
Mitochondrial Reactive Oxygen Species in Infection and Immunity

 ,
,  , and
, and
Abstract
:1. Introduction
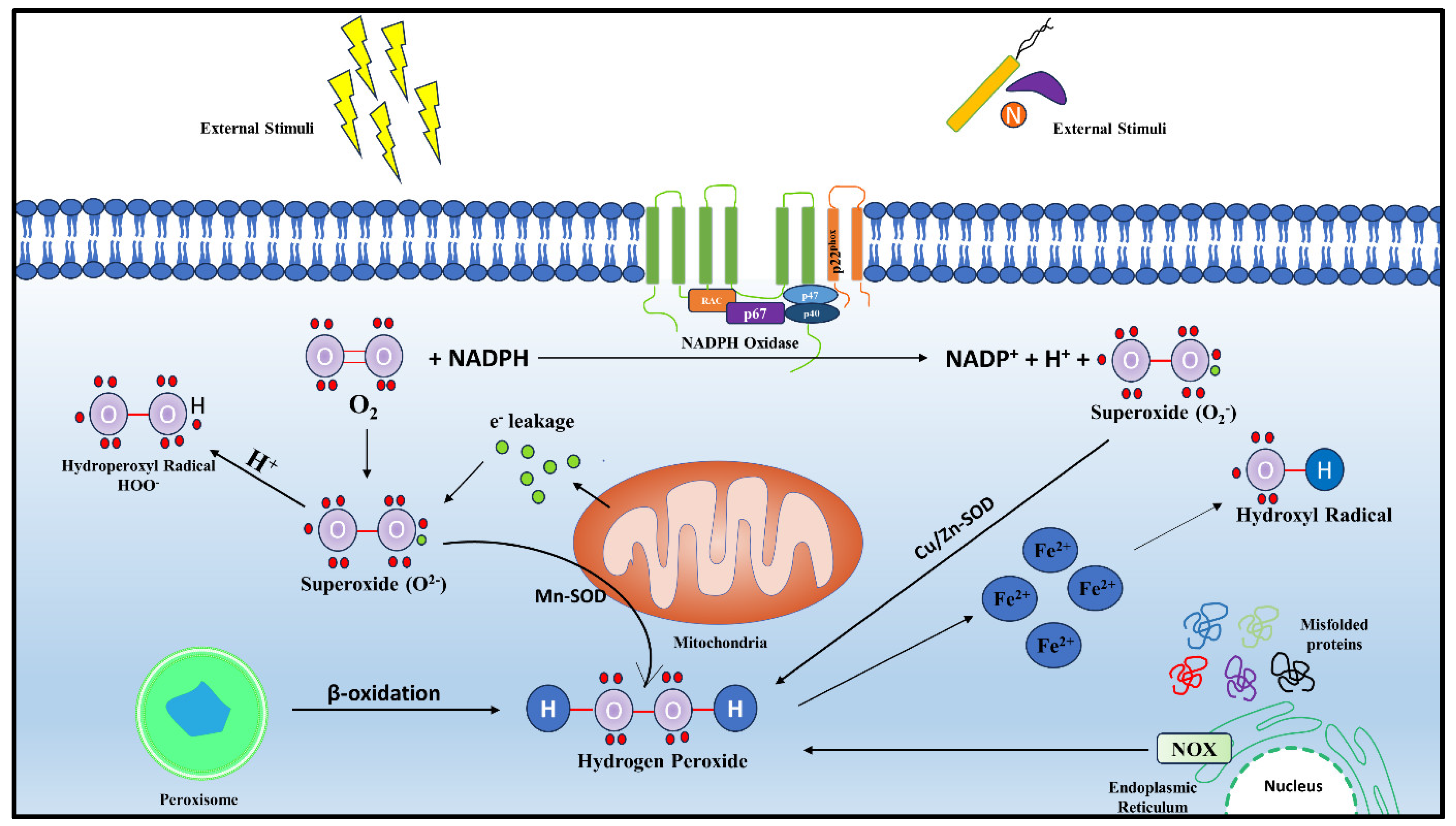
2. Generation and Regulation of Mitochondrial ROS
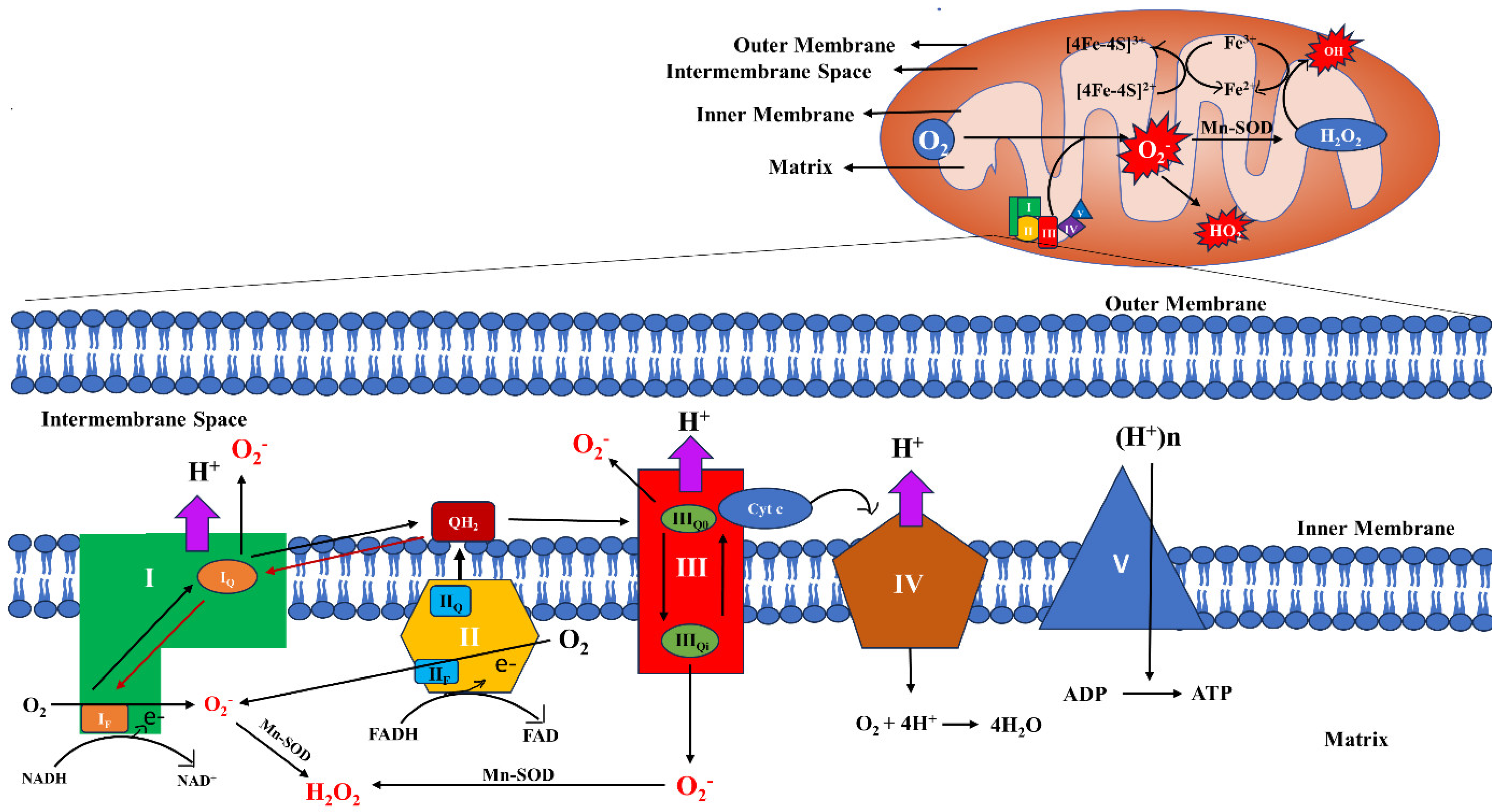
2.1. Sites and Regulation of mt-ROS Generation
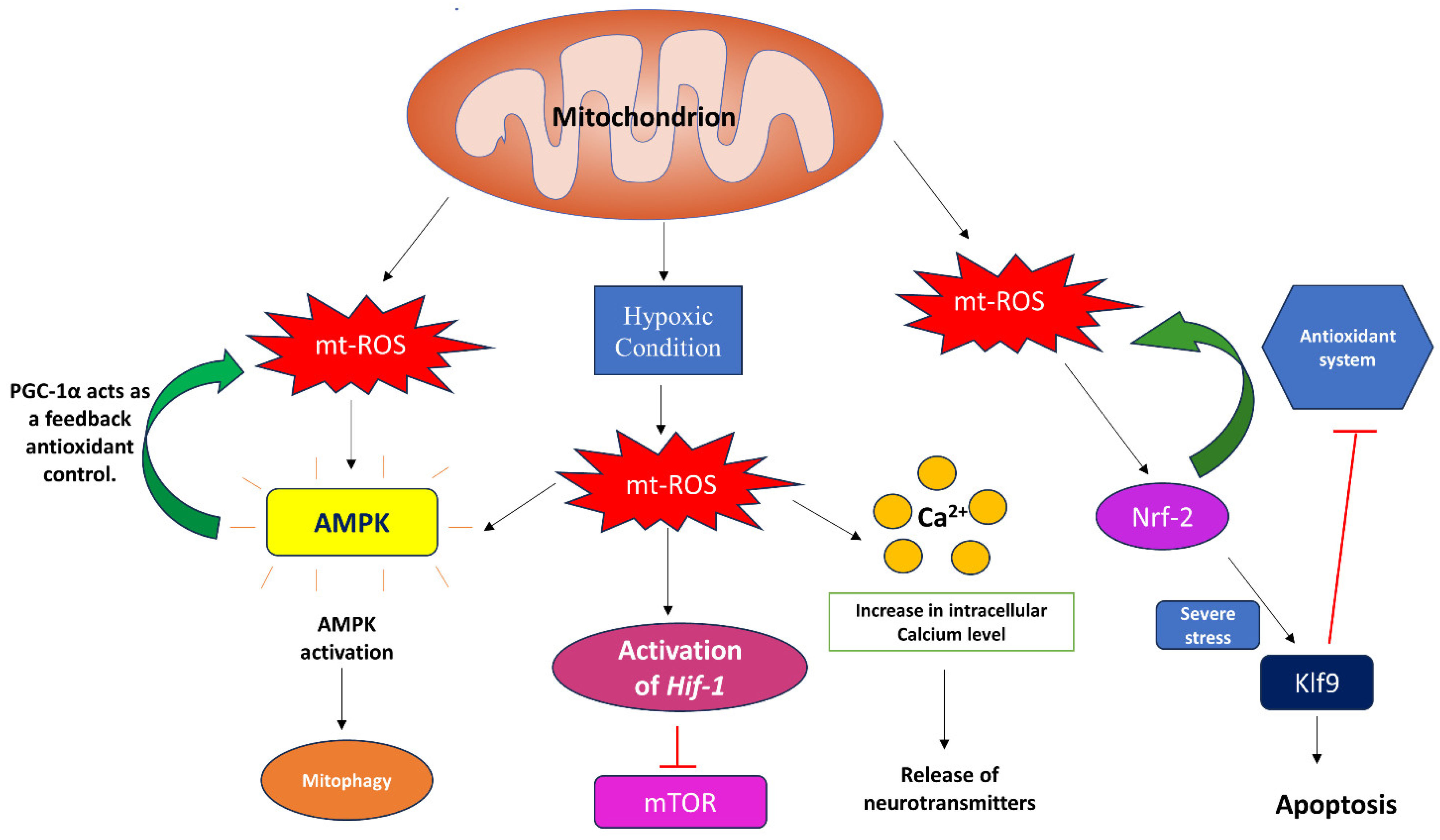
2.2. Regulation of mt-ROS
2.3. Regulators and Inhibitors of mt-ROS
3. Signaling through Mitochondrial Reactive Oxygen Species
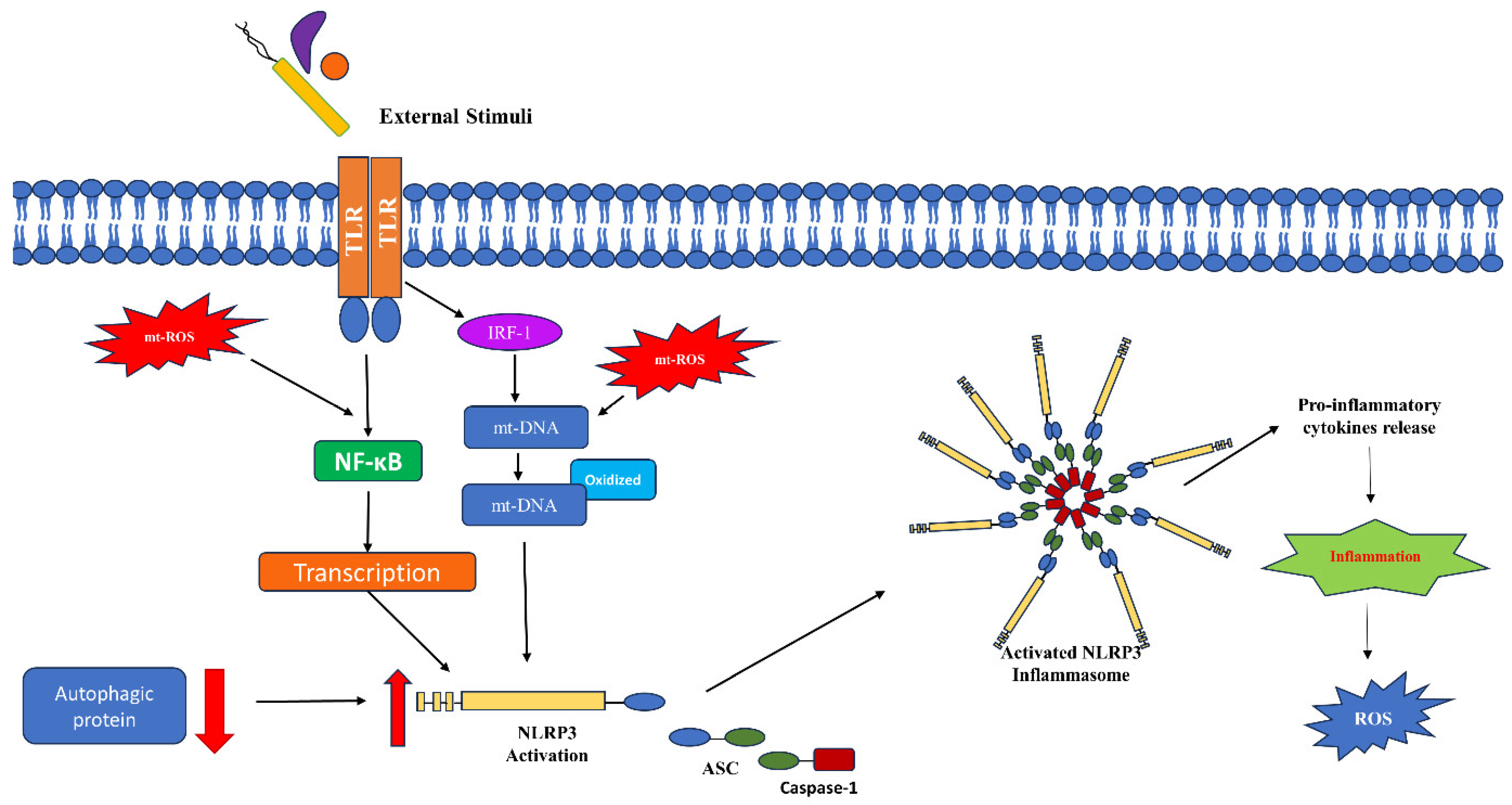
4. Mitochondrial ROS in Immune Signaling
5. Mitochondrial ROS in Bacterial Infection
6. Mitochondrial ROS in Protozoan Infection
7. Mitochondrial ROS in Viral Infection
8. Mitochondrial ROS in Fungal Infections
9. Future Perspectives: Targeting Mitochondrial ROS as a Therapeutic Approach
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pospisil, P.; Prasad, A.; Rac, M. Mechanism of the Formation of Electronically Excited Species by Oxidative Metabolic Processes: Role of Reactive Oxygen Species. Biomolecules 2019, 9, 258. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Zhang, Y.; Wong, H.S. Are mitochondria the main contributor of reactive oxygen species in cells? J. Exp. Biol. 2021, 224, jeb221606. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 2012, 12, 1–4. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal 2012, 16, 1077–1087. [Google Scholar] [CrossRef]
- Abdal Dayem, A.; Hossain, M.K.; Lee, S.B.; Kim, K.; Saha, S.K.; Yang, G.M.; Choi, H.Y.; Cho, S.G. The Role of Reactive Oxygen Species (ROS) in the Biological Activities of Metallic Nanoparticles. Int. J. Mol. Sci. 2017, 18, 120. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Filip-Ciubotaru, F.; Manciuc, C.; Stoleriu, G.; Foia, L. Nadph Oxidase: Structure and Activation Mechanisms (Review). Note I. Rev. Med. Chir. Soc. Med. Nat. Iasi 2016, 120, 29–33. [Google Scholar]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Yan, J.; Jiang, J.; He, L.; Chen, L. Mitochondrial superoxide/hydrogen peroxide: An emerging therapeutic target for metabolic diseases. Free Radic. Biol. Med. 2020, 152, 33–42. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Corpas, F.J.; Gonzalez-Gordo, S.; Palma, J.M. Plant Peroxisomes: A Factory of Reactive Species. Front. Plant Sci. 2020, 11, 853. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, Y.; Lu, Y.; Xiang, M. ROS and Endoplasmic Reticulum Stress in Pulmonary Disease. Front. Pharmacol. 2022, 13, 879204. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Oxidative reactions of hemoglobin. Methods Enzymol. 1990, 186, 265–272. [Google Scholar] [CrossRef]
- Aslan, M.; Ozben, T. Reactive oxygen and nitrogen species in Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 111–119. [Google Scholar] [CrossRef]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef]
- Akhigbe, R.; Ajayi, A. The impact of reactive oxygen species in the development of cardiometabolic disorders: A review. Lipids Health Dis. 2021, 20, 23. [Google Scholar] [CrossRef]
- Sun, K.; Xu, L.; Jing, Y.; Han, Z.; Chen, X.; Cai, C.; Zhao, P.; Zhao, X.; Yang, L.; Wei, L. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-kappaB-IL1alpha/beta-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017, 388, 198–207. [Google Scholar] [CrossRef]
- He, Z.; Zhang, C.; Liang, J.X.; Zheng, F.F.; Qi, X.Y.; Gao, F. Targeting Mitochondrial Oxidative Stress: Potential Neuroprotective Therapy for Spinal Cord Injury. J. Integr. Neurosci. 2023, 22, 153. [Google Scholar] [CrossRef]
- Agod, Z.; Fekete, T.; Budai, M.M.; Varga, A.; Szabo, A.; Moon, H.; Boldogh, I.; Biro, T.; Lanyi, A.; Bacsi, A.; et al. Regulation of type I interferon responses by mitochondria-derived reactive oxygen species in plasmacytoid dendritic cells. Redox Biol. 2017, 13, 633–645. [Google Scholar] [CrossRef]
- Dahiya, P.; Hussain, M.A.; Mazumder, S. mtROS Induced via TLR-2-SOCE Signaling Plays Proapoptotic and Bactericidal Role in Mycobacterium fortuitum-Infected Head Kidney Macrophages of Clarias gariepinus. Front. Immunol. 2021, 12, 748758. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Tavassolifar, M.J.; Vodjgani, M.; Salehi, Z.; Izad, M. The Influence of Reactive Oxygen Species in the Immune System and Pathogenesis of Multiple Sclerosis. Autoimmune Dis. 2020, 2020, 5793817. [Google Scholar] [CrossRef]
- Antonucci, S.; Di Lisa, F.; Kaludercic, N. Mitochondrial reactive oxygen species in physiology and disease. Cell Calcium 2021, 94, 102344. [Google Scholar] [CrossRef]
- Faas, M.M.; de Vos, P. Mitochondrial function in immune cells in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165845. [Google Scholar] [CrossRef]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Superoxide dismutase. Organelle specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [CrossRef]
