
Accurate Identification of Spatial Domain by Incorporating Global Spatial Proximity and Local Expression Proximity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
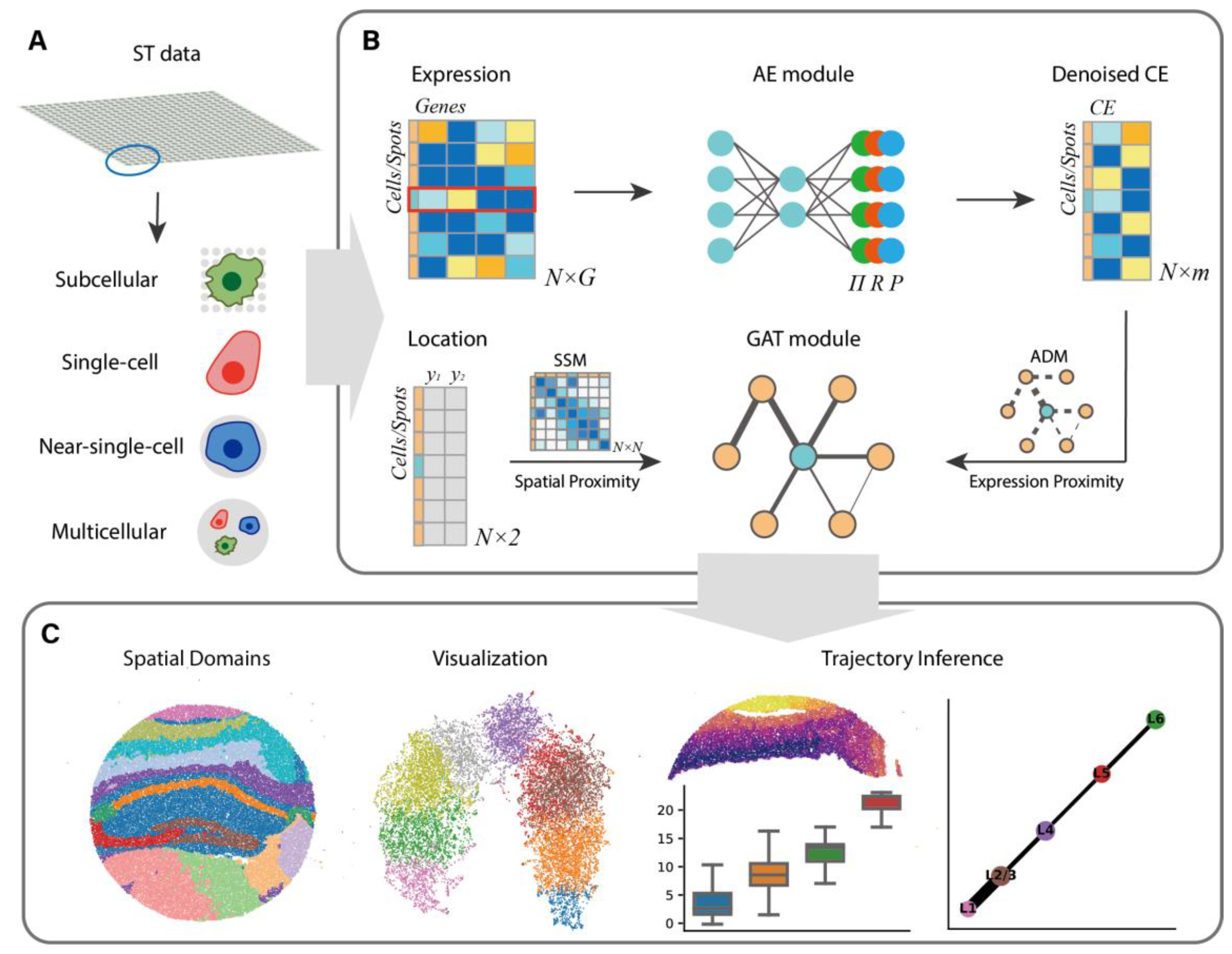
2.1. Architecture Overview
2.2. Extracting Expression Features with AE Module
2.3. Capturing Local and Global Relationships
2.4. Learning SE with GAT Module
2.5. Architecture of SECE
2.6. Datasets
2.7. Evaluation Metrics
2.8. Methods for Comparison
3. Results
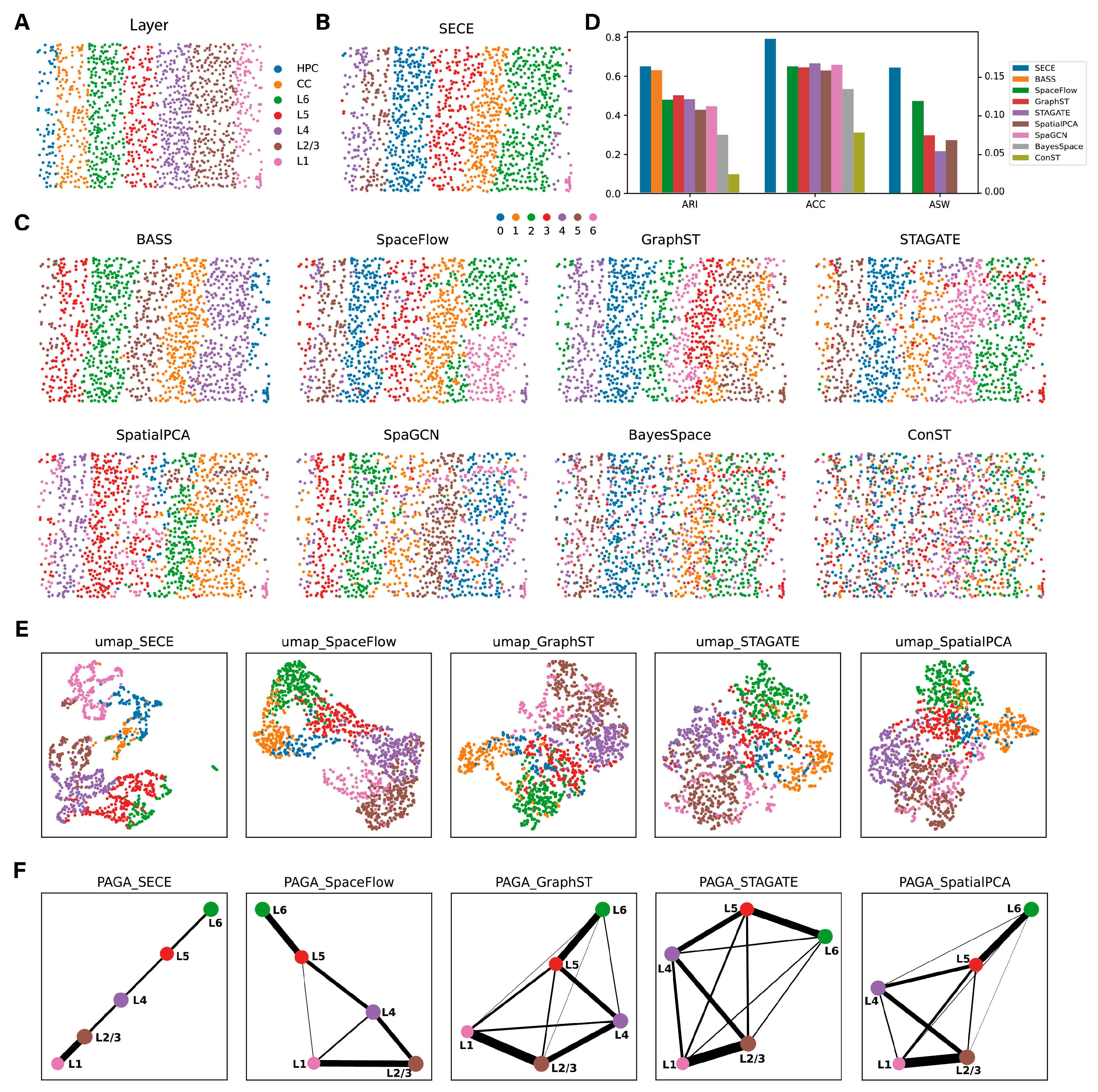
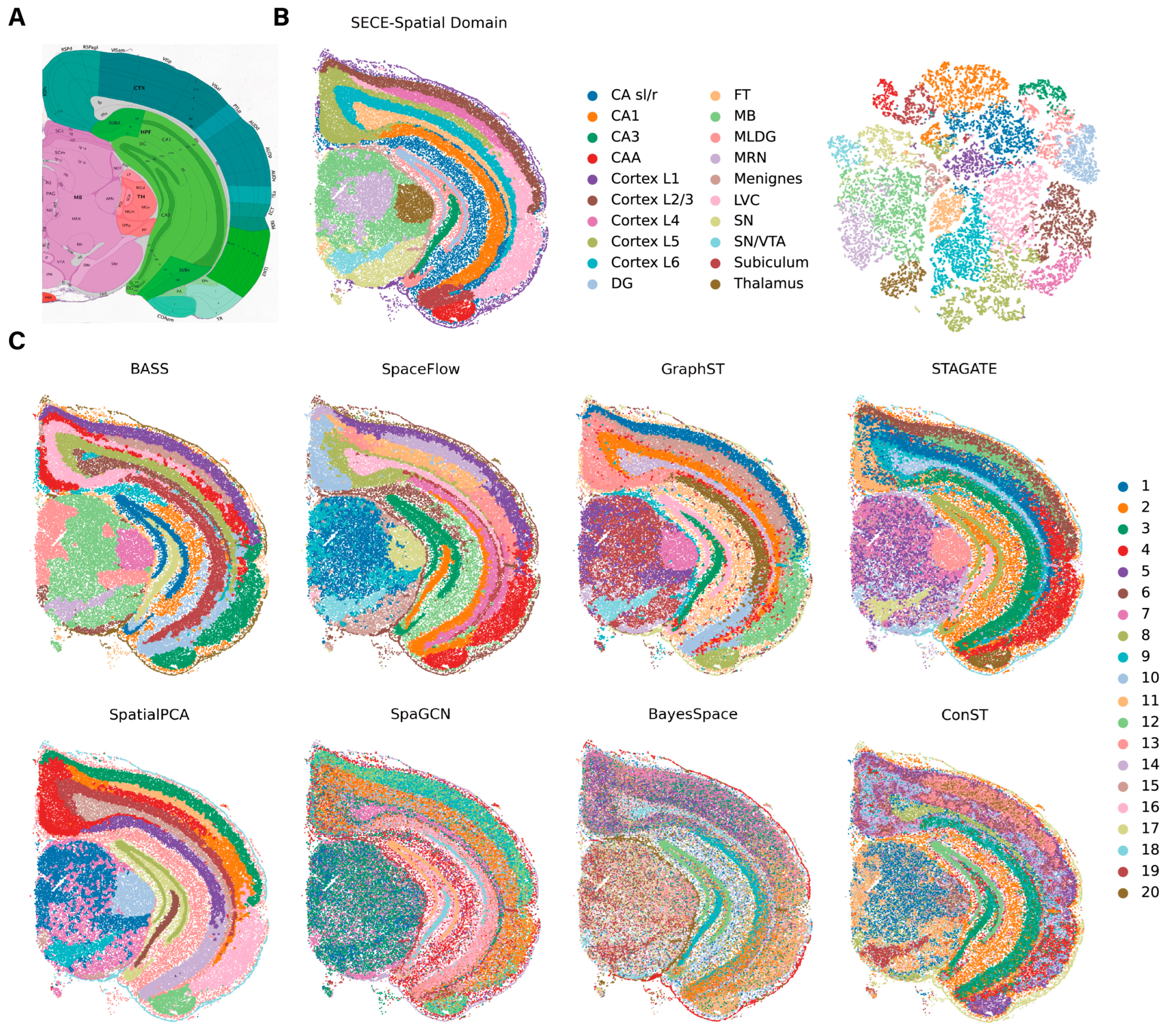
3.1. Application to STARmap Data
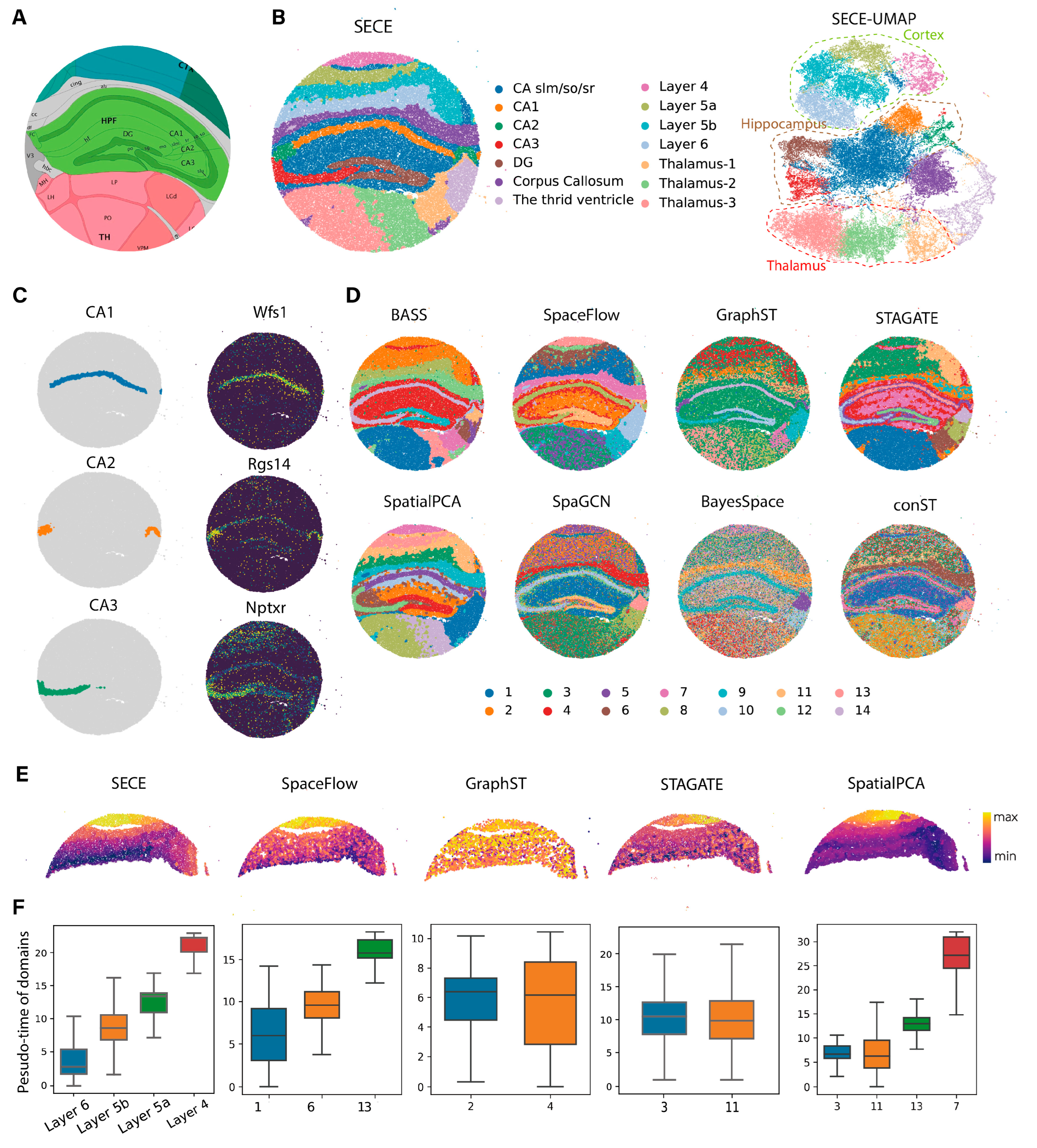
3.2. Application to Slide-seqV2 Data
3.3. Application to Stereo-Seq Data
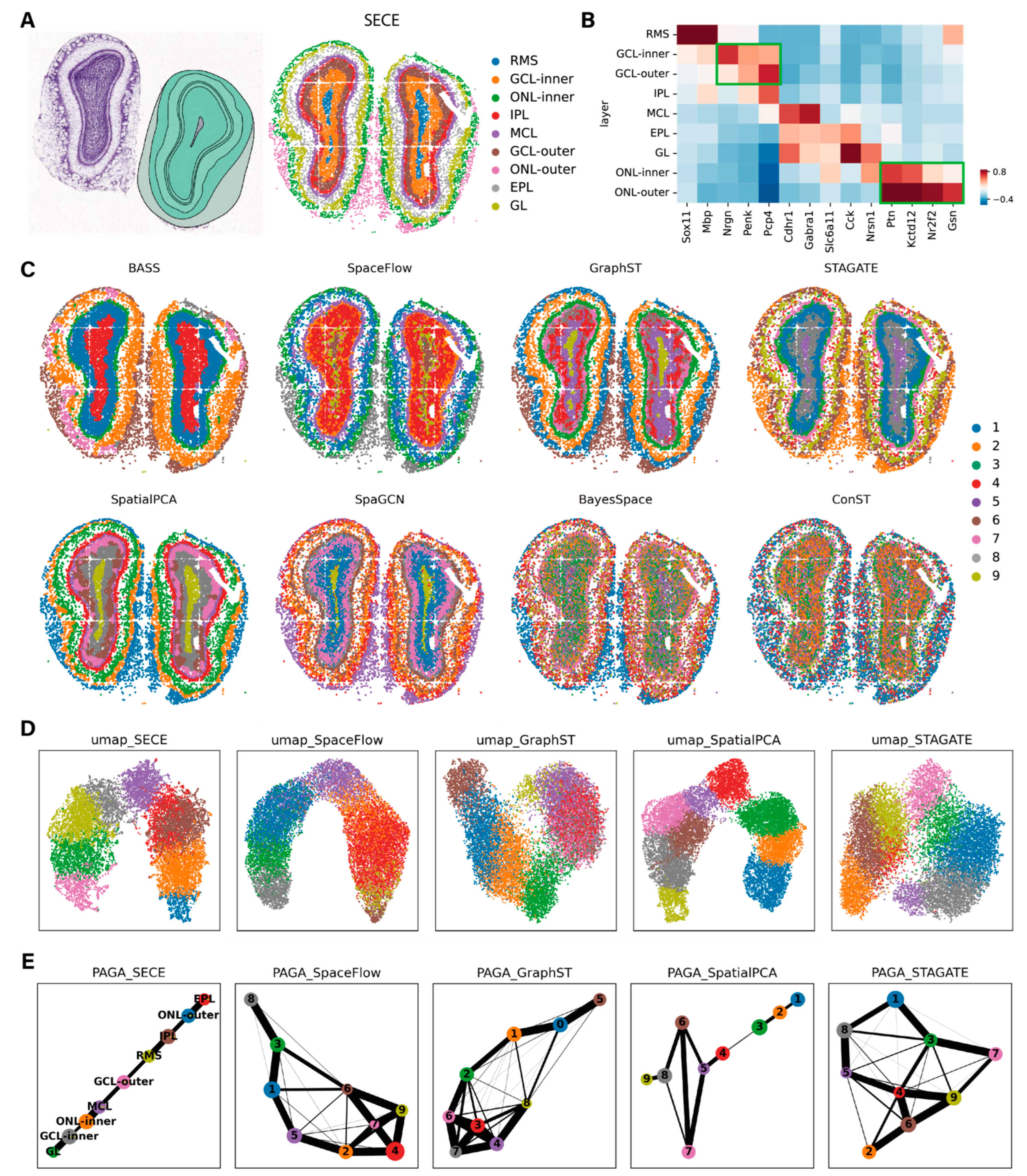
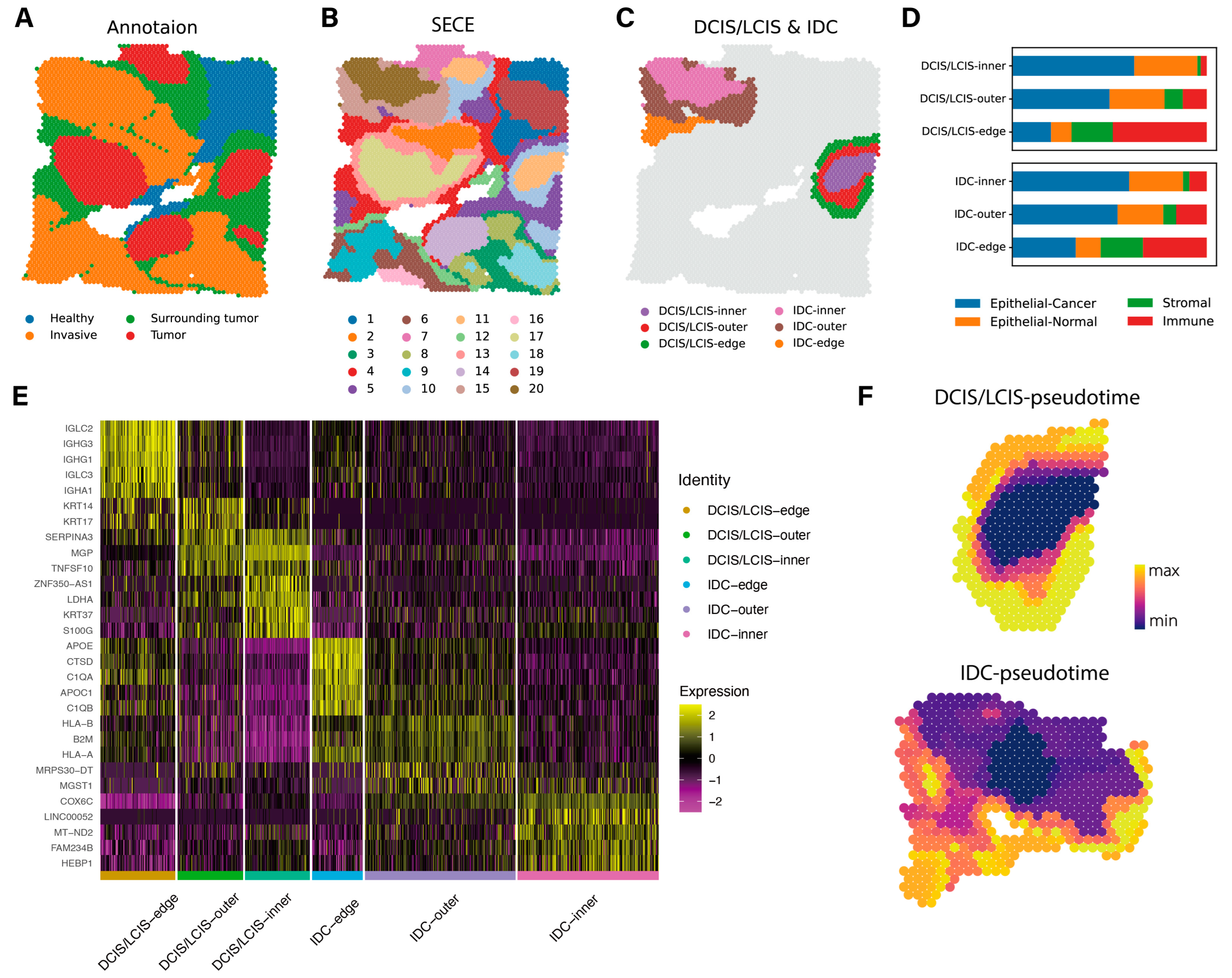
3.4. Application to Visium Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maynard, K.R.; Collado-Torres, L.; Weber, L.M.; Uytingco, C.; Barry, B.K.; Williams, S.R.; Catallini, J.L., 2nd; Tran, M.N.; Besich, Z.; Tippani, M.; et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 2021, 24, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hu, Q.; Lv, T.; Wang, Y.; Lan, Q.; Xiang, R.; Tu, Z.; Wei, Y.; Han, K.; Shi, C.; et al. High-resolution 3D spatiotemporal transcriptomic maps of developing Drosophila embryos and larvae. Dev. Cell 2022, 57, 1271–1283.e4. