

The Double-Edged Effects of MLN4924: Rethinking Anti-Cancer Drugs Targeting the Neddylation Pathway

Abstract
:1. Introduction
2. Neddylation Pathway and Cancer
2.1. Neddylation Substrates

{kind=link}
{kind=link}
{kind=link}
| Neddylation Substrates | Involvement in Biological Processes | Neddylation Effects | References |
|---|---|---|---|
| CRLs | Degrading anti-cancer factors | Promoting its activity | [1] |
| NIK | Inducing a non-canonical NF-κB pathway and accentuating inflammation | Promoting its ubiquitination and inhibiting its aberrant activation | [6] |
| PTEN | Inhibiting cancers via the PIK/FAK/MAPK signaling pathway | Reversing its function as a tumor suppressor | [7] |
| HER2 | Enhancing breast cancer cell proliferation, survival, migration, and polarity changes | Promoting its accumulation | [8] |
| IRF7 | Promoting host antiviral innate immunity against viruses | Promoting its nuclear translocation and preventing its dimerization with IRF5 | [11] |
| LANA | Repressing ORF50p and the onset of KSHV lytic reactivation in primary effusion lymphoma | Promoting its activity | [25] |
| p53 | Preventing the multiplication of damaged and potentially pre-cancerous cells | Inhibiting its transcriptional activity and function | [27] |
| SPH2 | Binding to the ITIM of SIRPα and promoting macrophage phagocytosis of cancer cells | Inhibiting its linkage with ITIM | [30,31] |
| β-catenin | Playing a crucial role in the Wnt signaling pathway and promoting cancer cell migration and adhesion | Promoting its fast degradation | [32] |
| Coro1a | Promoting the recruitment of Rab7 to multivesicular bodies to reduce extracellular vesicle secretion | Promoting its activity | [33] |
| mGlu7 | Modulating the maturation of excitatory presynaptic terminals | Promoting its ubiquitination and stabilizing its expression | [34] |
| TAK1 | Mediating the signaling transduction induced by TGF beta and morphogenetic protein | Promoting its nuclear import | [35] |
| Gadd45a | Responding to environmental stresses by activating the p38/JNK pathway via MTK1/MEKK4 kinase | Promoting its nuclear export | [35] |
| Cofilin | Influencing neuron growth and cell migration during brain development | Its site-specific neddylation modulates cytoskeletal actin dynamics and neuron development | [36] |
| c-Src | Promoting cancer progression by activating the PI3K-AKT pathway | Promoting its polyubiquitination and degradation | [37] |
| PCNA | Assisting DNA polymerase in mediating DNA replication | Antagonizing its ubiquitination and inhibiting its interaction with polη | [38] |
| MyD88 | Playing a central role in pro-cancer inflammation | Antagonizing its ubiquitination, reducing its dimerization, and suppressing NF-κB activity | [39] |
| MKK7 | Impeding breast cancer proliferation and EMT phenotype via JNK phosphorylation | Inhibiting its basal kinase activity | [40] |
| CXCR5 | Stimulating cell migration and motility | Targeting its location to the cell membrane | [41] |
| E2F-1 | Mediating the G1-to-S-phase transition | Downregulating its stability and transcriptional activity | [42] |
2.2. The Neddylation Pathways in Cancer Cells
2.3. The Neddylation Pathways in TME
2.4. Neddylation Pathways and Tumor Viruses
3. Anti-Cancer Effects of MLN4924
3.1. MLN4924 Inhibits Cancer Cell Proliferation by Accumulating Cancer Suppressors