- Pinegin, B.; Vorobjeva, N.; Pashenkov, M.; Chernyak, B. The role of mitochondrial ROS in antibacterial immunity. J. Cell Physiol. 2018, 233, 3745–3754. [Google Scholar] [CrossRef]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Ugun-Klusek, A.; Theodosi, T.S.; Fitzgerald, J.C.; Burte, F.; Ufer, C.; Boocock, D.J.; Yu-Wai-Man, P.; Bedford, L.; Billett, E.E. Monoamine oxidase-A promotes protective autophagy in human SH-SY5Y neuroblastoma cells through Bcl-2 phosphorylation. Redox Biol. 2019, 20, 167–181. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Venditti, P. Mitochondrial Management of Reactive Oxygen Species. Antioxidants 2021, 10, 1824. [Google Scholar] [CrossRef]
- Mailloux, R.J. An Update on Mitochondrial Reactive Oxygen Species Production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar] [CrossRef]
- Heo, S.; Kim, S.; Kang, D. The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle. Antioxidants 2020, 9, 280. [Google Scholar] [CrossRef]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Mitochondrial catalase and oxidative injury. Biol. Signals Recept. 2001, 10, 189–199. [Google Scholar] [CrossRef]
- Cao, Z.; Lindsay, J.G.; Isaacs, N.W. Mitochondrial peroxiredoxins. Subcell. Biochem. 2007, 44, 295–315. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef]
- Bai, J.; Rodriguez, A.M.; Melendez, J.A.; Cederbaum, A.I. Overexpression of catalase in cytosolic or mitochondrial compartment protects HepG2 cells against oxidative injury. J. Biol. Chem. 1999, 274, 26217–26224. [Google Scholar] [CrossRef]
- Santiago, A.P.; Chaves, E.A.; Oliveira, M.F.; Galina, A. Reactive oxygen species generation is modulated by mitochondrial kinases: Correlation with mitochondrial antioxidant peroxidases in rat tissues. Biochimie 2008, 90, 1566–1577. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, L.L.; Peng, Y.; Geng, X.X.; Xu, F. Alternative Oxidase Inhibition Impairs Tobacco Root Development and Root Hair Formation. Front. Plant Sci. 2021, 12, 664792. [Google Scholar] [CrossRef]
- Fiorani, F.; Umbach, A.L.; Siedow, J.N. The alternative oxidase of plant mitochondria is involved in the acclimation of shoot growth at low temperature. A study of Arabidopsis AOX1a transgenic plants. Plant Physiol. 2005, 139, 1795–1805. [Google Scholar] [CrossRef]
- Dinakar, C.; Vishwakarma, A.; Raghavendra, A.S.; Padmasree, K. Alternative Oxidase Pathway Optimizes Photosynthesis During Osmotic and Temperature Stress by Regulating Cellular ROS, Malate Valve and Antioxidative Systems. Front. Plant Sci. 2016, 7, 68. [Google Scholar] [CrossRef]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Ilari, S.; Giancotti, L.A.; Lauro, F.; Dagostino, C.; Gliozzi, M.; Malafoglia, V.; Sansone, L.; Palma, E.; Tafani, M.; Russo, M.A.; et al. Antioxidant modulation of sirtuin 3 during acute inflammatory pain: The ROS control. Pharmacol. Res. 2020, 157, 104851. [Google Scholar] [CrossRef]
- Roginsky, V.A.; Tashlitsky, V.N.; Skulachev, V.P. Chain-breaking antioxidant activity of reduced forms of mitochondria-targeted quinones, a novel type of geroprotectors. Aging 2009, 1, 481–489. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Anisimov, V.N.; Antonenko, Y.N.; Bakeeva, L.E.; Chernyak, B.V.; Erichev, V.P.; Filenko, O.F.; Kalinina, N.I.; Kapelko, V.I.; Kolosova, N.G.; et al. An attempt to prevent senescence: A mitochondrial approach. Biochim. Biophys. Acta 2009, 1787, 437–461. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef]
- El-Gammal, Z.; Nasr, M.A.; Elmehrath, A.O.; Salah, R.A.; Saad, S.M.; El-Badri, N. Regulation of mitochondrial temperature in health and disease. Pflugers Arch. 2022, 474, 1043–1051. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Nanayakkara, G.K.; Wang, H.; Yang, X. Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation—A novel concept. Arch. Biochem. Biophys. 2019, 662, 68–74. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Emerling, B.M.; Weinberg, F.; Snyder, C.; Burgess, Z.; Mutlu, G.M.; Viollet, B.; Budinger, G.R.; Chandel, N.S. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med. 2009, 46, 1386–1391. [Google Scholar] [CrossRef]
- Zhou, G.; Dada, L.A.; Wu, M.; Kelly, A.; Trejo, H.; Zhou, Q.; Varga, J.; Sznajder, J.I. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L1120–L1130. [Google Scholar] [CrossRef]
- Castejon-Vega, B.; Cordero, M.D.; Sanz, A. How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing. Antioxidants 2023, 12, 831. [Google Scholar] [CrossRef]
- Carriere, A.; Carmona, M.C.; Fernandez, Y.; Rigoulet, M.; Wenger, R.H.; Penicaud, L.; Casteilla, L. Mitochondrial reactive oxygen species control the transcription factor CHOP-10/GADD153 and adipocyte differentiation: A mechanism for hypoxia-dependent effect. J. Biol. Chem. 2004, 279, 40462–40469. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef]
- Khan, K.; Tran, H.C.; Mansuroglu, B.; Onsell, P.; Buratti, S.; Schwarzlander, M.; Costa, A.; Rasmusson, A.G.; Van Aken, O. Mitochondria-derived reactive oxygen species are the likely primary trigger of mitochondrial retrograde signaling in Arabidopsis. Curr. Biol. 2024, 34, 327–342.e324. [Google Scholar] [CrossRef]
- He, J.; Duan, Y.; Hua, D.; Fan, G.; Wang, L.; Liu, Y.; Chen, Z.; Han, L.; Qu, L.J.; Gong, Z. DEXH box RNA helicase-mediated mitochondrial reactive oxygen species production in Arabidopsis mediates crosstalk between abscisic acid and auxin signaling. Plant Cell 2012, 24, 1815–1833. [Google Scholar] [CrossRef]
