

Macrophage Perspectives in Liver Diseases: Programmed Death, Related Biomarkers, and Targeted Therapy
Abstract

1. Introduction
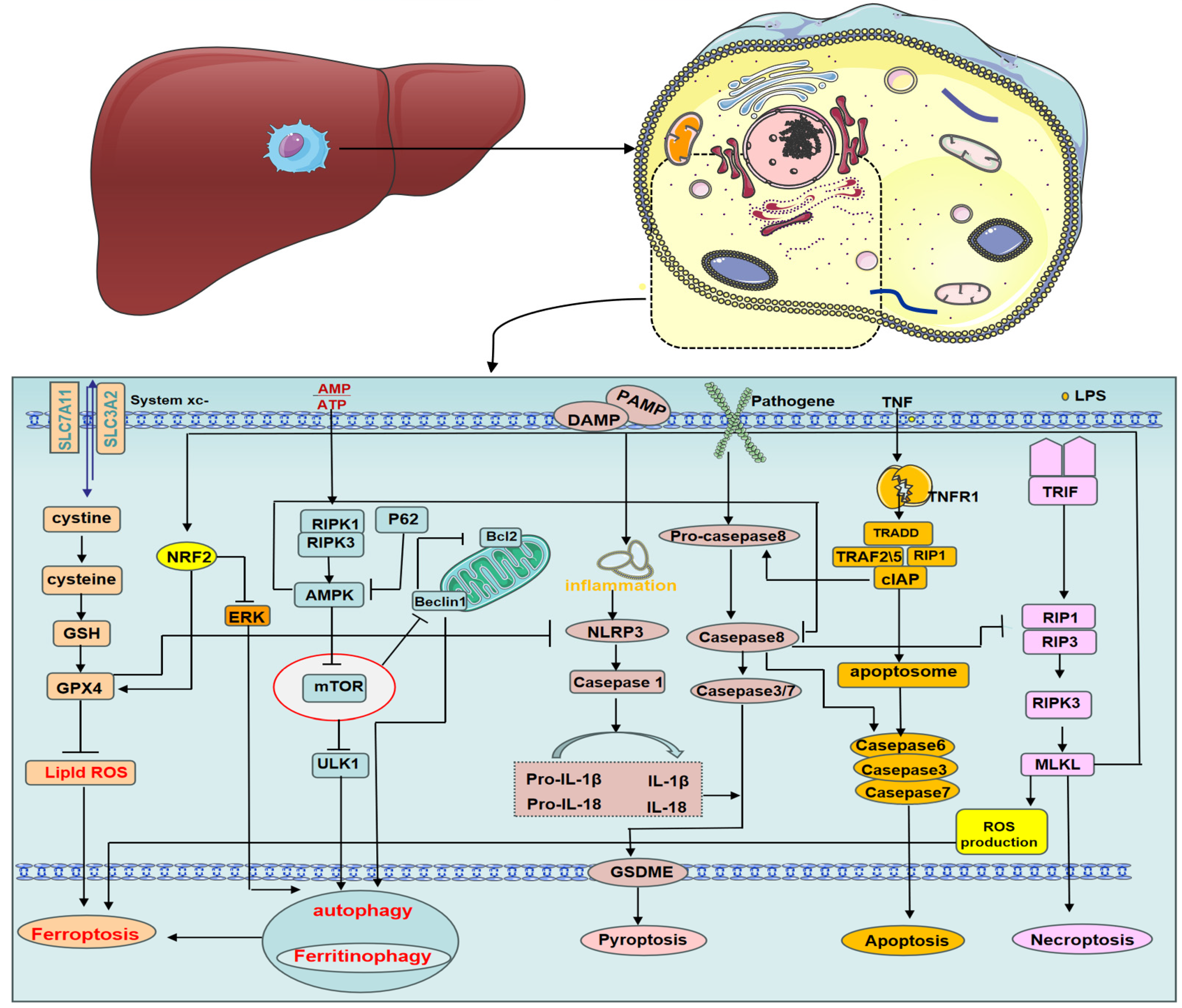
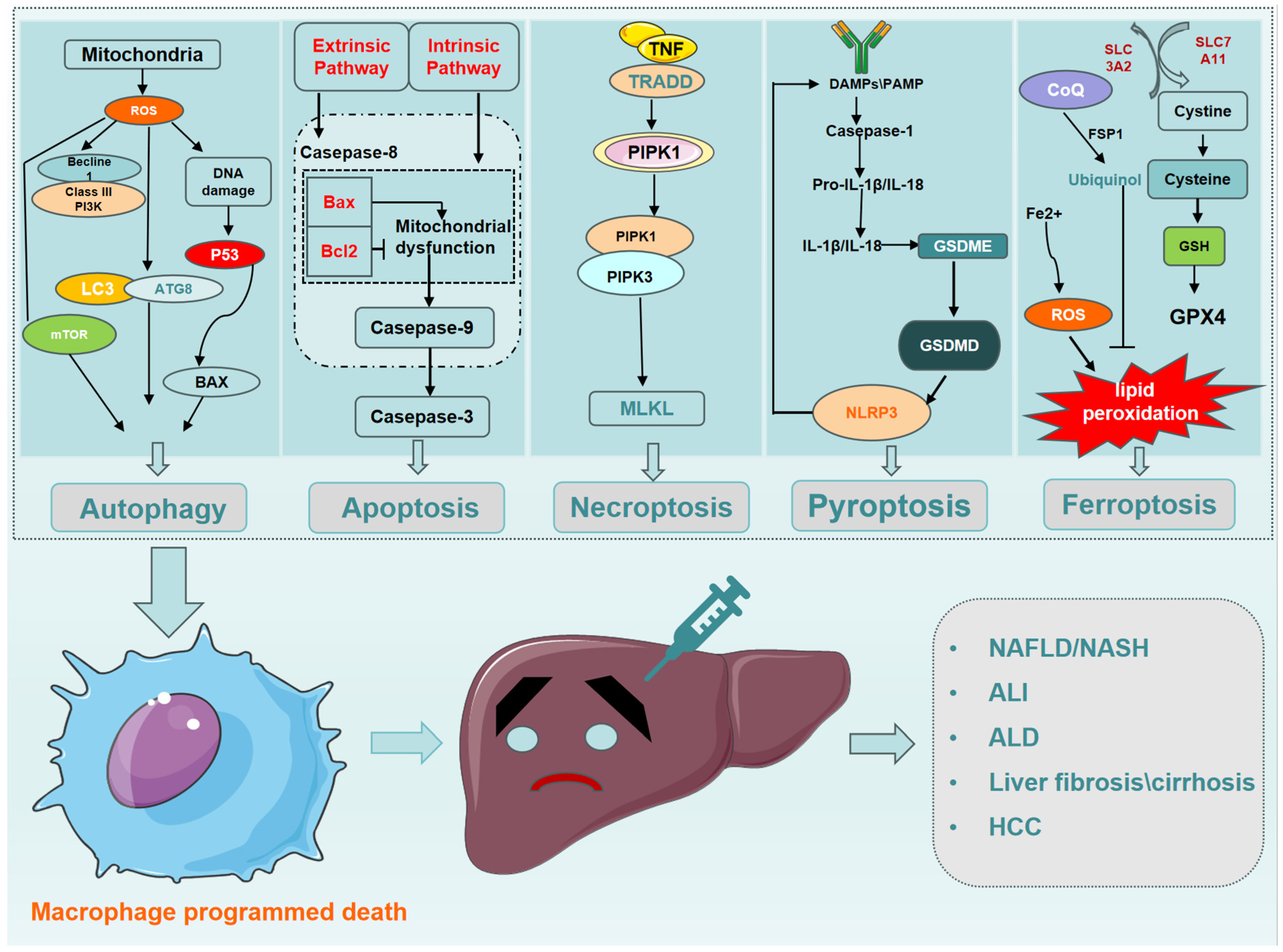
2. Macrophage Programmed Death
2.1. Macrophage Autophagy
2.2. Macrophage Apoptosis
2.3. Macrophage Necroptosis
2.4. Macrophage Pyroptosis
2.5. Macrophage Ferroptosis
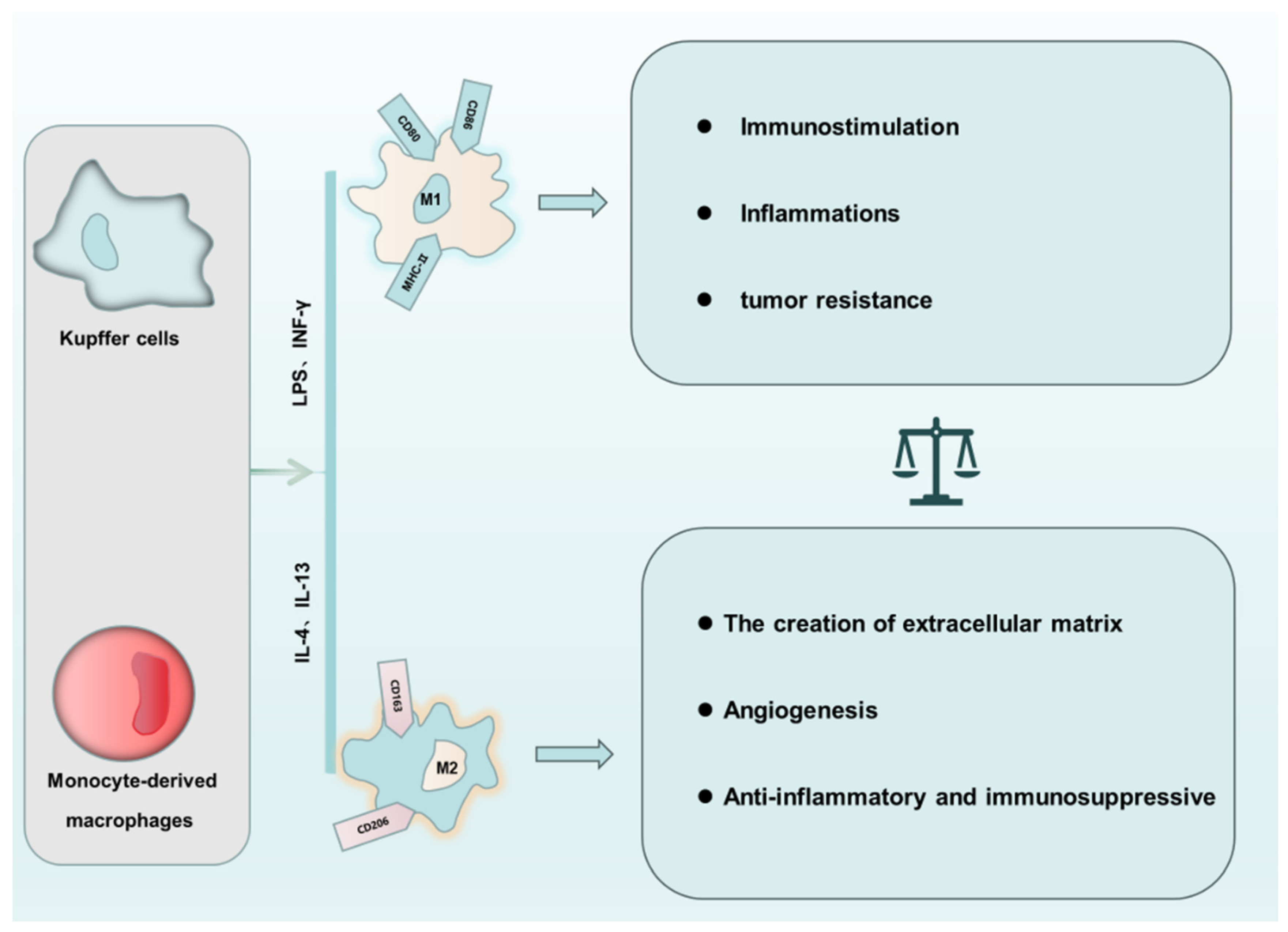
3. The “Love of Kill” between Macrophage Polarization and PCD
4. Interactions between Different Types of PCD in Macrophages
5. Effect of Macrophage PCD on Liver Disease
5.1. The Effect of Macrophage PCD on NAFLD/NASH
5.1.1. Autophagy
5.1.2. Apoptosis
5.1.3. Necroptosis
5.1.4. Pyroptosis
5.1.5. Ferroptosis
5.2. The Effect of Macrophage PCD on Alcoholic Liver Disease
5.2.1. Autophagy
5.2.2. Apoptosis
5.2.3. Necroptosis
5.3. The Effect of Macrophage PCD on Acute Liver Injury
5.3.1. Autophagy
5.3.2. Necroptosis
5.3.3. Pyroptosis
5.3.4. Ferroptosis
5.4. The Effect of Macrophage PCD on Liver Fibrosis/Cirrhosis
5.4.1. Autophagy
5.4.2. Apoptosis
5.4.3. Necroptosis
5.4.4. Pyroptosis
5.4.5. Ferroptosis
5.5. The Effect of Macrophage PCD on Hepatocellular Carcinoma
5.5.1. Autophagy
5.5.2. Apoptosis
5.5.3. Necroptosis
5.5.4. Pyroptosis
5.5.5. Ferroptosis
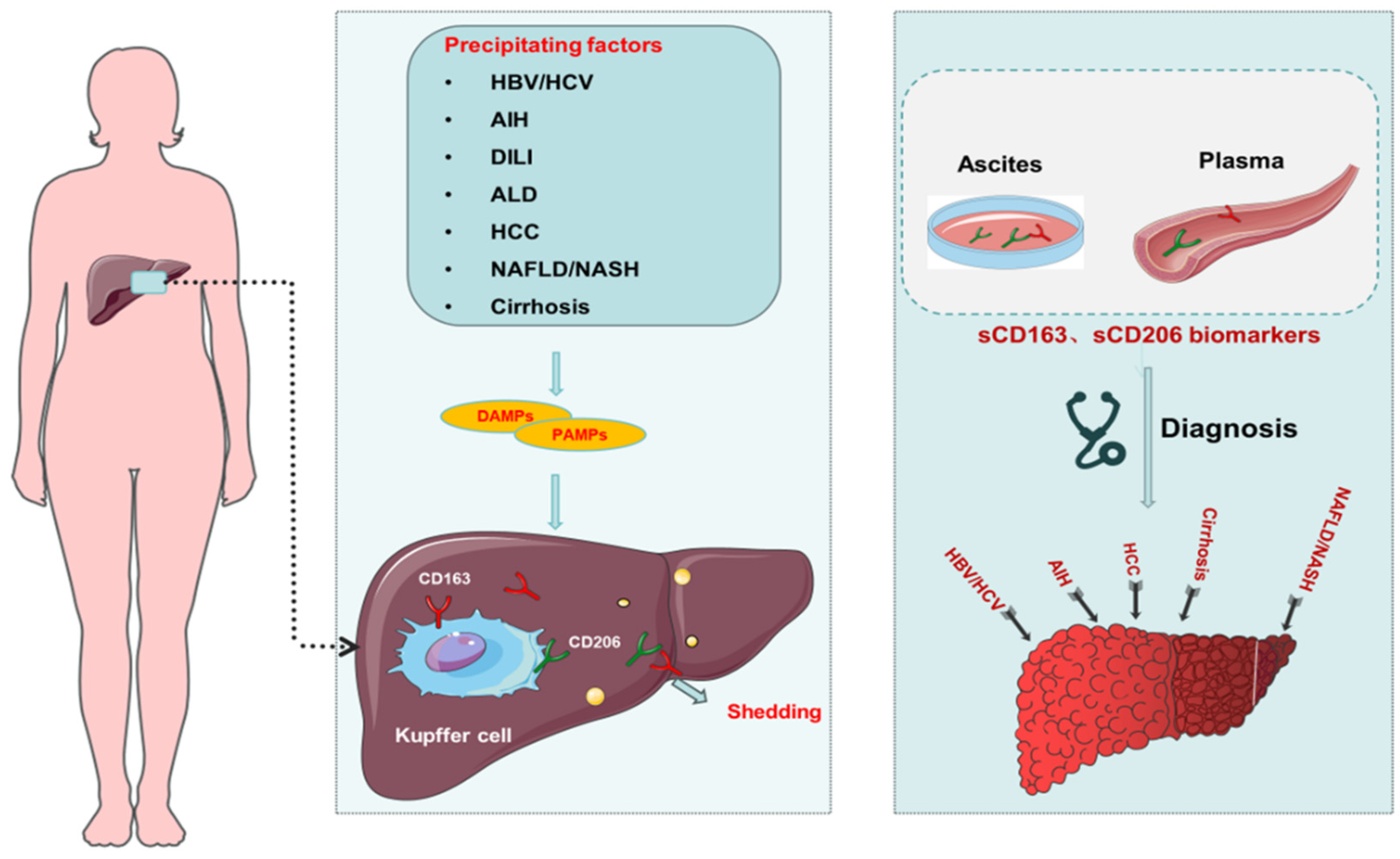
6. Macrophage-Related Biomarkers in Liver Disease
7. Mechanisms of Macrophage-Targeted Therapy for Liver Diseases and Development of Small Molecule Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Mechanism | Drug | Clinical Trial/Phase | Clinical Trial Number or Reference |
|---|---|---|---|---|
| ALD | ||||
| antibiotic | Attack intestinal bacteria and inhibit macrophage activation | Vancomycin, gentamicin, meropenem | I | NCT03157388 [153] |
| IL-1β antagonist | Inhibition of inflammasome activation in KCs | IL-1Ra | II | NCT01809132 [154] |
| NASH | ||||
| FXR agonists | Increased cholesterol transport in macrophages | Obeticholic acid | III | NCT02548351 [155] |
| CCR2/CCR5 antagonist | Inhibit monocyte recruitment | Cenicriviroc | II | NCT02217475 [156] |
| Galectin-3 antagonist | Inhibition of inflammatory macrophage function | GR-MD-02 | II | NCT02462967 [157] |
| PPARα/δ agonist | Promote differentiation of macrophages into anti-inflammatory subgroups | Elafibranor | III | NCT02704403 [158] |
| Viral hepatitis | ||||
| GM-CSF | GM-CSF promotes macrophage differentiation | Y peginterferon alpha-2b plus GM-CSF | II | NCT02332473 [159] |
| HCC | ||||
| PD-1/PD-L1 | Regulation of immune checkpoints in macrophages | CA-170 | II | NCT04343859 [160] |
| CSF1R | Multi-target inhibitor that suppresses angiogenesis-related kinases and decreases macrophage differentiation. | Chiauranib | I | NCT03245190 |
| CCR2/5 | CCR2/CCR5 antagonist (inhibits monocyte/macrophage infiltration) | Nivolumab plus CCR2/5 inhibitor | II | NCT04123379 |
8. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Shi, X.; He, X.; Gao, Y. Macrophage Phenotype and Function in Liver Disorder. Front. Immunol. 2020, 10, 3112. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage Polarization and Its Role in Liver Disease. Front. Immunol. 2021, 12, 803037. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kanai, T. Role of toll-like receptors in immune activation and tolerance in the liver. Front. Immunol. 2014, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Lamkanfi, M.; Núñez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Wang, Y.; Yang, L.; Zhang, Z.; Zhao, Y.; Shen, Z.; Han, X.; Du, X.; Jin, H.; Li, C.; et al. Rebalancing liver-infiltrating CCR3(+) and CD206(+) monocytes improves diet-induced NAFLD. Cell Rep. 2023, 42, 112753. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lu, Q.; Gao, W.; Yu, S. Recent advancement on development of drug-induced macrophage polarization in control of human diseases. Life Sci. 2021, 284, 119914. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Zhang, S.; Mizushima, N. Autophagy genes in biology and disease. Nat. Rev. Genet. 2023, 24, 382–400. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Z.; Huang, Y.; Bai, C.; Zhang, X.; Fang, M.; Ju, Z.; Liu, B. Membrane dynamics of ATG4B and LC3 in autophagosome formation. J Mol Cell Biol. 2022, 13, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qian, C.; Wang, Q.; Song, L.; He, Z.; Liu, W.; Wan, W. Deacetylation of ATG7 drives the induction of macroautophagy and LC3-associated microautophagy. Autophagy 2024, 20, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Wan, X.; Zou, X.; Sun, S.; Hao, X.; Liang, C.; Zhang, Z.; Zhang, F.; Sun, B.; Li, H.; et al. Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway. Cell Death Dis. 2021, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhou, K.; Liao, L.; Zhang, T.; Yang, M.; Sun, C. Lipoxin A4 receptor agonist BML-111 induces autophagy in alveolar macrophages and protects from acute lung injury by activating MAPK signaling. Respir. Res. 2018, 19, 243. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Yuk, J.M.; Lee, H.M.; Lee, S.-H.; Son, J.W.; Harding, C.V.; Kim, J.-M.; Modlin, R.L.; Jo, E.-K. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol. 2010, 12, 1648–1665. [Google Scholar] [CrossRef]