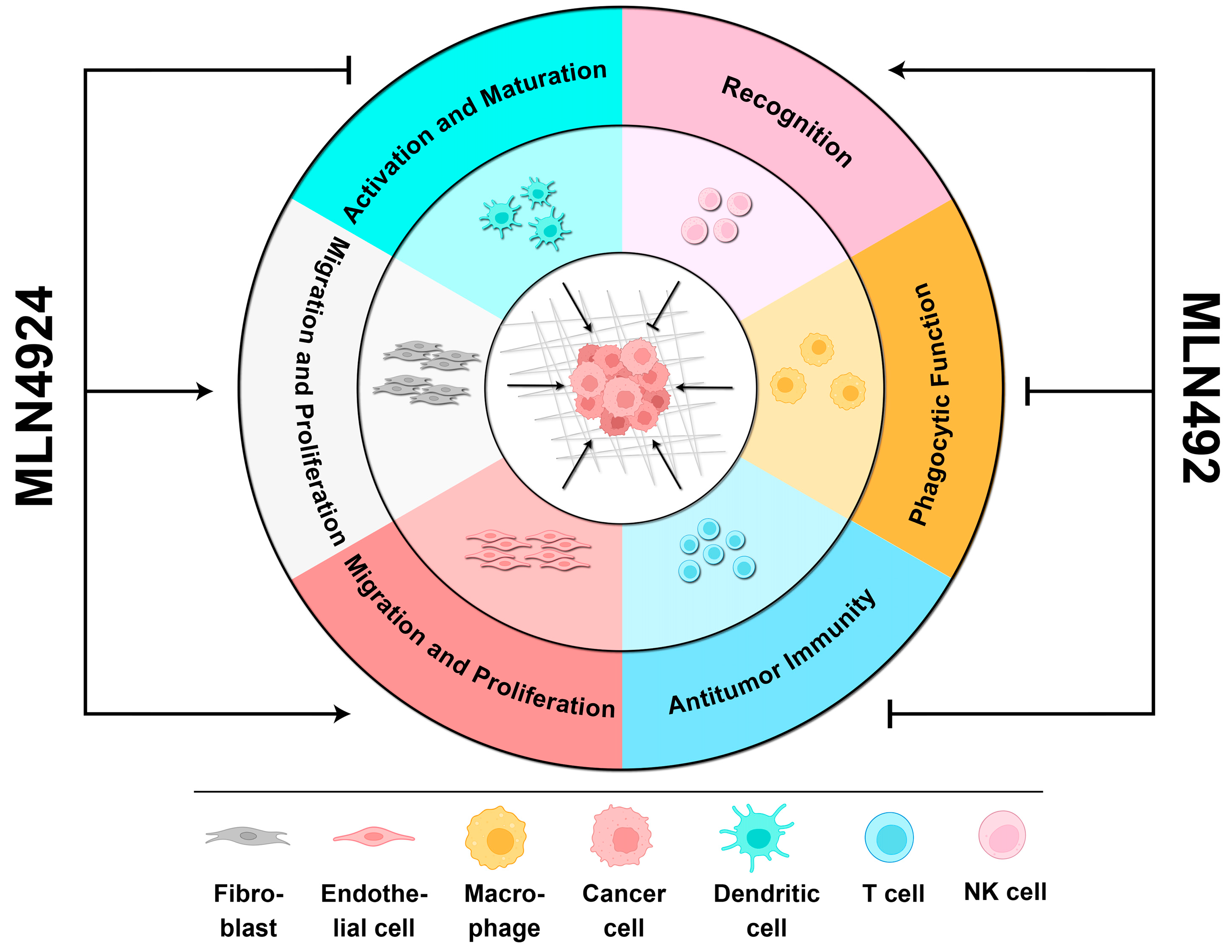
3.2. MLN4924 Represses the Reprogramming of the TME Mediated by Cancer Cells
3.3. MLN4924 Delays the Aggravation of Diseases Mediated by Tumor Viruses
3.4. MLN4924 Resensitizes Cancer Cells to Anti-Cancer Treatments
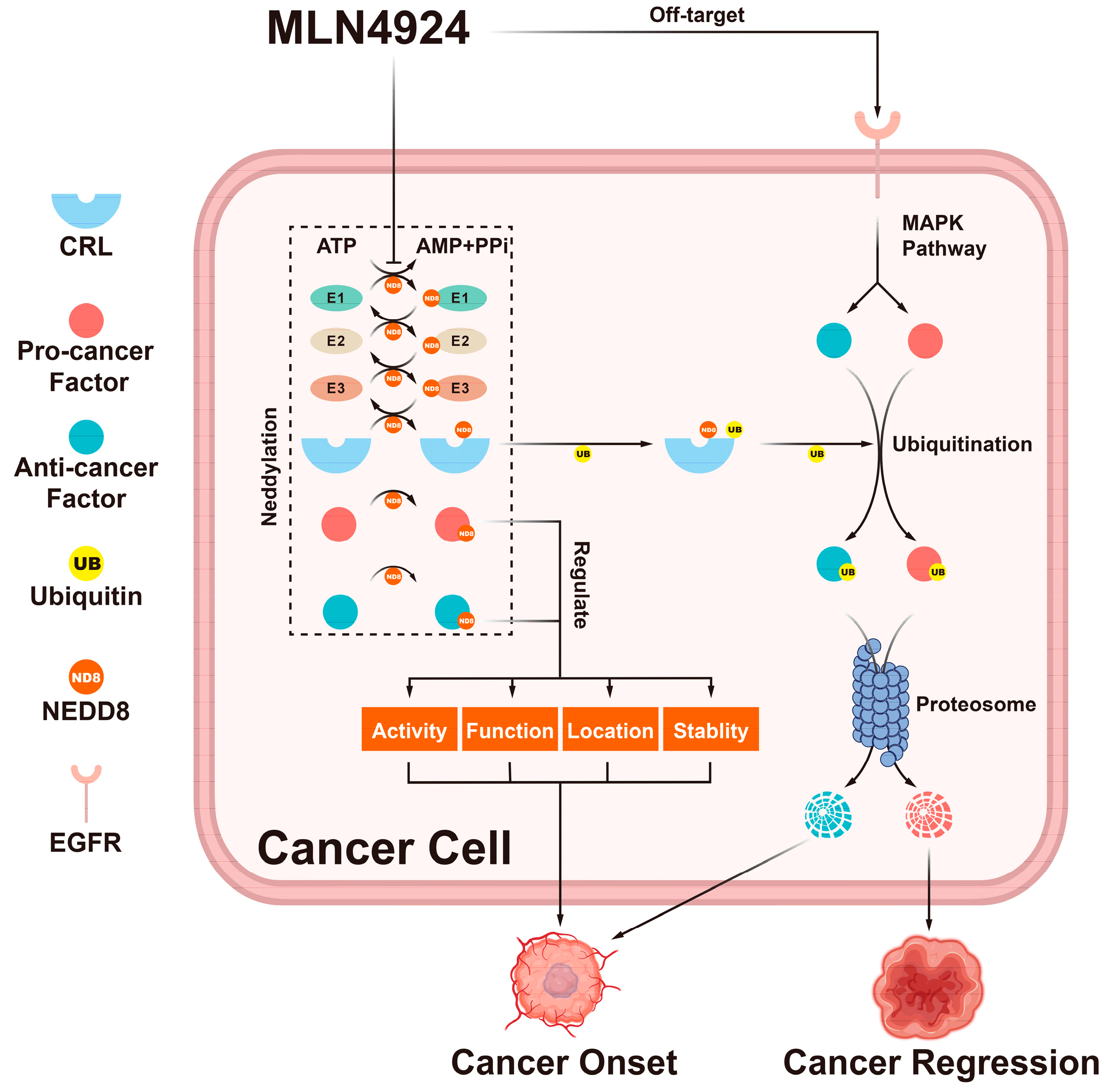
4. Pro-Cancer Side Effects of MLN4924
4.1. MLN4924 Enhances Cancer Onset by Stabilizing CRL Pro-Cancer Factors
4.1.1. c-Myc
4.1.2. Programmed Death-Ligand (PD-L1)
4.1.3. Alanine–Serine–Cysteine Transporter 2 (ASCT2)
4.1.4. Early Growth Response 1 (EGR1)
4.1.5. The Nuclear Factor-Erythroid 2 p45-Related Factor 2 (NRF2)
4.1.6. NF-κB-Inducing Kinase (NIK)
4.1.7. Hypoxia-Inducible Factors-1α (HIF-1α)
4.2. MLN4924 Supports the Formation of a Tumor-Comfortable Microenvironment
4.3. Rescue of MLN4924 Pro-Cancer Side Effects
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, Q.; Jiang, Y.; Sun, Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm. Sin. B 2020, 10, 746–765. [Google Scholar] [CrossRef] [PubMed]
- Kostrhon, S.; Prabu, J.R.; Baek, K.; Horn-Ghetko, D.; von Gronau, S.; Klügel, M.; Basquin, J.; Alpi, A.F.; Schulman, B.A. CUL5-ARIH2 E3-E3 ubiquitin ligase structure reveals cullin-specific NEDD8 activation. Nat. Chem. Biol. 2021, 17, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell. Signal. 2018, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Olaizola, P.; Lee-Law, P.Y.; Fernandez-Barrena, M.G.; Alvarez, L.; Cadamuro, M.; Azkargorta, M.; O’Rourke, C.J.; Caballero-Camino, F.J.; Olaizola, I.; Macias, R.I.R.; et al. Targeting NAE1-mediated protein hyper-NEDDylation halts cholangiocarcinogenesis and impacts on tumor-stroma crosstalk in experimental models. J. Hepatol. 2022, 77, 177–190. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, Q.; Li, Z.; Zhao, Y.; Sun, Y. Protein neddylation and its role in health and diseases. Signal Transduct. Target. Ther. 2024, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhou, H.; Jin, Y.; Sahay, K.; Robicsek, A.; Liu, Y.; Dong, K.; Zhou, J.; Barrett, A.; Su, H.; et al. Hepatic neddylation deficiency triggers fatal liver injury via inducing NF-κB-inducing kinase in mice. Nat. Commun. 2022, 13, 7782. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Peng, Z.; Chen, Y.; Li, H.; Du, M.; Tan, Y.; Zhang, X.; Lu, Z.; Cui, C.P.; Liu, C.H.; et al. Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res. 2021, 31, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Hu, T.; He, X.; Liu, Y.; Yu, C.; Kong, W.; Liao, Y.; Tang, D.; Liu, J.; Huang, H. Neddylation of HER2 Inhibits its Protein Degradation and promotes Breast Cancer Progression. Int. J. Biol. Sci. 2023, 19, 377–392. [Google Scholar] [CrossRef]
- Zhang, W.; Liang, Y.; Li, L.; Wang, X.; Yan, Z.; Dong, C.; Zeng, M.S.; Zhong, Q.; Liu, X.K.; Yu, J.; et al. The Nedd8-activating enzyme inhibitor MLN4924 (TAK-924/Pevonedistat) induces apoptosis via c-Myc-Noxa axis in head and neck squamous cell carcinoma. Cell Prolif. 2019, 52, e12536. [Google Scholar] [CrossRef]