- Postiglione, A.E.; Muday, G.K. Abscisic acid increases hydrogen peroxide in mitochondria to facilitate stomatal closure. Plant Physiol. 2023, 192, 469–487. [Google Scholar] [CrossRef]
- Santin, Y.; Fazal, L.; Sainte-Marie, Y.; Sicard, P.; Maggiorani, D.; Tortosa, F.; Yucel, Y.Y.; Teyssedre, L.; Rouquette, J.; Marcellin, M.; et al. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa(2+) overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020, 27, 1907–1923. [Google Scholar] [CrossRef]
- Yee, C.; Yang, W.; Hekimi, S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 2014, 157, 897–909. [Google Scholar] [CrossRef]
- Bonawitz, N.D.; Rodeheffer, M.S.; Shadel, G.S. Defective mitochondrial gene expression results in reactive oxygen species-mediated inhibition of respiration and reduction of yeast life span. Mol. Cell Biol. 2006, 26, 4818–4829. [Google Scholar] [CrossRef]
- Okoye, C.N.; Koren, S.A.; Wojtovich, A.P. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. 2023, 67, 102926. [Google Scholar] [CrossRef]
- Povea-Cabello, S.; Brischigliaro, M.; Fernandez-Vizarra, E. Emerging mechanisms in the redox regulation of mitochondrial cytochrome c oxidase assembly and function. Biochem. Soc. Trans. 2024, 52, 873–885. [Google Scholar] [CrossRef]
- Camello-Almaraz, C.; Gomez-Pinilla, P.J.; Pozo, M.J.; Camello, P.J. Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 2006, 291, C1082–C1088. [Google Scholar] [CrossRef]
- Zhang, X.; Lee, M.D.; Wilson, C.; McCarron, J.G. Hydrogen peroxide depolarizes mitochondria and inhibits IP(3)-evoked Ca(2+) release in the endothelium of intact arteries. Cell Calcium 2019, 84, 102108. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Dikalov, S.I. Mitochondrial ROS in the prohypertensive immune response. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R98–R100. [Google Scholar] [CrossRef]
- Murphy, M.P.; Siegel, R.M. Mitochondrial ROS fire up T cell activation. Immunity 2013, 38, 201–202. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Sauer, S.W.; Kaminski, M.; Opp, S.; Ruppert, T.; Grigaravicius, P.; Grudnik, P.; Grone, H.J.; Krammer, P.H.; Gulow, K. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2012, 2, 1300–1315. [Google Scholar] [CrossRef]
- Stocks, C.J.; Schembri, M.A.; Sweet, M.J.; Kapetanovic, R. For when bacterial infections persist: Toll-like receptor-inducible direct antimicrobial pathways in macrophages. J. Leukoc. Biol. 2018, 103, 35–51. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed. Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Geng, J.; Sun, X.; Wang, P.; Zhang, S.; Wang, X.; Wu, H.; Hong, L.; Xie, C.; Li, X.; Zhao, H.; et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 2015, 16, 1142–1152. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Prikhodko, A.; Galkin, I.; Pletjushkina, O.; Zinovkin, R.; Sud’ina, G.; Chernyak, B.; Pinegin, B. Mitochondrial reactive oxygen species are involved in chemoattractant-induced oxidative burst and degranulation of human neutrophils in vitro. Eur. J. Cell Biol. 2017, 96, 254–265. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Galkin, I.; Pletjushkina, O.; Golyshev, S.; Zinovkin, R.; Prikhodko, A.; Pinegin, V.; Kondratenko, I.; Pinegin, B.; Chernyak, B. Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165664. [Google Scholar] [CrossRef]
- Herb, M.; Gluschko, A.; Wiegmann, K.; Farid, A.; Wolf, A.; Utermohlen, O.; Krut, O.; Kronke, M.; Schramm, M. Mitochondrial reactive oxygen species enable proinflammatory signaling through disulfide linkage of NEMO. Sci. Signal 2019, 12, eaar5926. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Romaschenko, V.P.; Galkin, I.I.; Zakharova, V.V.; Pletjushkina, O.Y.; Chernyak, B.V.; Popova, E.N. Role of mitochondrial reactive oxygen species in age-related inflammatory activation of endothelium. Aging 2014, 6, 661–674. [Google Scholar] [CrossRef]
- Maurya, C.K.; Arha, D.; Rai, A.K.; Kumar, S.K.; Pandey, J.; Avisetti, D.R.; Kalivendi, S.V.; Klip, A.; Tamrakar, A.K. NOD2 activation induces oxidative stress contributing to mitochondrial dysfunction and insulin resistance in skeletal muscle cells. Free Radic. Biol. Med. 2015, 89, 158–169. [Google Scholar] [CrossRef]
- Tamrakar, A.K.; Schertzer, J.D.; Chiu, T.T.; Foley, K.P.; Bilan, P.J.; Philpott, D.J.; Klip, A. NOD2 activation induces muscle cell-autonomous innate immune responses and insulin resistance. Endocrinology 2010, 151, 5624–5637. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Girardin, S.E. Mitochondrial ROS fuel the inflammasome. Cell Res. 2011, 21, 558–560. [Google Scholar] [CrossRef]
- Dominic, A.; Le, N.T.; Takahashi, M. Loop Between NLRP3 Inflammasome and Reactive Oxygen Species. Antioxid. Redox Signal 2022, 36, 784–796. [Google Scholar] [CrossRef]
- Zhou, J.; Feng, D.; Li, X.; Chen, Y.; Zhang, M.; Wu, W.; Zhu, J.; Li, H.; Peng, X.; Zhang, T. L-Serine enables reducing the virulence of Acinetobacter baumannii and modulating the SIRT1 pathway to eliminate the pathogen. Microbiol. Spectr. 2024, 12, e0322623. [Google Scholar] [CrossRef]
