Virus Protein VP35 Natural Compounds. Biomedicines 2021, 9, 1796. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Byrne, B.M.; McCoy, A.M.; James, B.J.; Frank, J.D.; Oakley, G.G. In Silico and in Vitro Methods to Identify Ebola Virus VP35-dsRNA Inhibitors. Bioorg. Med. Chem. 2016, 24, 5388–5392. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Fanunza, E.; Salata, C.; Morwitzer, M.J.; Distinto, S.; Zinzula, L.; Sanna, C.; Frau, A.; Daino, G.L.; Quartu, M.; et al. Cynarin Blocks Ebola Virus Replication by Counteracting VP35 Inhibition of Interferon-Beta Production. Antiviral Res. 2022, 198, 105251. [Google Scholar] [CrossRef] [PubMed]
- Daino, G.L.; Frau, A.; Sanna, C.; Rigano, D.; Distinto, S.; Madau, V.; Esposito, F.; Fanunza, E.; Bianco, G.; Taglialatela-Scafati, O.; et al. Identification of Myricetin as an Ebola Virus VP35–Double-Stranded RNA Interaction Inhibitor through a Novel Fluorescence-Based Assay. Biochemistry 2018, 57, 6367–6378. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Jahrling, P.B. Differentiation of Filoviruses by Electron Microscopy. Virus Res. 1995, 39, 129–150. [Google Scholar] [CrossRef]
- Hoenen, T.; Volchkov, V.; Kolesnikova, L.; Mittler, E.; Timmins, J.; Ottmann, M.; Reynard, O.; Becker, S.; Weissenhorn, W. VP40 Octamers Are Essential for Ebola Virus Replication. J. Virol. 2005, 79, 1898–1905. [Google Scholar] [CrossRef]
- Han, Z.; Ruthel, G.; Dash, S.; Berry, C.T.; Freedman, B.D.; Harty, R.N.; Shtanko, O. Angiomotin Regulates Budding and Spread of Ebola Virus. J. Biol. Chem. 2020, 295, 8596–8601. [Google Scholar] [CrossRef] [PubMed]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping Motifs (PTAP and PPEY) within the Ebola Virus VP40 Protein Function Independently as Late Budding Domains: Involvement of Host Proteins TSG101 and VPS-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Bornholdt, Z.A.; Noda, T.; Abelson, D.M.; Halfmann, P.; Wood, M.R.; Kawaoka, Y.; Saphire, E.O. Structural Rearrangement of Ebola Virus VP40 Begets Multiple Functions in the Virus Life Cycle. Cell 2013, 154, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Adu-Gyamfi, E.; Soni, S.; Jee, C.; Digman, M.; Gratton, E.; Stahelin, R. A Loop Region in the N-Terminal Domain of Ebola Virus VP40 Is Important in Viral Assembly, Budding, and Egress. Viruses 2014, 6, 3837–3854. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Lu, J.; Liu, Y.; Davis, B.; Lee, M.S.; Olson, M.A.; Ruthel, G.; Freedman, B.D.; Schnell, M.J.; Wrobel, J.E.; et al. Small-Molecule Probes Targeting the Viral PPxY-Host Nedd4 Interface Block Egress of a Broad Range of RNA Viruses. J. Virol. 2014, 88, 7294–7306. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bexiga, M.; Palencia, A.; Corbi-Verge, C.; Martin-Malpartida, P.; Blanco, F.J.; Macias, M.J.; Cobos, E.S.; Luque, I. Binding Site Plasticity in Viral PPxY Late Domain Recognition by the Third WW Domain of Human NEDD4. Sci. Rep. 2019, 9, 15076. [Google Scholar] [CrossRef]
- Johnson, K.; Pokhrel, R.; Budicini, M.; Gerstman, B.; Chapagain, P.; Stahelin, R. A Conserved Tryptophan in the Ebola Virus Matrix Protein C-Terminal Domain Is Required for Efficient Virus-Like Particle Formation. Pathogens 2020, 9, 402. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Complexes | ΔE Binding | SASA | ΔE Electrostatic | ΔE Van der Waal |
|---|---|---|---|---|---|
| VP35 | Cystobactamid 934-2 | −214.49 | −22.22 | −38.14 | −289.45 |
| Cystobactamid 919-1 | −268.16 | −29.52 | −317.55 | −388.49 | |
| Cittilin A | −191.14 | −22.72 | −98.21 | −321.22 | |
| VP40 | 2-Hydroxysorangiadenosine | −231.83 | −28.99 | −189.37 | −346.24 |
| Enhypyrazinone B | −219.20 | −21.55 | −32.59 | −266.80 | |
| Sorangiadenosine | −211.25 | 23.20 | −36.68 | −281.38 |
| NPs | Physicochemical Properties (Lipinski Rule of Five) | Solubility | Pharmacokinetics | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MW | HBD | HBA | RB | Log P | Log S (ESOL) | Log S (Ali) | Log S (SILICOS-IT) | GI Absorption | CYP Enzyme Inhibitor | |
| Cystobactamid 934-2 | 934.90 | 8 | 15 | 25 | 3.89 | PS | NS | NS | low | CYP2C9 |
| Cystobactamid 919-1 | 917.87 | 8 | 14 | 23 | 3.44 | PS | NS | NS | low | CYP2C9 |
| Cittilin A | 630.69 | 6 | 9 | 4 | 1.62 | MS | MS | PS | Low | CYP3A4 |
| 2-Hydroxysorangiadenosine | 487.59 | 5 | 8 | 5 | 1.7 | MS | PS | S | Low | No |
| Enhypyrazinone B | 369.46 | 2 | 2 | 5 | 4.54 | MS | MS | PS | High | Yes |
| Sorangiadenosine | 471.59 | 4 | 7 | 5 | 2.5 | MS | PS | S | High | CYP2D6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayat, M.; Gao, T.; Cao, Y.; Rafiq, M.; Zhuo, L.; Li, Y.-Z. Identification of Prospective Ebola Virus VP35 and VP40 Protein Inhibitors from Myxobacterial Natural Products. Biomolecules 2024, 14, 660. https://doi.org/10.3390/biom14060660
Hayat M, Gao T, Cao Y, Rafiq M, Zhuo L, Li Y-Z. Identification of Prospective Ebola Virus VP35 and VP40 Protein Inhibitors from Myxobacterial Natural Products. Biomolecules. 2024; 14(6):660. https://doi.org/10.3390/biom14060660
Chicago/Turabian StyleHayat, Muhammad, Tian Gao, Ying Cao, Muhammad Rafiq, Li Zhuo, and Yue-Zhong Li. 2024. "Identification of Prospective Ebola Virus VP35 and VP40 Protein Inhibitors from Myxobacterial Natural Products" Biomolecules 14, no. 6: 660. https://doi.org/10.3390/biom14060660