- McGrail, D.J.; Garnett, J.; Yin, J.; Dai, H.; Shih, D.J.H.; Lam, T.N.A.; Li, Y.; Sun, C.; Li, Y.; Schmandt, R.; et al. Proteome Instability Is a Therapeutic Vulnerability in Mismatch Repair-Deficient Cancer. Cancer Cell 2020, 37, 371–386.e12. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, Y.; Yang, X.; Jin, J.; Shen, Z.; Feng, X.; Zou, T.; Deng, L.; Cheng, D.; Zhang, X.; et al. Myeloid neddylation targets IRF7 and promotes host innate immunity against RNA viruses. PLoS Pathog. 2021, 17, e1009901. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.J.; Elia, A.E.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.N.; Rush, J.; Hsu, P.W.; et al. Global identification of modular cullin-RING ligase substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Guo, H.; Lou, G.; Yao, J.; Liu, Y.; Sun, Y.; Yang, Z.; Zheng, M. Neddylation inhibitor MLN4924 has anti-HBV activity via modulating the ERK-HNF1α-C/EBPα-HNF4α axis. J. Cell Mol. Med. 2021, 25, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Zhang, J. Roles of neddylation against viral infections. Cell. Mol. Immunol. 2018, 15, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Wang, X.; Chen, C.; Vuong, D.; Rodriguez-Rodriguez, S.; Lam, V.; Roleder, C.; Wang, J.H.; Kambhampati, S.; Berger, A.; Pennock, N.; et al. Pharmacologic targeting of Nedd8-activating enzyme reinvigorates T-cell responses in lymphoid neoplasia. Leukemia 2023, 37, 1324–1335. [Google Scholar] [CrossRef]
- Mao, W.; Zhang, L.; Rong, Y.; Kuang, T.; Wang, D.; Xu, X.; Lou, W.; Li, J. NEDD8-Activating Enzyme Inhibitor MLN4924 Inhibits Both the Tumor Stroma and Angiogenesis in Pancreatic Cancer via Gli1 and REDD1. Dig. Dis. Sci. 2023, 68, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kuo, K.L.; Lin, W.C.; Chen, M.S.; Liu, S.H.; Liao, S.M.; Hsu, C.H.; Chang, Y.W.; Chang, H.C.; Huang, K.H. Neddylation inhibitor, MLN4924 suppresses angiogenesis in huvecs and solid cancers: In vitro and in vivo study. Am. J. Cancer Res. 2020, 10, 953–964. [Google Scholar]
- Qu, B.; Nebioglu, F.; Leuthold, M.M.; Ni, Y.; Mutz, P.; Beneke, J.; Erfle, H.; Vondran, F.W.R.; Bartenschlager, R.; Urban, S. Dual role of neddylation in transcription of hepatitis B virus RNAs from cccDNA and production of viral surface antigen. JHEP Rep. 2022, 4, 100551. [Google Scholar] [CrossRef]
- Zhou, Q.; Lin, W.; Wang, C.; Sun, F.; Ju, S.; Chen, Q.; Wang, Y.; Chen, Y.; Li, H.; Wang, L.; et al. Neddylation inhibition induces glutamine uptake and metabolism by targeting CRL3SPOP E3 ligase in cancer cells. Nat. Commun. 2022, 13, 3034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Tan, M.; Nyati, M.K.; Zhao, Y.; Wang, G.; Sun, Y. Blockage of neddylation modification stimulates tumor sphere formation in vitro and stem cell differentiation and wound healing in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, E2935–E2944. [Google Scholar] [CrossRef]
- Zhang, S.; You, X.; Xu, T.; Chen, Q.; Li, H.; Dou, L.; Sun, Y.; Xiong, X.; Meredith, M.A.; Sun, Y. PD-L1 induction via the MEK-JNK-AP1 axis by a neddylation inhibitor promotes cancer-associated immunosuppression. Cell Death Dis. 2022, 13, 844. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhao, X.; Yang, Z.; Yang, R.; Chen, C.; Zhao, K.; Wang, W.; Ma, Y.; Zhang, Q.; Wang, X. Neddylation inhibition upregulates PD-L1 expression and enhances the efficacy of immune checkpoint blockade in glioblastoma. Int. J. Cancer 2019, 145, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Chen, L.W.; Chen, L.Y.; Hung, C.H.; Shih, Y.J.; Wang, S.S. Effects of the NEDD8-Activating Enzyme Inhibitor MLN4924 on Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2017, 91, e00505-17. [Google Scholar] [CrossRef] [PubMed]