- An, Z.; Su, J. Acinetobacter baumannii outer membrane protein 34 elicits NLRP3 inflammasome activation via mitochondria-derived reactive oxygen species in RAW264.7 macrophages. Microbes Infect. 2019, 21, 143–153. [Google Scholar] [CrossRef]
- Xue, Y.; Du, M.; Zhu, M.J. Quercetin suppresses NLRP3 inflammasome activation in epithelial cells triggered by Escherichia coli O157:H7. Free Radic. Biol. Med. 2017, 108, 760–769. [Google Scholar] [CrossRef]
- Hwang, A.B.; Ryu, E.A.; Artan, M.; Chang, H.W.; Kabir, M.H.; Nam, H.J.; Lee, D.; Yang, J.S.; Kim, S.; Mair, W.B.; et al. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2014, 111, E4458–E4467. [Google Scholar] [CrossRef]
- Manago, A.; Becker, K.A.; Carpinteiro, A.; Wilker, B.; Soddemann, M.; Seitz, A.P.; Edwards, M.J.; Grassme, H.; Szabo, I.; Gulbins, E. Pseudomonas aeruginosa pyocyanin induces neutrophil death via mitochondrial reactive oxygen species and mitochondrial acid sphingomyelinase. Antioxid. Redox Signal 2015, 22, 1097–1110. [Google Scholar] [CrossRef]
- Roberts, J.S.; Atanasova, K.R.; Lee, J.; Diamond, G.; Deguzman, J.; Hee Choi, C.; Yilmaz, O. Opportunistic Pathogen Porphyromonas gingivalis Modulates Danger Signal ATP-Mediated Antibacterial NOX2 Pathways in Primary Epithelial Cells. Front. Cell Infect. Microbiol. 2017, 7, 291. [Google Scholar] [CrossRef]
- Wen, J.J.; Porter, C.; Garg, N.J. Inhibition of NFE2L2-Antioxidant Response Element Pathway by Mitochondrial Reactive Oxygen Species Contributes to Development of Cardiomyopathy and Left Ventricular Dysfunction in Chagas Disease. Antioxid. Redox Signal 2017, 27, 550–566. [Google Scholar] [CrossRef]
- Nogueira, N.P.; Saraiva, F.M.S.; Oliveira, M.P.; Mendonca, A.P.M.; Inacio, J.D.F.; Almeida-Amaral, E.E.; Menna-Barreto, R.F.; Laranja, G.A.T.; Torres, E.J.L.; Oliveira, M.F.; et al. Heme modulates Trypanosoma cruzi bioenergetics inducing mitochondrial ROS production. Free Radic. Biol. Med. 2017, 108, 183–191. [Google Scholar] [CrossRef]
- Syn, G.; Anderson, D.; Blackwell, J.M.; Jamieson, S.E. Toxoplasma gondii Infection Is Associated with Mitochondrial Dysfunction in-Vitro. Front. Cell Infect. Microbiol. 2017, 7, 512. [Google Scholar] [CrossRef]
- Goncalves, R.L.; Oliveira, J.H.; Oliveira, G.A.; Andersen, J.F.; Oliveira, M.F.; Oliveira, P.L.; Barillas-Mury, C. Mitochondrial reactive oxygen species modulate mosquito susceptibility to Plasmodium infection. PLoS ONE 2012, 7, e41083. [Google Scholar] [CrossRef]
- Diniz, S.Q.; Teixeira-Carvalho, A.; Figueiredo, M.M.; Costa, P.A.C.; Rocha, B.C.; Martins-Filho, O.A.; Goncalves, R.; Pereira, D.B.; Tada, M.S.; Oliveira, F.; et al. Plasmodium vivax Infection Alters Mitochondrial Metabolism in Human Monocytes. mBio 2021, 12, e0124721. [Google Scholar] [CrossRef]
- Ball, W.B.; Mukherjee, M.; Srivastav, S.; Das, P.K. Leishmania donovani activates uncoupling protein 2 transcription to suppress mitochondrial oxidative burst through differential modulation of SREBP2, Sp1 and USF1 transcription factors. Int. J. Biochem. Cell Biol. 2014, 48, 66–76. [Google Scholar] [CrossRef]
- Basu Ball, W.; Kar, S.; Mukherjee, M.; Chande, A.G.; Mukhopadhyaya, R.; Das, P.K. Uncoupling protein 2 negatively regulates mitochondrial reactive oxygen species generation and induces phosphatase-mediated anti-inflammatory response in experimental visceral leishmaniasis. J. Immunol. 2011, 187, 1322–1332. [Google Scholar] [CrossRef]
- Hu, M.; Schulze, K.E.; Ghildyal, R.; Henstridge, D.C.; Kolanowski, J.L.; New, E.J.; Hong, Y.; Hsu, A.C.; Hansbro, P.M.; Wark, P.A.; et al. Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. eLife 2019, 8, e42448. [Google Scholar] [CrossRef]
- Silwal, P.; Kim, J.K.; Kim, Y.J.; Jo, E.K. Mitochondrial Reactive Oxygen Species: Double-Edged Weapon in Host Defense and Pathological Inflammation During Infection. Front. Immunol. 2020, 11, 1649. [Google Scholar] [CrossRef]
- Ojeda, D.S.; Grasso, D.; Urquiza, J.; Till, A.; Vaccaro, M.I.; Quarleri, J. Cell Death Is Counteracted by Mitophagy in HIV-Productively Infected Astrocytes but Is Promoted by Inflammasome Activation Among Non-productively Infected Cells. Front. Immunol. 2018, 9, 2633. [Google Scholar] [CrossRef]
- Cheng, M.L.; Weng, S.F.; Kuo, C.H.; Ho, H.Y. Enterovirus 71 induces mitochondrial reactive oxygen species generation that is required for efficient replication. PLoS ONE 2014, 9, e113234. [Google Scholar] [CrossRef]
- Hatinguais, R.; Pradhan, A.; Brown, G.D.; Brown, A.J.P.; Warris, A.; Shekhova, E. Mitochondrial Reactive Oxygen Species Regulate Immune Responses of Macrophages to Aspergillus fumigatus. Front. Immunol. 2021, 12, 641495. [Google Scholar] [CrossRef]
- Govindan, J.A.; Jayamani, E.; Ruvkun, G. ROS-based lethality of Caenorhabditis elegans mitochondrial electron transport mutants grown on Escherichia coli siderophore iron release mutants. Proc. Natl. Acad. Sci. USA 2019, 116, 21651–21658. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Y.; Qiu, X.; Wang, G.; Hu, Z.; Chen, S.; Wu, Z.; Yuan, N.; Gao, H.; Wang, J.; et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 2019, 20, 433–446. [Google Scholar] [CrossRef]