- Chan, H.; Li, Q.; Wang, X.; Liu, W.Y.; Hu, W.; Zeng, J.; Xie, C.; Kwong, T.N.Y.; Ho, I.H.T.; Liu, X.; et al. Vitamin D(3) and carbamazepine protect against Clostridioides difficile infection in mice by restoring macrophage lysosome acidification. Autophagy 2022, 18, 2050–2067. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, D.; Rojas, M.; Hernández, I.; Radzioch, D.; García, L.F.; Barrera, L.F. Role of TLR2- and TLR4-mediated signaling in Mycobacterium tuberculosis-induced macrophage death. Cell Immunol. 2010, 260, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.-X.; Divangahi, M.; Remold, H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef]
- López, M.; Sly, L.M.; Luu, Y.; Young, D.; Cooper, H.; Reiner, N.E. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J. Immunol. 2003, 170, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Espinosa, P.; García, T.; Mancilla, R. The 19 kDa Mycobacterium tuberculosis lipoprotein (LpqH) induces macrophage apoptosis through extrinsic and intrinsic pathways: A role for the mitochondrial apoptosis-inducing factor. Clin. Dev. Immunol. 2012, 2012, 950503. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Espinosa, P.; Esparza, M.A.; Colon, M.; Bernal, G.; Mancilla, R. Mycobacterium tuberculosis 38-kDa lipoprotein is apoptogenic for human monocyte-derived macrophages. Scand. J. Immunol. 2009, 69, 20–28. [Google Scholar] [CrossRef]
- Ramón-Vázquez, A.; de la Rosa, J.V.; Tabraue, C.; Castrillo, A. Bone Marrow-Derived Macrophage Immortalization of LXR Nuclear Receptor-Deficient Cells. Methods Mol. Biol. 2019, 1951, 75–85. [Google Scholar] [PubMed]
- Che, X.; Xiao, Q.; Song, W.; Zhang, H.; Sun, B.; Geng, N.; Tao, Z.; Shao, Q.; Pu, J. Protective Functions of Liver X Receptor α in Established Vulnerable Plaques: Involvement of Regulating Endoplasmic Reticulum-Mediated Macrophage Apoptosis and Efferocytosis. J. Am. Heart Assoc. 2021, 10, e018455. [Google Scholar] [CrossRef] [PubMed]
- Hilbi, H.; Zychlinsky, A.; Sansonetti, P.J. Macrophage apoptosis in microbial infections. Parasitology 1997, 115, S79–S87. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD(+) Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium tuberculosis. Cell Rep. 2018, 24, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell. 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Wang, J.; Li, Y.; Wang, Y.; Shi, F.; Hong, L.; Li, L.; Diao, H. zVAD alleviates experimental autoimmune hepatitis in mice by increasing the sensitivity of macrophage to TNFR1-dependent necroptosis. J. Autoimmun. 2022, 133, 102904. [Google Scholar] [CrossRef]
- Yi, Y.S. Caspase-11 non-canonical inflammasome: A critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 2017, 152, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.M.; Riestra, A.M.; Ali, S.R.; Fong, J.J.; Liu, J.Z.; Hughes, G.; Varki, A.; Nizet, V. Siglec-14 Enhances NLRP3-Inflammasome Activation in Macrophages. J. Innate Immun. 2020, 12, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ma, W.; Gao, W.; Xing, Y.; Chen, L.; Xia, Z.; Zhang, Z.; Dai, Z. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019, 10, 542. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Bao, X.; Weng, X.; Bai, X.; Feng, Y.; Huang, J.; Liu, S.; Jia, H.; Yu, B. The protective effect of quercetin on macrophage pyroptosis via TLR2/Myd88/NF-κB and ROS/AMPK pathway. Life Sci. 2022, 291, 120064. [Google Scholar] [CrossRef] [PubMed]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 2017, 24, 507–514.e4. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Iron-induced oxidant stress in alcoholic liver fibrogenesis. Alcohol 2003, 30, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Youssef, L.A.; Rebbaa, A.; Pampou, S.; Weisberg, S.P.; Stockwell, B.R.; Hod, E.A.; Spitalnik, S.L. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood 2018, 131, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Liang, Y.; Wei, T.; Zou, L.; Huang, X.; Kong, L.; Tang, M.; Zhang, T. The role of ferroptosis mediated by NRF2/ERK-regulated ferritinophagy in CdTe QDs-induced inflammation in macrophage. J. Hazard. Mater. 2022, 436, 129043. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Che, M.; Li, C.; Li, Y.; Zhang, T.; Li, X.; Sun, C. PP1A prevents ROS-induced pyroptosis by inhibiting MAPK/caspase-3 in mouse adipose tissue. FEBS J. 2022, 289, 3839–3853. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; E Tanaka, K.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Idell, S.; Tang, H. M1 Macrophages Are More Susceptible to Necroptosis. J. Cell Immunol. 2021, 3, 97–102. [Google Scholar] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, J.; Geng, C.; Wang, X.; Wang, Y.; Sun, N.; Wang, P.; Han, L.; Li, Z.; Fan, H.; et al. Myoglobin promotes macrophage polarization to M1 type and pyroptosis via the RIG-I/Caspase1/GSDMD signaling pathway in CS-AKI. Cell Death Discov. 2022, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Shotland, A.M.; Fontenot, A.P.; McKee, A.S. Pulmonary Macrophage Cell Death in Lung Health and Disease. Am. J. Respir. Cell Mol. Biol. 2021, 64, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013, 27, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef]
- Newton, K.; Dugger, D.L.; Wickliffe, K.E.; Kapoor, N.; de Almagro, M.C.; Vucic, D.; Komuves, L.; Ferrando, R.E.; French, D.M.; Webster, J.; et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014, 343, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.-D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, X.; Sun, Y.; Berleth, N.; Deitersen, J.; Schlütermann, D.; Stuhldreier, F.; Wallot-Hieke, N.; Mendiburo, M.J.; Cox, J.; et al. TNF-induced necroptosis initiates early autophagy events via RIPK3-dependent AMPK activation, but inhibits late autophagy. Autophagy 2021, 17, 3992–4009. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tao, K.; Wang, Y.; Huang, Y.; Duan, C.; Wang, T.; Li, C.; Zhang, P.; Yin, Y.; Gao, J.; et al. Necrosulfonamide ameliorates intestinal inflammation via inhibiting GSDMD-medicated pyroptosis and MLKL-mediated necroptosis. Biochem. Pharmacol. 2022, 206, 115338. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Gram, A.M.; Booty, L.M.; Bryant, C.E. Chopping GSDMD: Caspase-8 has joined the team of pyroptosis-mediating caspases. EMBO J. 2019, 38, e102065. [Google Scholar] [CrossRef]
- Zhang, Y.; Swanda, R.V.; Nie, L.; Liu, X.; Wang, C.; Lee, H.; Lei, G.; Mao, C.; Koppula, P.; Cheng, W.; et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 2021, 12, 1589. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, Q.; Lv, M.; Ma, W.; Sun, J.; Zhong, X.; Hu, R.; Ma, M.; Han, Z.; Zhang, W.; et al. Role of liensinine in sensitivity of activated macrophages to ferroptosis and in acute liver injury. Cell Death Discov. 2023, 9, 189. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, J.; Huang, F.; Yao, Y.; Xu, L. Leptin induces NAFLD progression through infiltrated CD8+ T lymphocytes mediating pyroptotic-like cell death of hepatocytes and macrophages. Dig. Liver Dis. 2021, 53, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Yang, L.; Yang, Y.; Xu, J. Inhibition of keratinocyte ferroptosis suppresses psoriatic inflammation. Cell Death Dis. 2021, 12, 1009. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wang, Y.; He, Y.; Zhao, B.; Zhang, Q.; Li, S. MiR-1656 targets GPX4 to trigger pyroptosis in broilers kidney tissues by activating NLRP3 inflammasome under Se deficiency. J. Nutr. Biochem. 2022, 105, 109001. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Huang, B.; Cao, J.; Wang, Y.; Xiao, H.; Zhu, Y.; Zhang, H. ROS fine-tunes the function and fate of immune cells. Int. Immunopharmacol. 2023, 119, 110069. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; de Carvalho Ribeiro, M.; Iracheta-Vellve, A.; Lowe, P.; Ambade, A.; Satishchandran, A.; Bukong, T.; Catalano, D.; Kodys, K.; Szabo, G. Macrophage-Specific Hypoxia-Inducible Factor-1α Contributes to Impaired Autophagic Flux in Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Deust, A.; Chobert, M.N.; Demontant, V.; Gricourt, G.; Denaës, T.; Thiolat, A.; Ruiz, I.; Rodriguez, C.; Pawlotsky, J.-M.; Teixeira-Clerc, F. Macrophage autophagy protects against hepatocellular carcinogenesis in mice. Sci. Rep. 2021, 11, 18809. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Luo, Y.; Lai, X. The comprehensive role of apoptosis inhibitor of macrophage (AIM) in pathological conditions. Clin. Exp. Immunol. 2023, 212, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Yi, Y.; Chen, Y.; Zhang, H.; Orning, P.; Lien, E.; Jie, J.; Zhang, W.; Xu, Q.; Li, Y.; et al. RIP1 kinase activity promotes steatohepatitis through mediating cell death and inflammation in macrophages. Cell Death Differ. 2021, 28, 1418–1433. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ni, M.; Tian, Y.; Wang, H.; Qiu, J.; You, W.; Wei, S.; Shi, Y.; Zhou, J.; Cheng, F.; et al. Myeloid Nrf2 deficiency aggravates non-alcoholic steatohepatitis progression by regulating YAP-mediated NLRP3 inflammasome signaling. iScience 2021, 24, 102427. [Google Scholar] [CrossRef] [PubMed]