- El-Mesery, M.; Rosenthal, T.; Rauert-Wunderlich, H.; Schreder, M.; Stühmer, T.; Leich, E.; Schlosser, A.; Ehrenschwender, M.; Wajant, H.; Siegmund, D. The NEDD8-activating enzyme inhibitor MLN4924 sensitizes a TNFR1+ subgroup of multiple myeloma cells for TNF-induced cell death. Cell Death Dis. 2019, 10, 611. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W. FBXO11 promotes the Neddylation of p53 and inhibits its transcriptional activity. J. Biol. Chem. 2007, 282, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, J.; Sun, Y. Early growth response-1 is a new substrate of the GSK3β-FBXW7 axis. Neoplasia 2022, 34, 100839. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xiong, X.; Jia, L.; Sun, Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012, 3, e386. [Google Scholar] [CrossRef]
- Huntoon, K.; Jiang, W.; Kim, B.Y. Waking immune-resistant tumors with neddylation. J. Clin. Investig. 2023, 133, e167894. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, H.; Liu, P.; Lv, D.; Shi, Y.; Tang, B.; Xu, J.; Zhong, T.; Xu, W.; Zhang, J.; et al. SHP2 deneddylation mediates tumor immunosuppression in colon cancer via the CD47/SIRPα axis. J. Clin. Investig. 2023, 133, e162870. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, T.; Zhu, H.; Yan, R.; Li, X.; Zhang, C.; Tao, W.; Ke, X.; Hao, P.; Qu, Y. Neddylation is essential for β-catenin degradation in Wnt signaling pathway. Cell Rep. 2022, 38, 110538. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Li, Z.; Yang, D.; Kong, X.; Lu, X.; Shen, Y.; Li, X.; Xie, S.; Wang, J.; Zhao, Y.; et al. Neddylation of Coro1a determines the fate of multivesicular bodies and biogenesis of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12153. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Lee, D.; Song, J.M.; Park, S.; Park, D.H.; Lee, S.; Suh, Y.H. Neddylation is required for presynaptic clustering of mGlu7 and maturation of presynaptic terminals. Exp. Mol. Med. 2021, 53, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fang, W.; Cui, Y.; Shi, H.; Chen, J.; Li, L.; Zhang, L.; Zhang, X. Neddylation promotes protein translocation between the cytoplasm and nucleus. Biochem. Biophys. Res. Commun. 2020, 529, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Vogl, A.M.; Phu, L.; Becerra, R.; Giusti, S.A.; Verschueren, E.; Hinkle, T.B.; Bordenave, M.D.; Adrian, M.; Heidersbach, A.; Yankilevich, P.; et al. Global site-specific neddylation profiling reveals that NEDDylated cofilin regulates actin dynamics. Nat. Struct. Mol. Biol. 2020, 27, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.W.; Park, J.B.; Park, S.Y.; Seo, J.; Shin, S.H.; Park, J.W.; Kim, S.J.; Watanabe, M.; Chun, Y.S. The E3 ligase C-CBL inhibits cancer cell migration by neddylating the proto-oncogene c-Src. Oncogene 2018, 37, 5552–5568. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Yu, S.; Zheng, X. NEDDylation antagonizes ubiquitination of proliferating cell nuclear antigen and regulates the recruitment of polymerase η in response to oxidative DNA damage. Protein Cell 2018, 9, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Guan, J.; Peng, Y.; Zheng, X. MyD88 NEDDylation negatively regulates MyD88-dependent NF-κB signaling through antagonizing its ubiquitination. Biochem. Biophys. Res. Commun. 2017, 482, 632–637. [Google Scholar] [CrossRef]