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.V.; Moncada, R.; Weiss, J.M.; Yanai, I.; White, R.M. Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface. Nat. Commun. 2021, 12, 6278. [Google Scholar] [CrossRef]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J.; et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Lubeck, E.; Zhou, W.; Cai, L. In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron 2016, 92, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.-H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.R.; Bambah-Mukku, D.; Eichhorn, S.W.; Vaughn, E.; Shekhar, K.; Perez, J.D.; Rubinstein, N.D.; Hao, J.; Regev, A.; Dulac, C.; et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 2018, 362, eaau5324. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.F.; Li, C.; Amamoto, R.; et al. Highly multiplexed subcellular RNA sequencing in situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef]
- Moses, L.; Pachter, L. Museum of spatial transcriptomics. Nat. Methods 2022, 19, 534–546. [Google Scholar] [CrossRef]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Vickovic, S.; Eraslan, G.; Salmen, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Aijo, T.; Bonneau, R.; Bergenstrahle, L.; Navarro, J.F.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.-S.; Xi, J.; Si, Y.; Park, S.-R.; Hsu, J.-E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 2021, 184, 3559–3572.e22. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, C.; Navarro, J.F.; Jurek, A.; Martin, A.; Lundeberg, J.; Meletis, K. Molecular atlas of the adult mouse brain. Sci. Adv. 2020, 6, eabb3446. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Hochgerner, H.; Lonnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Haring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Stone, M.R.; Ren, X.; Guenthoer, J.; Smythe, K.S.; Pulliam, T.; Williams, S.R.; Uytingco, C.R.; Taylor, S.E.B.; Nghiem, P.; et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 2021, 39, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, X. BASS: Multi-scale and multi-sample analysis enables accurate cell type clustering and spatial domain detection in spatial transcriptomic studies. Genome Biol. 2022, 23, 168. [Google Scholar] [CrossRef]
- Hu, J.; Li, X.; Coleman, K.; Schroeder, A.; Ma, N.; Irwin, D.J.; Lee, E.B.; Shinohara, R.T.; Li, M. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 2021, 18, 1342–1351. [Google Scholar] [CrossRef]
- Dong, K.; Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat. Commun. 2022, 13, 1739. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Ang, K.S.; Li, M.; Chong, K.L.K.; Sethi, R.; Zhong, C.; Xu, H.; Ong, Z.; Sachaphibulkij, K.; Chen, A.; et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat. Commun. 2023, 14, 1155. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Walker, B.L.; Cang, Z.; Nie, Q. Identifying multicellular spatiotemporal organization of cells with SpaceFlow. Nat. Commun. 2022, 13, 4076. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Yu, T.; Wang, X.; Wang, Y.; Hu, Z.; Li, Y. conST: An interpretable multi-modal contrastive learning framework for spatial transcriptomics. bioRxiv 2022. [Google Scholar] [CrossRef]
- Shang, L.; Zhou, X. Spatially aware dimension reduction for spatial transcriptomics. Nat. Commun. 2022, 13, 7203. [Google Scholar] [CrossRef] [PubMed]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016, 8, 289–317. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar] [CrossRef]
- Wolf, F.A.; Hamey, F.K.; Plass, M.; Solana, J.; Dahlin, J.S.; Göttgens, B.; Rajewsky, N.; Simon, L.; Theis, F.J. PAGA: Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Xu, H.; Fu, H.; Long, Y.; Ang, K.S.; Sethi, R.; Chong, K.; Li, M.; Uddamvathanak, R.; Lee, H.K.; Ling, J.; et al. Unsupervised spatially embedded deep representation of spatial transcriptomics. Genome Medicine 2024, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Frisen, J.; Lundeberg, J. Spatially resolved transcriptomics adds a new dimension to genomics. Nat. Methods 2021, 18, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Munkres, J. Algorithms for the Assignment and Transportation Problems. J. Soc. Ind. Appl. Math. 1957, 5, 32–38. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e16. [Google Scholar] [CrossRef] [PubMed]