- Drummer, C.; Saaoud, F.; Jhala, N.C.; Cueto, R.; Sun, Y.; Xu, K.; Shao, Y.; Lu, Y.; Shen, H.; Yang, L.; et al. Caspase-11 promotes high-fat diet-induced NAFLD by increasing glycolysis, OXPHOS, and pyroptosis in macrophages. Front. Immunol. 2023, 14, 1113883. [Google Scholar] [CrossRef] [PubMed]
- Rametta, R.; Fracanzani, A.L.; Fargion, S.; Dongiovanni, P. Dysmetabolic Hyperferritinemia and Dysmetabolic Iron Overload Syndrome (DIOS): Two Related Conditions or Different Entities? Curr. Pharm. Des. 2020, 26, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, Y.; Tanaka, M.; Itoh, M.; Ochi, K.; Ito, A.; Hidaka, I.; Sakaida, I.; Ogawa, Y.; Suganami, T. Iron-rich Kupffer cells exhibit phenotypic changes during the development of liver fibrosis in NASH. iScience 2021, 24, 102032. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e5. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Kok, C.Y.-Y.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Babuta, M.; Furi, I.; Bala, S.; Bukong, T.N.; Lowe, P.; Catalano, D.; Calenda, C.; Kodys, K.; Szabo, G. Dysregulated Autophagy and Lysosome Function Are Linked to Exosome Production by Micro-RNA 155 in Alcoholic Liver Disease. Hepatology 2019, 70, 2123–2141. [Google Scholar] [CrossRef] [PubMed]
- Denaës, T.; Lodder, J.; Chobert, M.N.; Ruiz, I.; Pawlotsky, J.-M.; Lotersztajn, S.; Teixeira-Clerc, F. The Cannabinoid Receptor 2 Protects Against Alcoholic Liver Disease Via a Macrophage Autophagy-Dependent Pathway. Sci. Rep. 2016, 6, 28806. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhong, Z.; Kim, S.Y.; Uchiyama, R.; Roh, Y.S.; Matsushita, H.; Gottlieb, R.A.; Seki, E. Murine macrophage autophagy protects against alcohol-induced liver injury by degrading interferon regulatory factor 1 (IRF1) and removing damaged mitochondria. J. Biol. Chem. 2019, 294, 12359–12369. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, G.; Cingolani, F.; Zhao, E.; Tanaka, K.; Czaja, M.J. Decreased Macrophage Autophagy Promotes Liver Injury and Inflammation from Alcohol. Alcohol Clin. Exp. Res. 2019, 43, 1403–1413. [Google Scholar] [CrossRef]
- Zhao, N.; Xia, G.; Cai, J.; Li, Z.; Lv, X.W. Adenosine receptor A2B mediates alcoholic hepatitis by regulating cAMP levels and the NF-KB pathway. Toxicol. Lett. 2022, 359, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Singhal, P.C.; Reddy, K.; Ding, G.; Kapasi, A.; Franki, N.; Ranjan, R.; Nwakoby, I.E.; Gibbons, N. Ethanol-induced macrophage apoptosis: The role of TGF-beta. J. Immunol. 1999, 162, 3031–3036. [Google Scholar] [CrossRef] [PubMed]
- Roca, F.J.; Ramakrishnan, L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.; Zhao, C.; Li, F.; Gu, Z.; Liu, L.; Zhang, L.; Wang, Y.; He, L.; Liu, Y.; Liu, Q.; et al. Intestinal HIF-1α deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J. Hepatol. 2018, 69, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Shibamoto, A.; Kaji, K.; Nishimura, N.; Kubo, T.; Iwai, S.; Tomooka, F.; Suzuki, J.; Tsuji, Y.; Fujinaga, Y.; Kawaratani, H.; et al. Vitamin D deficiency exacerbates alcohol-related liver injury via gut barrier disruption and hepatic overload of endotoxin. J. Nutr. Biochem. 2023, 122, 109450. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.D. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res. Health 2003, 27, 300–306. [Google Scholar] [PubMed]
- Lin, X.; Cui, M.; Xu, D.; Hong, D.; Xia, Y.; Xu, C.; Li, R.; Zhang, X.; Lou, Y.; He, Q.; et al. Liver-specific deletion of Eva1a/Tmem166 aggravates acute liver injury by impairing autophagy. Cell Death Dis. 2018, 9, 768. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, G.; Zhao, E.; Liu, K.; Lin, Y.; Tesfa, L.; Tanaka, K.E.; Czaja, M.J. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1β. J. Hepatol. 2016, 64, 118–127. [Google Scholar] [CrossRef] [PubMed]
- He, C.G.; Piao, Y.J.; Hu, L.M. Association of receptor-mediated endocytosis and autophagy with apoptosis. Di Yi Jun Yi Da Xue Xue Bao 2003, 23, 1025–1027. [Google Scholar] [PubMed]