- Yuan, J.; Zheng, Z.; Wang, L.; Ran, H.; Tang, X.; Xie, X.; Li, F.; Liu, F.; Wang, X.; Zhang, J.; et al. The Dynll1-Cox4i1 Complex Regulates Intracellular Pathogen Clearance via Release of Mitochondrial Reactive Oxygen Species. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef]
- La Pietra, L.; Hudel, M.; Pillich, H.; Abu Mraheil, M.; Berisha, B.; Aden, S.; Hodnik, V.; Lochnit, G.; Rafiq, A.; Perniss, A.; et al. Phosphocholine Antagonizes Listeriolysin O-Induced Host Cell Responses of Listeria monocytogenes. J. Infect. Dis. 2020, 222, 1505–1516. [Google Scholar] [CrossRef]
- Cohen, T.S.; Boland, M.L.; Boland, B.B.; Takahashi, V.; Tovchigrechko, A.; Lee, Y.; Wilde, A.D.; Mazaitis, M.J.; Jones-Nelson, O.; Tkaczyk, C.; et al. S. aureus Evades Macrophage Killing through NLRP3-Dependent Effects on Mitochondrial Trafficking. Cell Rep. 2018, 22, 2431–2441. [Google Scholar] [CrossRef]
- Dunham-Snary, K.J.; Surewaard, B.G.; Mewburn, J.D.; Bentley, R.E.; Martin, A.Y.; Jones, O.; Al-Qazazi, R.; Lima, P.A.; Kubes, P.; Archer, S.L. Mitochondria in human neutrophils mediate killing of Staphylococcus aureus. Redox Biol. 2022, 49, 102225. [Google Scholar] [CrossRef]
- Abuaita, B.H.; Schultz, T.L.; O’Riordan, M.X. Mitochondria-Derived Vesicles Deliver Antimicrobial Reactive Oxygen Species to Control Phagosome-Localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636.E5. [Google Scholar] [CrossRef]
- Krause, K.; Daily, K.; Estfanous, S.; Hamilton, K.; Badr, A.; Abu Khweek, A.; Hegazi, R.; Anne, M.N.; Klamer, B.; Zhang, X.; et al. Caspase-11 counteracts mitochondrial ROS-mediated clearance of Staphylococcus aureus in macrophages. EMBO Rep. 2019, 20, e48109. [Google Scholar] [CrossRef]
- Cook, G.M.; Berney, M.; Gebhard, S.; Heinemann, M.; Cox, R.A.; Danilchanka, O.; Niederweis, M. Physiology of mycobacteria. Adv. Microb. Physiol. 2009, 55, 81–182, 318–319. [Google Scholar] [CrossRef]
- Roca, F.J.; Ramakrishnan, L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef]
- Roca, F.J.; Whitworth, L.J.; Prag, H.A.; Murphy, M.P.; Ramakrishnan, L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376, eabh2841. [Google Scholar] [CrossRef]
- Chandra, P.; He, L.; Zimmerman, M.; Yang, G.; Koster, S.; Ouimet, M.; Wang, H.; Moore, K.J.; Dartois, V.; Schilling, J.D.; et al. Inhibition of Fatty Acid Oxidation Promotes Macrophage Control of Mycobacterium tuberculosis. mBio 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Ruan, H.; Zhang, Z.; Tian, L.; Wang, S.; Hu, S.; Qiao, J.J. The Salmonella effector SopB prevents ROS-induced apoptosis of epithelial cells by retarding TRAF6 recruitment to mitochondria. Biochem. Biophys. Res. Commun. 2016, 478, 618–623. [Google Scholar] [CrossRef]
- Ariffin, J.K.; das Gupta, K.; Kapetanovic, R.; Iyer, A.; Reid, R.C.; Fairlie, D.P.; Sweet, M.J. Histone Deacetylase Inhibitors Promote Mitochondrial Reactive Oxygen Species Production and Bacterial Clearance by Human Macrophages. Antimicrob. Agents Chemother. 2015, 60, 1521–1529. [Google Scholar] [CrossRef]
- Li, B.; Wan, Z.; Wang, Z.; Zuo, J.; Xu, Y.; Han, X.; Phouthapane, V.; Miao, J. TLR2 Signaling Pathway Combats Streptococcus uberis Infection by Inducing Mitochondrial Reactive Oxygen Species Production. Cells 2020, 9, 494. [Google Scholar] [CrossRef]
- Yaeger, R.G. Protozoa: Structure, Classification, Growth, and Development. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Seed, J.R. Protozoa: Pathogenesis and Defenses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Muller, J.; Hemphill, A. Drug target identification in protozoan parasites. Expert. Opin. Drug Discov. 2016, 11, 815–824. [Google Scholar] [CrossRef]
- Gupta, S.; Bhatia, V.; Wen, J.J.; Wu, Y.; Huang, M.H.; Garg, N.J. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic. Biol. Med. 2009, 47, 1414–1421. [Google Scholar] [CrossRef]
- Ana, Y.; Rojas Marquez, J.D.; Fozzatti, L.; Baigorri, R.E.; Marin, C.; Maletto, B.A.; Cerban, F.M.; Radi, R.; Piacenza, L.; Stempin, C.C. An exacerbated metabolism and mitochondrial reactive oxygen species contribute to mitochondrial alterations and apoptosis in CD4 T cells during the acute phase of Trypanosoma cruzi infection. Free Radic. Biol. Med. 2021, 163, 268–280. [Google Scholar] [CrossRef]
- Rojas Marquez, J.D.; Ana, Y.; Baigorri, R.E.; Stempin, C.C.; Cerban, F.M. Mammalian Target of Rapamycin Inhibition in Trypanosoma cruzi-Infected Macrophages Leads to an Intracellular Profile That Is Detrimental for Infection. Front. Immunol. 2018, 9, 313. [Google Scholar] [CrossRef]
- Moreira-Souza, A.C.A.; Almeida-da-Silva, C.L.C.; Rangel, T.P.; Rocha, G.D.C.; Bellio, M.; Zamboni, D.S.; Vommaro, R.C.; Coutinho-Silva, R. The P2X7 Receptor Mediates Toxoplasma gondii Control in Macrophages through Canonical NLRP3 Inflammasome Activation and Reactive Oxygen Species Production. Front. Immunol. 2017, 8, 1257. [Google Scholar] [CrossRef]
- Pernas, L.; Bean, C.; Boothroyd, J.C.; Scorrano, L. Mitochondria Restrict Growth of the Intracellular Parasite Toxoplasma gondii by Limiting Its Uptake of Fatty Acids. Cell Metab. 2018, 27, 886–897.e884. [Google Scholar] [CrossRef]