- Zhu, T.; Wang, J.; Pei, Y.; Wang, Q.; Wu, Y.; Qiu, G.; Zhang, D.; Lv, M.; Li, W.; Zhang, J. Neddylation controls basal MKK7 kinase activity in breast cancer cells. Oncogene 2016, 35, 2624–2633. [Google Scholar] [CrossRef]
- Renaudin, X.; Guervilly, J.H.; Aoufouchi, S.; Rosselli, F. Proteomic analysis reveals a FANCA-modulated neddylation pathway involved in CXCR5 membrane targeting and cell mobility. J. Cell Sci. 2014, 127 Pt 16, 3546–3554. [Google Scholar] [PubMed]
- Loftus, S.J.; Liu, G.; Carr, S.M.; Munro, S.; La Thangue, N.B. NEDDylation regulates E2F-1-dependent transcription. EMBO Rep. 2012, 13, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Petillo, S.; Capuano, C.; Molfetta, R.; Fionda, C.; Mekhloufi, A.; Pighi, C.; Antonangeli, F.; Zingoni, A.; Soriani, A.; Petrucci, M.T.; et al. Immunomodulatory effect of NEDD8-activating enzyme inhibition in Multiple Myeloma: Upregulation of NKG2D ligands and sensitization to Natural Killer cell recognition. Cell Death Dis. 2021, 12, 836. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.M.; Espitia, C.M.; Ooi, A.; Bauman, J.E.; Carew, J.S.; Nawrocki, S.T. Targeted CUL4A inhibition synergizes with cisplatin to yield long-term survival in models of head and neck squamous cell carcinoma through a DDB2-mediated mechanism. Cell Death Dis. 2022, 13, 350. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lin, X.; Zhang, L.; Chen, S.; Chen, J.; Zhou, Z.; Tang, A.; Ruan, J.; Wang, X.; Chen, B. Neddylation pathway promotes myeloid-derived suppressor cell infiltration via NF-κB-mCXCL5 signaling in lung cancer. Int. Immunopharmacol. 2022, 113 Pt A, 109329. [Google Scholar]
- Wei, D.; Li, H.; Yu, J.; Sebolt, J.T.; Zhao, L.; Lawrence, T.S.; Smith, P.G.; Morgan, M.A.; Sun, Y. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012, 72, 282–293. [Google Scholar] [PubMed]
- Pflug, K.M.; Sitcheran, R. Targeting NF-κB-Inducing Kinase (NIK) in Immunity, Inflammation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8470. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Edwards, J.; Pepper, C.; Mackay, S. Inhibitory-κB Kinase (IKK) α and Nuclear Factor-κB (NFκB)-Inducing Kinase (NIK) as Anti-Cancer Drug Targets. Cells 2018, 7, 176. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Haecker, H.; Karin, M.; Ciechanover, A. Mechanism of processing of the NF-kappa B2 p100 precursor: Identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene 2004, 23, 2540–2547. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, Y.; Liu, X.; Li, L.; Yang, X.; Dong, C.; Liu, X.; Lin, Y.; Li, Y.; Yu, J.; et al. Promotion of tumor-associated macrophages infiltration by elevated neddylation pathway via NF-κB-CCL2 signaling in lung cancer. Oncogene 2019, 38, 5792–5804. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Best, S.; Lam, V.; Liu, T.; Bruss, N.; Kittai, A.; Danilova, O.V.; Murray, S.; Berger, A.; Pennock, N.D.; Lind, E.F.; et al. Immunomodulatory effects of pevonedistat, a NEDD8-activating enzyme inhibitor, in chronic lymphocytic leukemia-derived T cells. Leukemia 2021, 35, 156–168. [Google Scholar] [CrossRef] [PubMed]
- da Silva, S.R.; de Oliveira, D.E. HIV, EBV and KSHV: Viral cooperation in the pathogenesis of human malignancies. Cancer Lett. 2011, 305, 175–185. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Berman, T.A.; Schiller, J.T. Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer 2017, 123, 2219–2229. [Google Scholar] [CrossRef]