- Deutsch, M.; Graffeo, C.S.; Rokosh, R.; Pansari, M.; Ochi, A.; Levie, E.M.; Van Heerden, E.; Tippens, D.M.; Greco, S.; Barilla, R.; et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015, 6, e1759. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Hamesch, K.; Weiskirchen, R.; Tacke, F. The concanavalin A model of acute hepatitis in mice. Lab. Anim. 2015, 49 (Suppl. S1), 12–20. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Wang, W.; Hu, K.; Liu, W.; Pan, S.; Lv, Q.; Xu, G.; Yu, Q. EuHD1 protects against inflammatory injury driven by NLRP3 inflammasome. Int. Immunopharmacol. 2023, 115, 109712. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, X.K.; Cheng, Y.J.; Zhang, Q.; Wu, J.; Lu, S.; Zhang, W.; Liu, Y.; Zhou, M.Y.; Wang, Y.; et al. Gasdermin D-mediated hepatocyte pyroptosis expands inflammatory responses that aggravate acute liver failure by upregulating monocyte chemotactic protein 1/CC chemokine receptor-2 to recruit macrophages. World J. Gastroenterol. 2019, 25, 6527–6540. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Tacke, F.; Koch, A.; Trautwein, C. Liver—Guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zang, K.; Shang, F.; Guo, S.; Gao, L.; Zhang, X. HMGB1 mediates acute liver injury in sepsis through pyroptosis of liver macrophages. Int. J. Burns Trauma 2020, 10, 60–67. [Google Scholar] [PubMed]
- Chen, Q.; Zhang, Q.; Cao, P.; Shi, C.; Zhang, L.; Wang, L.; Gong, Z. NOD2-mediated HDAC6/NF-κb signalling pathway regulates ferroptosis induced by extracellular histone H3 in acute liver failure. J. Cell Mol. Med. 2022, 26, 5528–5538. [Google Scholar] [CrossRef] [PubMed]
- Caballería, L.; Pera, G.; Arteaga, I.; Rodríguez, L.; Alumà, A.; Morillas, R.M.; de la Ossa, N.; Díaz, A.; Expósito, C.; Miranda, D.; et al. High Prevalence of Liver Fibrosis Among European Adults With Unknown Liver Disease: A Population-Based Study. Clin. Gastroenterol. Hepatol. 2018, 16, 1138–1145.e5. [Google Scholar] [CrossRef] [PubMed]
- Lodder, J.; Denaës, T.; Chobert, M.N.; Wan, J.; El-Benna, J.; Pawlotsky, J.-M.; Lotersztajn, S.; Teixeira-Clerc, F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015, 11, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, G.; Wang, J.; Deng, M.; Yuan, F.; Gong, J. TIM-4 interference in Kupffer cells against CCL4-induced liver fibrosis by mediating Akt1/Mitophagy signalling pathway. Cell Prolif. 2020, 53, e12731. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Weiss, E.; Ben Mkaddem, S.; Mabire, M.; Choinier, P.-M.; Thibault-Sogorb, T.; Hegde, P.; Bens, M.; Broer, L.; Gilgenkrantz, H.; et al. LC3-associated phagocytosis in myeloid cells, a fireman that restrains inflammation and liver fibrosis, via immunoreceptor inhibitory signaling. Autophagy 2020, 16, 1526–1528. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Yamashina, S.; Arakawa, A.; Taniguchi, G.; Aoyama, T.; Uchiyama, A.; Kon, K.; Ikejima, K.; Watanabe, S. Formation of p62-positive inclusion body is associated with macrophage polarization in non-alcoholic fatty liver disease. Hepatol. Res. 2018, 48, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Xu, L.; Jing, Y.; Han, Z.; Chen, X.; Cai, C.; Zhao, P.; Zhao, X.; Yang, L.; Wei, L. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-κB-IL1α/β-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017, 388, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Ge, Y.J.; Wang, E.J.; Liao, Q.; Ren, Z.; Yu, Y.; Zhu, G.; Liu, C.; Zhang, M.; Su, H.; et al. Enhancement of efferocytosis through biased FPR2 signaling attenuates intestinal inflammation. EMBO Mol. Med. 2023, 15, e17815. [Google Scholar] [CrossRef] [PubMed]
- Secchi, M.F.; Crescenzi, M.; Masola, V.; Russo, F.P.; Floreani, A.; Onisto, M. Heparanase and macrophage interplay in the onset of liver fibrosis. Sci. Rep. 2017, 7, 14956. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, M.; Tomita, K.; Sugihara, N.; Nakashima, H.; Furuhashi, H.; Nishikawa, M.; Inaba, K.; Wada, A.; Horiuchi, K.; Hanawa, Y.; et al. Chitinase 3-like 1 deficiency ameliorates liver fibrosis by promoting hepatic macrophage apoptosis. Hepatol. Res. 2019, 49, 1316–1328. [Google Scholar] [CrossRef]
- Mera, K.; Uto, H.; Mawatari, S.; Ido, A.; Yoshimine, Y.; Nosaki, T.; Oda, K.; Tabu, K.; Kumagai, K.; Tamai, T.; et al. Serum levels of apoptosis inhibitor of macrophage are associated with hepatic fibrosis in patients with chronic hepatitis C. BMC Gastroenterol. 2014, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kropp, E.M.; Clavo, V.F.; Kobrossi, C.R.; Han, J.; Mauer, A.S.; Yong, J.; Kaufman, R.J. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J. Biol. Chem. 2013, 288, 18624–18642. [Google Scholar] [CrossRef] [PubMed]
- Airik, M.; McCourt, B.; Ozturk, T.T.; Huynh, A.B.; Zhang, X.; Tometich, J.T.; Topaloglu, R.; Ozen, H.; Orhan, D.; Monga, S.P.; et al. Mitigation of portal fibrosis and cholestatic liver disease in ANKS6-deficient livers by macrophage depletion. FASEB J. 2022, 36, e22157. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Thadathil, N.; Selvarani, R.; Nicklas, E.H.; Wang, D.; Miller, B.F.; Richardson, A.; Deepa, S.S. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 2021, 20, e13512. [Google Scholar] [CrossRef] [PubMed]