- Beiersdorfer, A.; Wolburg, H.; Grawe, J.; Scheller, A.; Kirchhoff, F.; Lohr, C. Sublamina-specific organization of the blood brain barrier in the mouse olfactory nerve layer. Glia 2020, 68, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Kleshchevnikov, V.; Shmatko, A.; Dann, E.; Aivazidis, A.; King, H.W.; Li, T.; Elmentaite, R.; Lomakin, A.; Kedlian, V.; Gayoso, A.; et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 2022, 40, 661–671. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Bilandzic, M.; Rainczuk, A.; Green, E.; Fairweather, N.; Jobling, T.W.; Plebanski, M.; Stephens, A.N. Keratin-14 (KRT14) Positive Leader Cells Mediate Mesothelial Clearance and Invasion by Ovarian Cancer Cells. Cancers 2019, 11, 1228. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ordonez, A.; Seoane, S.; Avila, L.; Eiro, N.; Macia, M.; Arias, E.; Pereira, F.; Garcia-Caballero, T.; Gomez-Lado, N.; Aguiar, P.; et al. POU1F1 transcription factor induces metabolic reprogramming and breast cancer progression via LDHA regulation. Oncogene 2021, 40, 2725–2740. [Google Scholar] [CrossRef] [PubMed]
- Nalio Ramos, R.; Missolo-Koussou, Y.; Gerber-Ferder, Y.; Bromley, C.P.; Bugatti, M.; Nunez, N.G.; Tosello Boari, J.; Richer, W.; Menger, L.; Denizeau, J.; et al. Tissue-resident FOLR2+ macrophages associate with CD8+ T cell infiltration in human breast cancer. Cell 2022, 185, 1189–1207.e25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Liu, C.; Li, W.; Zhou, F.; Wang, X.; Zheng, J. The Apolipoprotein C1 is involved in breast cancer progression via EMT and MAPK/JNK pathway. Pathol. Res. Pract. 2022, 229, 153746. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.U.; Woo, S.M.; Im, S.-S.; Jang, Y.; Han, E.; Kim, S.H.; Lee, H.; Lee, H.-S.; Nam, J.-O.; Gabrielson, E.; et al. Cathepsin D as a potential therapeutic target to enhance anticancer drug-induced apoptosis via RNF183-mediated destabilization of Bcl-xL in cancer cells. Cell Death Dis. 2022, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, M.; Song, S. Cathepsin D enhances breast cancer invasion and metastasis through promoting hepsin ubiquitin-proteasome degradation. Cancer Lett. 2018, 438, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Revel, M.; Sautes-Fridman, C.; Fridman, W.-H.; Roumenina, L.T. C1q+ macrophages: Passengers or drivers of cancer progression. Trends Cancer 2022, 8, 517–526. [Google Scholar] [CrossRef]
- Noblejas-Lopez, M.D.M.; Nieto-Jimenez, C.; Morcillo Garcia, S.; Perez-Pena, J.; Nuncia-Cantarero, M.; Andres-Pretel, F.; Galan-Moya, E.M.; Amir, E.; Pandiella, A.; Gyorffy, B.; et al. Expression of MHC class I, HLA-A and HLA-B identifies immune-activated breast tumors with favorable outcome. Oncoimmunology 2019, 8, e1629780. [Google Scholar] [CrossRef]