- Ding, M.; Kwok, L.Y.; Schluter, D.; Clayton, C.; Soldati, D. The antioxidant systems in Toxoplasma gondii and the role of cytosolic catalase in defence against oxidative injury. Mol. Microbiol. 2004, 51, 47–61. [Google Scholar] [CrossRef]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Res 2017, 6, 750. [Google Scholar] [CrossRef]
- Gupta, A.K.; Ghosh, K.; Palit, S.; Barua, J.; Das, P.K.; Ukil, A. Leishmania donovani inhibits inflammasome-dependent macrophage activation by exploiting the negative regulatory proteins A20 and UCP2. FASEB J. 2017, 31, 5087–5101. [Google Scholar] [CrossRef]
- Saha, S.; Basu, M.; Guin, S.; Gupta, P.; Mitterstiller, A.M.; Weiss, G.; Jana, K.; Ukil, A. Leishmania donovani Exploits Macrophage Heme Oxygenase-1 To Neutralize Oxidative Burst and TLR Signaling-Dependent Host Defense. J. Immunol. 2019, 202, 827–840. [Google Scholar] [CrossRef]
- Chakrabarty, Y.; Bhattacharyya, S.N. Leishmania donovani restricts mitochondrial dynamics to enhance miRNP stability and target RNA repression in host macrophages. Mol. Biol. Cell 2017, 28, 2091–2105. [Google Scholar] [CrossRef]
- Michel, G.; Pomares, C.; Ferrua, B.; Marty, P. Importance of worldwide asymptomatic carriers of Leishmania infantum (L. chagasi) in human. Acta Trop. 2011, 119, 69–75. [Google Scholar] [CrossRef]
- Pessoa-Pereira, D.; Scorza, B.M.; Cyndari, K.I.; Beasley, E.A.; Petersen, C.A. Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi. Pathogens 2023, 12, 1128. [Google Scholar] [CrossRef]
- Liu, L.; Fang, R.; Wei, Z.; Wu, J.; Li, X.; Li, W. Giardia duodenalis Induces Apoptosis in Intestinal Epithelial Cells via Reactive Oxygen Species-Mediated Mitochondrial Pathway In Vitro. Pathogens 2020, 9, 693. [Google Scholar] [CrossRef]
- Quan, J.H.; Kang, B.H.; Yang, J.B.; Rhee, Y.E.; Noh, H.T.; Choi, I.W.; Cha, G.H.; Yuk, J.M.; Lee, Y.H. Trichomonas vaginalis Induces SiHa Cell Apoptosis by NF-kappaB Inactivation via Reactive Oxygen Species. Biomed. Res. Int. 2017, 2017, 3904870. [Google Scholar] [CrossRef]
- Pellett, P.E.; Mitra, S.; Holland, T.C. Basics of virology. Handb. Clin. Neurol. 2014, 123, 45–66. [Google Scholar] [CrossRef]
- Ren, Z.; Ding, T.; Zuo, Z.; Xu, Z.; Deng, J.; Wei, Z. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front. Immunol. 2020, 11, 1030. [Google Scholar] [CrossRef]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Lin, X.; Ren, C.; Zhao, J.; Wang, F.; Gao, X.; Xiao, R.; Zhao, L.; Chen, H.; et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy 2019, 15, 1163–1181. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, C.H.; Ryu, J.H.; Kim, M.J.; Park, C.Y.; Lee, J.M.; Holtzman, M.J.; Yoon, J.H. Reactive oxygen species induce antiviral innate immune response through IFN-lambda regulation in human nasal epithelial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 855–865. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.J.; Park, D.Y.; Chung, H.J.; Kim, C.H.; Yoon, J.H.; Kim, H.J. Mitochondrial reactive oxygen species modulate innate immune response to influenza A virus in human nasal epithelium. Antivir. Res. 2015, 119, 78–83. [Google Scholar] [CrossRef]
- To, E.E.; Erlich, J.R.; Liong, F.; Luong, R.; Liong, S.; Esaq, F.; Oseghale, O.; Anthony, D.; McQualter, J.; Bozinovski, S.; et al. Mitochondrial Reactive Oxygen Species Contribute to Pathological Inflammation During Influenza A Virus Infection in Mice. Antioxid. Redox Signal 2020, 32, 929–942. [Google Scholar] [CrossRef]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Subversion of Host Cell Mitochondria by RSV to Favor Virus Production is Dependent on Inhibition of Mitochondrial Complex I and ROS Generation. Cells 2019, 8, 1417. [Google Scholar] [CrossRef]
- Clippinger, A.J.; Bouchard, M.J. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 2008, 82, 6798–6811. [Google Scholar] [CrossRef]
- Yang, B.; Bouchard, M.J. The hepatitis B virus X protein elevates cytosolic calcium signals by modulating mitochondrial calcium uptake. J. Virol. 2012, 86, 313–327. [Google Scholar] [CrossRef]
- Rahmani, Z.; Huh, K.W.; Lasher, R.; Siddiqui, A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef]
- Li, J.; Ma, C.; Long, F.; Yang, D.; Liu, X.; Hu, Y.; Wu, C.; Wang, B.; Wang, M.; Chen, Y.; et al. Parkin Impairs Antiviral Immunity by Suppressing the Mitochondrial Reactive Oxygen Species-Nlrp3 Axis and Antiviral Inflammation. iScience 2019, 16, 468–484. [Google Scholar] [CrossRef]
- Piccoli, C.; Scrima, R.; Quarato, G.; D’Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Moradpour, D.; Capitanio, N. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 2007, 46, 58–65. [Google Scholar] [CrossRef]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef]
- Guerrero, C.A.; Acosta, O. Inflammatory and oxidative stress in rotavirus infection. World J. Virol. 2016, 5, 38–62. [Google Scholar] [CrossRef]