- Blériot, C.; Dupuis, T.; Jouvion, G.; Eberl, G.; Disson, O.; Lecuit, M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 2015, 42, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kong, X.; You, Y.; Xiang, L.; Zhang, Y.; Wu, R.; Zhou, L.; Duan, L. S100A8-Mediated NLRP3 Inflammasome-Dependent Pyroptosis in Macrophages Facilitates Liver Fibrosis Progression. Cells 2022, 11, 3579. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Zhang, W.; Huang, C.; Jian, J.; Zhang, Y.; Liu, Q.; Chen, P.; Zhu, X. Ursolic acid alleviates Kupffer cells pyroptosis in liver fibrosis by the NOX2/NLRP3 inflammasome signaling pathway. Int. Immunopharmacol. 2022, 113, 109321. [Google Scholar] [CrossRef] [PubMed]
- Herranz-Itúrbide, M.; Peñuelas-Haro, I.; Espinosa-Sotelo, R.; Bertran, E.; Fabregat, I. The TGF-β/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells 2021, 10, 2312. [Google Scholar] [CrossRef]
- Nauseef, W.M. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 2019, 60, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Boaru, S.G.; Borkham-Kamphorst, E.; Tihaa, L.; Haas, U.; Weiskirchen, R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J. Inflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Zheng, J.; Li, Z.; Yu, Y.; Chen, Z.; Wang, Q. ACSL4 inhibition prevents macrophage ferroptosis and alleviates fibrosis in bleomycin-induced systemic sclerosis model. Arthr. Res. Ther. 2023, 25, 212. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.L.; Caldas, C.; Santos, A.G.; Silva-Gomes, S.; Santos-Gonçalves, A.; Martins, M.J.; Porto, G.; Lopes, J.M. Genetic disruption of NRF2 promotes the development of necroinflammation and liver fibrosis in a mouse model of HFE-hereditary hemochromatosis. Redox Biol. 2017, 11, 157–169. [Google Scholar] [CrossRef]
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; D’Angelica, M.I.; Abbott, D.E.; Abrams, T.A.; Alberts, S.R.; Anaya, D.A.; Anders, R.; Are, C.; Brown, D.; Chang, D.T.; et al. Guidelines Insights: Hepatobiliary Cancers, Version 2.2019. J. Natl. Compr. Cancer Netw. 2019, 17, 302–310. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F.; Bsc, M.F.B.; Me, J.F.; Soerjomataram, M.I.; et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Han, J.; Wang, B.; Liu, W.; Wang, S.; Chen, R.; Chen, M.; Fu, Z. Declining disease burden of HCC in the United States, 1992–2017: A population-based analysis. Hepatology 2022, 76, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.Y.; Wang, N.; Man, K.; Tsao, S.W.; Che, C.M.; Feng, Y. Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 2015, 6, e1942. [Google Scholar] [CrossRef] [PubMed]
- Sugisawa, R.; Komatsu, G.; Hiramoto, E.; Takeda, N.; Yamamura, K.-I.; Arai, S.; Miyazaki, T. Independent modes of disease repair by AIM protein distinguished in AIM-felinized mice. Sci. Rep. 2018, 8, 13157. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Maehara, N.; Kai, T.; Arai, S.; Miyazaki, T. Dietary fructose-induced hepatocellular carcinoma development manifested in mice lacking apoptosis inhibitor of macrophage (AIM). Genes Cells 2016, 21, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Yamazaki, T.; Kanetsuki, Y.; Hirota, J.; Asai, T.; Mitsumoto, Y.; Mizuno, M.; Shima, T.; Kanbara, Y.; Arai, S.; et al. Activation of apoptosis inhibitor of macrophage is a sensitive diagnostic marker for NASH-associated hepatocellular carcinoma. J. Gastroenterol. 2018, 53, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 Orchestrates Fatty Acid Metabolism in Tumor-Associated Macrophages and Hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Steffani, M.; Steffani, M.; Stöß, C.; Ankerst, D.; Friess, H.; Hüser, N.; Hartmann, D. Novel Risk Classification Based on Pyroptosis-Related Genes Defines Immune Microenvironment and Pharmaceutical Landscape for Hepatocellular Carcinoma. Cancers 2022, 14, 447. [Google Scholar] [CrossRef] [PubMed]
- Hage, C.; Hoves, S.; Strauss, L.; Bissinger, S.; Prinz, Y.; Pöschinger, T.; Kiessling, F.; Ries, C.H. Sorafenib Induces Pyroptosis in Macrophages and Triggers Natural Killer Cell–Mediated Cytotoxicity Against Hepatocellular Carcinoma. Hepatology 2019, 70, 1280–1297. [Google Scholar] [CrossRef]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.-W.; You, Y.; Hsu, J.-M.; Nie, L.; Chen, Y.; Wang, Y.-C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef]
- Tang, B.; Zhu, J.; Wang, Y.; Chen, W.; Fang, S.; Mao, W.; Xu, Z.; Yang, Y.; Weng, Q.; Zhao, Z.; et al. Targeted xCT-mediated Ferroptosis and Protumoral Polarization of Macrophages Is Effective against HCC and Enhances the Efficacy of the Anti-PD-1/L1 Response. Adv. Sci. 2022, 10, e2203973. [Google Scholar] [CrossRef]