- Nomura, T.; Huang, W.-C.; Zhau, H.E.; Josson, S.; Mimata, H.; WK Chung, L. β2-Microglobulin-mediated signaling as a target for cancer therapy. Anti-Cancer Agents Med. Chem. 2014, 14, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Pan, Y.; Liu, G.; Yang, T.; Jin, Y.; Zhou, F.; Wei, Y. MRPS30-DT Knockdown Inhibits Breast Cancer Progression by Targeting Jab1/Cops5. Front. Oncol. 2019, 9, 1170. [Google Scholar] [CrossRef]
- Zeng, B.; Ge, C.; Li, R.; Zhang, Z.; Fu, Q.; Li, Z.; Lin, Z.; Liu, L.; Xue, Y.; Xu, Y.; et al. Knockdown of microsomal glutathione S-transferase 1 inhibits lung adenocarcinoma cell proliferation and induces apoptosis. Biomed. Pharmacother. 2020, 121, 109562. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yu, J.; Lai, S.; Li, Z.; Qu, F.; Fu, X.; Li, Q.; Zhong, X.; Zhang, D.; Li, H. Long Non-Coding RNA LINC00052 Targets miR-548p/Notch2/Pyk2 to Modulate Tumor Budding and Metastasis of Human Breast Cancer. Biochem. Genet. 2022, 61, 336–353. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Wang, D.; Chen, Y. Role of the long non-coding RNA LINC00052 in tumors. Oncol. Lett. 2021, 21, 316. [Google Scholar] [CrossRef]
- Salameh, A.; Fan, X.; Choi, B.K.; Zhang, S.; Zhang, N.; An, Z. HER3 and LINC00052 interplay promotes tumor growth in breast cancer. Oncotarget 2017, 8, 6526–6539. [Google Scholar] [CrossRef]