- Kozaki, T.; Komano, J.; Kanbayashi, D.; Takahama, M.; Misawa, T.; Satoh, T.; Takeuchi, O.; Kawai, T.; Shimizu, S.; Matsuura, Y.; et al. Mitochondrial damage elicits a TCDD-inducible poly(ADP-ribose) polymerase-mediated antiviral response. Proc. Natl. Acad. Sci. USA 2017, 114, 2681–2686. [Google Scholar] [CrossRef]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef]
- Shim, J.H.; Cho, K.J.; Lee, K.A.; Kim, S.H.; Myung, P.K.; Choe, Y.K.; Yoon, D.Y. E7-expressing HaCaT keratinocyte cells are resistant to oxidative stress-induced cell death via the induction of catalase. Proteomics 2005, 5, 2112–2122. [Google Scholar] [CrossRef]
- Qadri, I.; Iwahashi, M.; Capasso, J.M.; Hopken, M.W.; Flores, S.; Schaack, J.; Simon, F.R. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: Role of JNK, p38 MAPK and AP-1. Biochem. J. 2004, 378, 919–928. [Google Scholar] [CrossRef]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef]
- Wasilenko, S.T.; Stewart, T.L.; Meyers, A.F.; Barry, M. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 14345–14350. [Google Scholar] [CrossRef]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef]
- Shlezinger, N.; Hohl, T.M. Mitochondrial Reactive Oxygen Species Enhance Alveolar Macrophage Activity against Aspergillus fumigatus but Are Dispensable for Host Protection. mSphere 2021, 6, e0026021. [Google Scholar] [CrossRef]
- Lyu, L.; Bi, Y.; Li, S.; Xue, H.; Zhang, Z.; Prusky, D.B. Early Defense Responses Involved in Mitochondrial Energy Metabolism and Reactive Oxygen Species Accumulation in Harvested Muskmelons Infected by Trichothecium roseum. J. Agric. Food Chem. 2019, 67, 4337–4345. [Google Scholar] [CrossRef]
- Okamoto, M.; Nakano, K.; Takahashi-Nakaguchi, A.; Sasamoto, K.; Yamaguchi, M.; Teixeira, M.C.; Chibana, H. In Candida glabrata, ERMES Component GEM1 Controls Mitochondrial Morphology, mtROS, and Drug Efflux Pump Expression, Resulting in Azole Susceptibility. J. Fungi 2023, 9, 240. [Google Scholar] [CrossRef]
- Duvenage, L.; Walker, L.A.; Bojarczuk, A.; Johnston, S.A.; MacCallum, D.M.; Munro, C.A.; Gourlay, C.W. Inhibition of Classical and Alternative Modes of Respiration in Candida albicans Leads to Cell Wall Remodeling and Increased Macrophage Recognition. mBio 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Zaragoza, O.; Chrisman, C.J.; Castelli, M.V.; Frases, S.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L.; Casadevall, A. Capsule enlargement in Cryptococcus neoformans confers resistance to oxidative stress suggesting a mechanism for intracellular survival. Cell Microbiol. 2008, 10, 2043–2057. [Google Scholar] [CrossRef]
- Trevijano-Contador, N.; Rossi, S.A.; Alves, E.; Landin-Ferreiroa, S.; Zaragoza, O. Capsule Enlargement in Cryptococcus neoformans Is Dependent on Mitochondrial Activity. Front. Microbiol. 2017, 8, 1423. [Google Scholar] [CrossRef]
- Gao, X.; Fu, Y.; Sun, S.; Gu, T.; Li, Y.; Sun, T.; Li, H.; Du, W.; Suo, C.; Li, C.; et al. Cryptococcal Hsf3 controls intramitochondrial ROS homeostasis by regulating the respiratory process. Nat. Commun. 2022, 13, 5407. [Google Scholar] [CrossRef]
- Pradhan, A.; Avelar, G.M.; Bain, J.M.; Childers, D.S.; Larcombe, D.E.; Netea, M.G.; Shekhova, E.; Munro, C.A.; Brown, G.D.; Erwig, L.P.; et al. Hypoxia Promotes Immune Evasion by Triggering beta-Glucan Masking on the Candida albicans Cell Surface via Mitochondrial and cAMP-Protein Kinase A Signaling. mBio 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Cui, S.; Li, M.; Hassan, R.Y.A.; Heintz-Buschart, A.; Wang, J.; Bilitewski, U. Inhibition of Respiration of Candida albicans by Small Molecules Increases Phagocytosis Efficacy by Macrophages. mSphere 2020, 5, 1–14. [Google Scholar] [CrossRef]
- Grahl, N.; Dinamarco, T.M.; Willger, S.D.; Goldman, G.H.; Cramer, R.A. Aspergillus fumigatus mitochondrial electron transport chain mediates oxidative stress homeostasis, hypoxia responses and fungal pathogenesis. Mol. Microbiol. 2012, 84, 383–399. [Google Scholar] [CrossRef]
- Akhter, S.; McDade, H.C.; Gorlach, J.M.; Heinrich, G.; Cox, G.M.; Perfect, J.R. Role of alternative oxidase gene in pathogenesis of Cryptococcus neoformans. Infect. Immun. 2003, 71, 5794–5802. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Zhang, Y.; Niu, L.; Li, W.; Lu, W.; Li, J.; Schafer, P.; Meng, Y.; Shan, W. A mitochondrial RNA processing protein mediates plant immunity to a broad spectrum of pathogens by modulating the mitochondrial oxidative burst. Plant Cell 2022, 34, 2343–2363. [Google Scholar] [CrossRef]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Strasfeld, L.; Chou, S. Antiviral drug resistance: Mechanisms and clinical implications. Infect. Dis. Clin. N. Am. 2010, 24, 413–437. [Google Scholar] [CrossRef]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Bruggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef]
- Capela, R.; Moreira, R.; Lopes, F. An Overview of Drug Resistance in Protozoal Diseases. Int. J. Mol. Sci. 2019, 20, 5748. [Google Scholar] [CrossRef]
- Abugri, D.A.; Wijerathne, S.V.T.; Sharma, H.N.; Ayariga, J.A.; Napier, A.; Robertson, B.K. Quercetin inhibits Toxoplasma gondii tachyzoite proliferation and acts synergically with azithromycin. Parasit. Vectors 2023, 16, 261. [Google Scholar] [CrossRef]