- Kraus, R.J.; Cordes, B.A.; Sathiamoorthi, S.; Patel, P.; Yuan, X.; Iempridee, T.; Yu, X.; Lee, D.L.; Lambert, P.F.; Mertz, J.E. Reactivation of Epstein-Barr Virus by HIF-1α Requires p53. J. Virol. 2020, 94, e00722-20. [Google Scholar] [CrossRef] [PubMed]
- Nekorchuk, M.D.; Sharifi, H.J.; Furuya, A.K.; Jellinger, R.; de Noronha, C.M. HIV relies on neddylation for ubiquitin ligase-mediated functions. Retrovirology 2013, 10, 138. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Benamar, M.; Guessous, F.; Du, K.; Corbett, P.; Obeid, J.; Gioeli, D.; Slingluff, C.L., Jr.; Abbas, T. Inactivation of the CRL4-CDT2-SET8/p21 ubiquitylation and degradation axis underlies the therapeutic efficacy of pevonedistat in melanoma. EBioMedicine 2016, 10, 85–100. [Google Scholar] [CrossRef]
- Luo, Z.; Pan, Y.; Jeong, L.S.; Liu, J.; Jia, L. Inactivation of the Cullin (CUL)-RING E3 ligase by the NEDD8-activating enzyme inhibitor MLN4924 triggers protective autophagy in cancer cells. Autophagy 2012, 8, 1677–1679. [Google Scholar] [CrossRef]
- Luo, Z.; Yu, G.; Lee, H.W.; Li, L.; Wang, L.; Yang, D.; Pan, Y.; Ding, C.; Qian, J.; Wu, L.; et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012, 72, 3360–3371. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, P.; Wang, Y.; Jin, B.; Zhou, J.; Zhang, J.; Pan, J. Neddylation Blockade Diminishes Hepatic Metastasis by Dampening Cancer Stem-like Cells and Angiogenesis in Uveal Melanoma. Clin. Cancer Res. 2018, 24, 3741–3754. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Kelly, K.R.; Smith, P.G.; Espitia, C.M.; Possemato, A.; Beausoleil, S.A.; Milhollen, M.; Blakemore, S.; Thomas, M.; Berger, A.; et al. Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin. Cancer Res. 2013, 19, 3577–3590. [Google Scholar] [CrossRef] [PubMed]
- Garcia, K.; Blank, J.L.; Bouck, D.C.; Liu, X.J.; Sappal, D.S.; Hather, G.; Cosmopoulos, K.; Thomas, M.P.; Kuranda, M.; Pickard, M.D.; et al. Nedd8-activating enzyme inhibitor MLN4924 provides synergy with mitomycin C through interactions with ATR, BRCA1/BRCA2, and chromatin dynamics pathways. Mol. Cancer Ther. 2014, 13, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Umberger, P.A.; Ogden, S.K. SPOP and CUL3 Modulate the Sonic Hedgehog Signal Response through Controlled Degradation of GLI Family Transcription Factors. Front. Cell Dev. Biol. 2021, 9, 710295. [Google Scholar] [CrossRef] [PubMed]
- Joost, S.; Almada, L.L.; Rohnalter, V.; Holz, P.S.; Vrabel, A.M.; Fernandez-Barrena, M.G.; McWilliams, R.R.; Krause, M.; Fernandez-Zapico, M.E.; Lauth, M. GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells. Cancer Res. 2012, 72, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sun, Y. Neddylation-CRLs regulate the functions of Treg immune cells. Bioessays 2023, 45, e2200222. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Curtis, V.F.; Ehrentraut, S.F.; Campbell, E.L.; Glover, L.E.; Bayless, A.; Kelly, C.J.; Kominsky, D.J.; Colgan, S.P. Stabilization of HIF through inhibition of Cullin-2 neddylation is protective in mucosal inflammatory responses. FASEB J. 2015, 29, 208–215. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef]