- Hao, X.; Zheng, Z.; Liu, H.; Zhang, Y.; Kang, J.; Kong, X.; Rong, D.; Sun, G.; Sun, G.; Liu, L.; et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022, 56, 102463. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yin, Y.; Jiang, J.; Yan, C.; Wang, Y.; Wang, D.; Li, L. Exosomal miR-142-3p secreted by hepatitis B virus (HBV)-hepatocellular carcinoma (HCC) cells promotes ferroptosis of M1-type macrophages through SLC3A2 and the mechanism of HCC progression. J. Gastrointest. Oncol. 2022, 13, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.C.; Hvidbjerg Gantzel, R.; Clària, J.; Trebicka, J.; Møller, H.J.; Grønbæk, H. Macrophage Activation Markers, CD163 and CD206, in Acute-on-Chronic Liver Failure. Cells 2020, 9, 1175. [Google Scholar] [CrossRef] [PubMed]
- Stengel, S.; Quickert, S.; Lutz, P.; Ibidapo-Obe, O.; Steube, A.; Köse-Vogel, N.; Yarbakht, M.; Reuken, P.A.; Busch, M.; Brandt, A.; et al. Peritoneal Level of CD206 Associates With Mortality and an Inflammatory Macrophage Phenotype in Patients With Decompensated Cirrhosis and Spontaneous Bacterial Peritonitis. Gastroenterology 2020, 158, 1745–1761. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, L.; Xiang, Z.; Ren, Y.; Zheng, X.; Zhao, Q.; Zhou, Q.; Zhou, Y.; Xu, L.; Wang, Y. Microcystin-LR ameliorates pulmonary fibrosis via modulating CD206(+) M2-like macrophage polarization. Cell Death Dis. 2020, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Barrera, F.; Møller, H.J.; Rosso, C.; Bugianesi, E.; David, E.; Jouness, R.I.K.; Esmaili, S.; Eslam, M.; McLeod, D.; et al. The macrophage activation marker sCD163 is associated with morphological disease stages in patients with non-alcoholic fatty liver disease. Liver Int. 2016, 36, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Barrera, F.; Møller, H.J.; Bibby, B.M.; Vilstrup, H.; George, J.; Grønbaek, H. Soluble CD163, a macrophage activation marker, is independently associated with fibrosis in patients with chronic viral hepatitis B and C. Hepatology 2014, 60, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Grønbæk, H.; Rødgaard-Hansen, S.; Aagaard, N.K.; Arroyo, V.; Moestrup, S.K.; Garcia, E.; Solà, E.; Domenicali, M.; Piano, S.; Vilstrup, H.; et al. Macrophage activation markers predict mortality in patients with liver cirrhosis without or with acute-on-chronic liver failure (ACLF). J. Hepatol. 2016, 64, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Grønbaek, H.; Kreutzfeldt, M.; Kazankov, K.; Jessen, N.; Sandahl, T.; Hamilton-Dutoit, S.; Vilstrup, H.; Møller, H.J. Single-centre experience of the macrophage activation marker soluble (s)CD163—Associations with disease activity and treatment response in patients with autoimmune hepatitis. Aliment. Pharmacol. Ther. 2016, 44, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Sandahl, T.D.; Støy, S.H.; Laursen, T.L.; Rødgaard-Hansen, S.; Møller, H.J.; Møller, S.; Vilstrup, H.; Grønbæk, H. The soluble mannose receptor (sMR) is elevated in alcoholic liver disease and associated with disease severity, portal hypertension, and mortality in cirrhosis patients. PLoS ONE 2017, 12, e0189345. [Google Scholar] [CrossRef]
- Rainer, F.; Horvath, A.; Sandahl, T.D.; Leber, B.; Schmerboeck, B.; Blesl, A.; Groselj-Strele, A.; Stauber, R.E.; Fickert, P.; Stiegler, P.; et al. Soluble CD163 and soluble mannose receptor predict survival and decompensation in patients with liver cirrhosis, and correlate with gut permeability and bacterial translocation. Aliment. Pharmacol. Ther. 2018, 47, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Waidmann, O.; Brunner, F.; Herrmann, E.; Zeuzem, S.; Piiper, A.; Kronenberger, B. Macrophage activation is a prognostic parameter for variceal bleeding and overall survival in patients with liver cirrhosis. J. Hepatol. 2013, 58, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Rode, A.; Nicoll, A.; Møller, H.J.; Lim, L.; Angus, P.W.; Kronborg, I.; Arachchi, N.; Gorelik, A.; Liew, D.; Kazankov, K.; et al. Hepatic macrophage activation predicts clinical decompensation in chronic liver disease. Gut 2013, 62, 1231–1232. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Tornai, D.; Kodys, K.; Adejumo, A.; Lowe, P.; McClain, C.; Mitchell, M.; McCullough, A.; Dasarathy, S.; Kroll-Desrosiers, A.; et al. Biomarkers of Macrophage Activation and Immune Danger Signals Predict Clinical Outcomes in Alcoholic Hepatitis. Hepatology 2019, 70, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Holland-Fischer, P.; Grønbæk, H.; Sandahl, T.D.; Moestrup, S.K.; Riggio, O.; Ridola, L.; Aagaard, N.K.; Møller, H.J.; Vilstrup, H. Kupffer cells are activated in cirrhotic portal hypertension and not normalised by TIPS. Gut 2011, 60, 1389–1393. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kurniawan, D.W.; Jajoriya, A.K.; Dhawan, G.; Mishra, D.; Argemi, J.; Bataller, R.; Storm, G.; Mishra, D.P.; Prakash, J.; Bansal, R. Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis. J. Control. Release 2018, 288, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Postigo-Fernandez, J.; Kim, K.; Zhu, C.; Yu, J.; Meroni, M.; Mayfield, B.; Bartolomé, A.; Dapito, D.H.; Ferrante, A.W.; et al. Notch-mediated hepatocyte MCP-1 secretion causes liver fibrosis. JCI Insight 2023, 8, e165369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Zhan, D.L.; Chen, Y.Y.; Wang, W.-H.; He, C.-Y.; Lin, Y.; Lin, Y.-C.; Lin, Z.-N. Aflatoxin B1 enhances pyroptosis of hepatocytes and activation of Kupffer cells to promote liver inflammatory injury via dephosphorylation of cyclooxygenase-2: An in vitro, ex vivo and in vivo study. Arch. Toxicol. 2019, 93, 3305–3320. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Q.; Qin, L.; Zhao, S.; Wang, J.; Chen, X. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol. 2011, 12, 43. [Google Scholar] [CrossRef]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Pimienta, M.; Seki, E. Alcoholic liver disease: A current molecular and clinical perspective. Liver Res. 2018, 2, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.; Gao, B. Therapeutic potential of interleukin 1 inhibitors in the treatment of alcoholic liver disease. Hepatology 2013, 57, 2078–2080. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Marri, S.R.; Chalasani, N.; Kohli, R.; Aronstein, W.; Thompson, G.A.; Irish, W.; Miles, M.V.; Xanthakos, S.A.; Lawitz, E.; et al. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment. Pharmacol. Ther. 2016, 44, 1183–1198. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Kuang, W.; Jia, H.; Lu, Y.; Zhang, X.; Ye, C.; Gu, J.; Lv, Y.; Yu, J.; Zhang, Y.; et al. Pegylated interferon-α-2b combined with tenofovir disoproxil fumarate, granulocyte-macrophage colony-stimulating factor, and hepatitis B vaccine treatment for naïve HBeAg-positive chronic hepatitis B patients: A prospective, multicenter, randomized controlled study. J. Med. Virol. 2022, 94, 5475–5483. [Google Scholar] [PubMed]
- Wu, C.; Cao, X.; Zhang, X. VISTA inhibitors in cancer immunotherapy: A short perspective on recent progresses. RSC Med. Chem. 2021, 12, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
| Cell Death Type | Death Pathway/Mechanism | Key Factor | Feature | Inducer |
|---|---|---|---|---|
| Autophagy | 1. mTOR pathway 2. MAPK pathway 3. Beclin 1 pathway 4. PI3K/Akt pathway 5. ROS pathway 6. NF-κB pathway | LC3 Beclin-1 | Autophagosome formation | 1. Hungry 2. Oxygen deficit 3. Bacterial or viral infections |
| Apoptosis | 1. Extrinsic pathway induced by TNF or TRAIL 2. Intrinsic (mitochondrial) pathway mediated by pro-apoptotic Bcl-2 family proteins 3. Endoplasmic reticulum stress-induced pathway | Caspase3 Caspase8 DISC Caspase 9 MOMP | 1. Cell shrinkage, but cell membrane integrity 2. Chromatin condensation 3. DNA fragmentation 4. Apoptotic body formation | 1. Bacterial infections 2. Hypoxia 3. Chemicals |
| Necroptosis | 1. TNFR1-RIPK1 pathway 2. RIPK3-MLKL pathway | RIPK1 RIPK3 MLKL | 1. Cell membrane rupture 2. Cell content extravasation 3. Inflammatory response | 1. Bacterial toxin 2. Oxidative stress 3. Cell trauma 4. Pathogen 5. Nutritional deficiency 6. ATP depletion 7. Mitochondrial permeability transition |
| Pyroptosis | 1. Caspase1-dependent pathway 2. Caspase11/4/5-independent pathway | GSDMD | 1. Cell swelling 2. Cell membrane rupture 3. Release of IL-1β 4. ASC-mediated inflammasome Formation 5. Create pores in the cell membrane | 1. Cytosolic dsDNA 2. Anthrax lethal toxin 3. Membrane damage 4. Toxoplasma 5. Chemotherapy drugs |
| Ferroptosis | Excess Fe3+ in cell is reduced to Fe2+ to form hydroxyl radicals, leading to accumulation of lipid peroxides, resulting in increased ROS | GPX4 | 1. Smaller outer membrane of mitochondria ruptures 2. MPK mediated Beclin-1 phosphorylation | 1. GSH depletion 2. Inactivation of GPX4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Z.; Xiong, W.; Mao, X.; Li, J. Macrophage Perspectives in Liver Diseases: Programmed Death, Related Biomarkers, and Targeted Therapy. Biomolecules 2024, 14, 700. https://doi.org/10.3390/biom14060700
Qian Z, Xiong W, Mao X, Li J. Macrophage Perspectives in Liver Diseases: Programmed Death, Related Biomarkers, and Targeted Therapy. Biomolecules. 2024; 14(6):700. https://doi.org/10.3390/biom14060700
Chicago/Turabian StyleQian, Zibing, Wanyuan Xiong, Xiaorong Mao, and Junfeng Li. 2024. "Macrophage Perspectives in Liver Diseases: Programmed Death, Related Biomarkers, and Targeted Therapy" Biomolecules 14, no. 6: 700. https://doi.org/10.3390/biom14060700
APA StyleQian, Z., Xiong, W., Mao, X., & Li, J. (2024). Macrophage Perspectives in Liver Diseases: Programmed Death, Related Biomarkers, and Targeted Therapy. Biomolecules, 14(6), 700. https://doi.org/10.3390/biom14060700