- Grzybowska-Szatkowska, L.; Slaska, B. Mitochondrial NADH dehydrogenase polymorphisms are associated with breast cancer in Poland. J. Appl. Genet. 2014, 55, 173–181. [Google Scholar] [CrossRef]
- Lyu, L.; Wang, M.; Zheng, Y.; Tian, T.; Deng, Y.; Xu, P.; Lin, S.; Yang, S.; Zhou, L.; Hao, Q.; et al. Overexpression of FAM234B Predicts Poor Prognosis in Patients with Luminal Breast Cancer. Cancer Manag. Res. 2020, 12, 12457–12471. [Google Scholar] [CrossRef]
- Biancalani, T.; Scalia, G.; Buffoni, L.; Avasthi, R.; Lu, Z.; Sanger, A.; Tokcan, N.; Vanderburg, C.R.; Segerstolpe, A.; Zhang, M.; et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 2021, 18, 1352–1362. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; He, Y.; Xie, Z. Accurate Identification of Spatial Domain by Incorporating Global Spatial Proximity and Local Expression Proximity. Biomolecules 2024, 14, 674. https://doi.org/10.3390/biom14060674
Yu Y, He Y, Xie Z. Accurate Identification of Spatial Domain by Incorporating Global Spatial Proximity and Local Expression Proximity. Biomolecules. 2024; 14(6):674. https://doi.org/10.3390/biom14060674
Chicago/Turabian StyleYu, Yuanyuan, Yao He, and Zhi Xie. 2024. "Accurate Identification of Spatial Domain by Incorporating Global Spatial Proximity and Local Expression Proximity" Biomolecules 14, no. 6: 674. https://doi.org/10.3390/biom14060674