- Zamble, D.B.; Lippard, S.J. Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem. Sci. 1995, 20, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Yang, J.P.; Cao, Y.; Peng, L.X.; Zheng, L.S.; Sun, R.; Meng, D.F.; Wang, M.Y.; Mei, Y.; Qiang, Y.Y.; et al. Promoting tumorigenesis in nasopharyngeal carcinoma, NEDD8 serves as a potential theranostic target. Cell Death Dis. 2017, 8, e2834. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Belisario, D.C.; Bironzo, P.; Ananthanarayanan, P.; Ricci, L.; Digiovanni, S.; Fontana, S.; Napoli, F.; Sandri, A.; Facolmatà, C.; et al. SKP2 drives the sensitivity to neddylation inhibitors and cisplatin in malignant pleural mesothelioma. J. Exp. Clin. Cancer Res. 2022, 41, 75. [Google Scholar] [CrossRef] [PubMed]
- El-Mesery, M.; Seher, A.; Stühmer, T.; Siegmund, D.; Wajant, H. MLN4924 sensitizes monocytes and maturing dendritic cells for TNF-dependent and -independent necroptosis. Br. J. Pharmacol. 2015, 172, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Paiva, C.; Godbersen, J.C.; Berger, A. Targeting neddylation induces DNA damage and checkpoint activation and sensitizes chronic lymphocytic leukemia B cells to alkylating agents. Cell Death Dis. 2015, 6, e1807. [Google Scholar] [CrossRef]
- Meroni, A.; Grosser, J.; Agashe, S.; Ramakrishnan, N.; Jackson, J.; Verma, P.; Baranello, L.; Vindigni, A. NEDDylated Cullin 3 mediates the adaptive response to topoisomerase 1 inhibitors. Sci. Adv. 2022, 8, eabq0648. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Tan, M.; Wang, G.; Sun, Y. The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS ONE 2012, 7, e34079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Ochiiwa, H.; Ailiken, G.; Yokoyama, M.; Yamagata, K.; Nagano, H.; Yoshimura, C.; Muraoka, H.; Ishida, K.; Haruma, T.; Nakayama, A.; et al. TAS4464, a NEDD8-activating enzyme inhibitor, activates both intrinsic and extrinsic apoptotic pathways via c-Myc-mediated regulation in acute myeloid leukemia. Oncogene 2021, 40, 1217–1230. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Filippova, N.; Yang, X.; An, Z.; Nabors, L.B.; Pereboeva, L. Blocking PD1/PDL1 Interactions Together with MLN4924 Therapy is a Potential Strategy for Glioma Treatment. J. Cancer Sci. Ther. 2018, 10, 190–197. [Google Scholar] [CrossRef]
- Wang, B.; Guo, H.; Yu, H.; Chen, Y.; Xu, H.; Zhao, G. The Role of the Transcription Factor EGR1 in Cancer. Front. Oncol. 2021, 11, 642547. [Google Scholar] [CrossRef] [PubMed]
- Joslin, J.M.; Fernald, A.A.; Tennant, T.R.; Davis, E.M.; Kogan, S.C.; Anastasi, J.; Crispino, J.D.; Le Beau, M.M. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood 2007, 110, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Krones-Herzig, A.; Mittal, S.; Yule, K.; Liang, H.; English, C.; Urcis, R.; Soni, T.; Adamson, E.D.; Mercola, D. Early growth response 1 acts as a tumor suppressor in vivo and in vitro via regulation of p53. Cancer Res. 2005, 65, 5133–5143. [Google Scholar] [CrossRef]