- Das, R.; Roy, A.; Dutta, N.; Majumder, H.K. Reactive oxygen species and imbalance of calcium homeostasis contributes to curcumin induced programmed cell death in Leishmania donovani. Apoptosis 2008, 13, 867–882. [Google Scholar] [CrossRef]
- Andrade, J.T.; Fantini de Figueiredo, G.; Cruz, L.F.; Eliza de Morais, S.; Souza, C.D.F.; Pinto, F.C.H.; Ferreira, J.M.S.; Araujo, M.G.F. Efficacy of curcumin in the treatment of experimental vulvovaginal candidiasis. Rev. Iberoam. Micol. 2019, 36, 192–199. [Google Scholar] [CrossRef]
- De Sarkar, S.; Sarkar, D.; Sarkar, A.; Dighal, A.; Staniek, K.; Gille, L.; Chatterjee, M. Berberine chloride mediates its antileishmanial activity by inhibiting Leishmania mitochondria. Parasitol. Res. 2019, 118, 335–345. [Google Scholar] [CrossRef]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control Release 2011, 156, 128–145. [Google Scholar] [CrossRef]
- Dourado, D.; Silva Medeiros, T.; do Nascimento Alencar, E.; Matos Sales, E.; Formiga, F.R. Curcumin-loaded nanostructured systems for treatment of leishmaniasis: A review. Beilstein J. Nanotechnol. 2024, 15, 37–50. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathogenic Microbes | Model | Effect on mt-ROS | Effect on | Action | Reference | ||
|---|---|---|---|---|---|---|---|
| Bacteria | Host | Pathogen | |||||
| (1) | A. baumannii | Beas 2B cells; RAW264.7cells | ↓ ↑ | + +/− | − −/+ |
| [102,103] |
| (2) | E. coli O157:H7 | Caco-2 cells | ↑ | − | + |
| [104] |
| (3) | Enterococcus faecalis | C. elegans | ↑ | + | isp-1 mutation in C. elegans, which reduces mitochondrial respiration, increases mt-ROS via AMPK and Hif-1 pathways and aids in attaining resistance against E. faecalis. | [105] | |
| (4) | P. aeruginosa | Neutrophils | ↑ | − | + | Bacterial toxin pyocyanin; releases cytochrome c, induces mt-ROS, activates sphingomyelinase, and aids in neutrophil elimination. | [106] |
| (5) | P. gingivalis | Gingival epithelial cells (GECs) | ↓ | − | + | Attenuates eATP/NOX-2-ROS pathway, which is one of the antibacterial pathways. | [107] |
| Protozoan parasites: | |||||||
| (6) | T. cruzi | C57B6 WT mice. HeLa cells, C57B6 MnSODtg | ↑ ↓ | − + | + − | Attenuates NFE2L2/ARE pathway induces fibrotic gene expression and leads to cardiomyopathy. Activates NFE2L3/ARE pathway reduces fibrotic gene expression, inhibits cardiac failure. | [108] |
| (7) | T. cruzi | Epimastigotes | ↑ | NA | + | Mitochondrial membrane potential is modulated via heme, which induces mt-ROS generation and aids in proliferation and survival of parasites. | [109] |
| (8) | Toxoplasma gondii | Human foreskin fibroblast (HFF) cells | ↑ | − | + | Hampers host mitochondrial function for its survival. It includes deregulation of OXPHOS, production of mt-ROS, and reduced expression of complex IV, I, and II, respectively. | [110] |
| (9) | Plasmodium berghei | Anopheles gambiae | ↓ | − | + | AgMC1 in Anopheles gambiae mosquitoes reduces membrane potential and increases proton leak and uncoupling, which ultimately reduces mt-ROS in midgut and increases the plasmodium susceptibility. | [111] |
| (10) | Plasmodium vivax | Monocytes | ↑ | + |
| [112] | |
| (11) | Leishmania donovani | Murine macrophages | ↓ | − | + |
| [113,114] |
| Viruses | |||||||
| (12) | Respiratory syncytial virus (RSV) | A549 cells, vero cells, BCi-NS1 cells, pBECs | ↑ | − | + |
| [115] |
| (13) | Human immunodeficiency virus (HIV) | Astrocytes | Productive infection: ↓ Abortive infection:↑ | + | + | With reduced mt-ROS, cell death resistance occurs. Mitochondrial damage and inflammasome activation. | [116,117] |
| (14) | Enterovirus 71 (EV71) | SF268 glioblastoma cells | ↑ | - | + | Decrease in ATP production. Increased mt-ROS. mtROS activates PKR and PERK, which in turn activates eIF2a phosphorylation, crucial for viral replication as inhibiting this reduces viral titer. | [118] |
| Fungi: | |||||||
| (15) | Aspergillus fumigatus | Murine bone marrow-derived macrophages (BMDMs) | ↑ | + | - | mt-ROS is produced via RET and complex II. Aids in cytokine production. Has an antifungal effect. | [119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, A.; Ghosh, K.K.; Chakrabortty, S.; Gulyás, B.; Padmanabhan, P.; Ball, W.B. Mitochondrial Reactive Oxygen Species in Infection and Immunity. Biomolecules 2024, 14, 670. https://doi.org/10.3390/biom14060670
Mukherjee A, Ghosh KK, Chakrabortty S, Gulyás B, Padmanabhan P, Ball WB. Mitochondrial Reactive Oxygen Species in Infection and Immunity. Biomolecules. 2024; 14(6):670. https://doi.org/10.3390/biom14060670
Chicago/Turabian StyleMukherjee, Arunima, Krishna Kanta Ghosh, Sabyasachi Chakrabortty, Balázs Gulyás, Parasuraman Padmanabhan, and Writoban Basu Ball. 2024. "Mitochondrial Reactive Oxygen Species in Infection and Immunity" Biomolecules 14, no. 6: 670. https://doi.org/10.3390/biom14060670
APA StyleMukherjee, A., Ghosh, K. K., Chakrabortty, S., Gulyás, B., Padmanabhan, P., & Ball, W. B. (2024). Mitochondrial Reactive Oxygen Species in Infection and Immunity. Biomolecules, 14(6), 670. https://doi.org/10.3390/biom14060670