- Kim, J.; Kang, H.S.; Lee, Y.J.; Lee, H.J.; Yun, J.; Shin, J.H.; Lee, C.W.; Kwon, B.M.; Hong, S.H. EGR1-dependent PTEN upregulation by 2-benzoyloxycinnamaldehyde attenuates cell invasion and EMT in colon cancer. Cancer Lett. 2014, 349, 35–44. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Sells, S.F.; Venkatasubbarao, K.; Fruitwala, S.M.; Muthukkumar, S.; Harp, C.; Mohiuddin, M.; Rangnekar, V.M. Ionizing radiation-inducible apoptosis in the absence of p53 linked to transcription factor EGR-1. J. Biol. Chem. 1997, 272, 33056–33061. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; He, Y.; Li, S.J.; Zhang, G.G.; Yu, W.; Yang, J.; Huang, Z.J.; Zheng, C.C.; He, Q.Y.; Li, Y.; et al. Anti-HIV Drug Elvitegravir Suppresses Cancer Metastasis via Increased Proteasomal Degradation of m6A Methyltransferase METTL3. Cancer Res. 2022, 82, 2444–2457. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ameri, A.H.; Wang, S.; Jansson, K.H.; Casey, O.M.; Yang, Q.; Beshiri, M.L.; Fang, L.; Lake, R.G.; Agarwal, S.; et al. EGR1 regulates angiogenic and osteoclastogenic factors in prostate cancer and promotes metastasis. Oncogene 2019, 38, 6241–6255. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Mehine, M.; Ahvenainen, T.; Khamaiseh, S.; Härkönen, J.; Reinikka, S.; Heikkinen, T.; Äyräväinen, A.; Pakarinen, P.; Härkki, P.; Pasanen, A.; et al. A novel uterine leiomyoma subtype exhibits NRF2 activation and mutations in genes associated with neddylation of the Cullin 3-RING E3 ligase. Oncogenesis 2022, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Gao, Z.; Zhang, Y.; Fan, K.; Wang, F.; Li, Y.; Zhong, J.; Fan, H.Y.; Cao, Q.; Zhou, J.; et al. CRL4DCAF2 negatively regulates IL-23 production in dendritic cells and limits the development of psoriasis. J. Exp. Med. 2018, 215, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xie, X.; Jie, Z.; Zhu, L.; Yang, J.Y.; Ko, C.J.; Gao, T.; Jain, A.; Jung, S.Y.; Baran, N.; et al. DYRK1a mediates BAFF-induced noncanonical NF-κB activation to promote autoimmunity and B-cell leukemogenesis. Blood 2021, 138, 2360–2371. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, Y.; Feng, W.; Hua, R.; Zhang, J.; Huo, Y.; Jiang, H.; Yin, B.; Yang, X. Neddylation pathway alleviates chronic pancreatitis by reducing HIF1α-CCL5-dependent macrophage infiltration. Cell Death Dis. 2021, 12, 273. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhuang, C.; Xu, X.; Li, J.; Wang, J.; Min, X.; Zhang, W.; Zhang, H.; Miao, Z. Discovery of benzothiazole derivatives as novel non-sulfamide NEDD8 activating enzyme inhibitors by target-based virtual screening. Eur. J. Med. Chem. 2017, 133, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.C.; Guo, Y.J.; Wang, B.; Wang, C.; Mamun, M.A.A.; Gao, Y.; Liu, H.M. Targeting neddylation E2s: A novel therapeutic strategy in cancer. J. Hematol. Oncol. 2021, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lu, J.; Chinnaswamy, K.; Stuckey, J.A.; Liu, L.; McEachern, D.; Yang, C.Y.; Bernard, D.; Shen, H.; Rui, L.; et al. Selective inhibition of cullin 3 neddylation through covalent targeting DCN1 protects mice from acetaminophen-induced liver toxicity. Nat. Commun. 2021, 12, 2621. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Liu, R.Z.; Ying, W.W.; Yang, Y.N.; Xiang, S.F.; Shao, X.J.; Cao, J.; Zhang, Y.Q.; Yang, B.; He, Q.J.; et al. Arctigenin impairs UBC12 enzyme activity and cullin neddylation to attenuate cancer cells. Acta Pharmacol. Sin. 2023, 44, 661–669. [Google Scholar] [CrossRef]
- Xu, T.; Ma, Q.; Li, Y.; Yu, Q.; Pan, P.; Zheng, Y.; Li, Z.; Xiong, X.; Hou, T.; Yu, B.; et al. A small molecule inhibitor of the UBE2F-CRL5 axis induces apoptosis and radiosensitization in lung cancer. Signal Transduct. Target. Ther. 2022, 7, 354. [Google Scholar] [CrossRef]
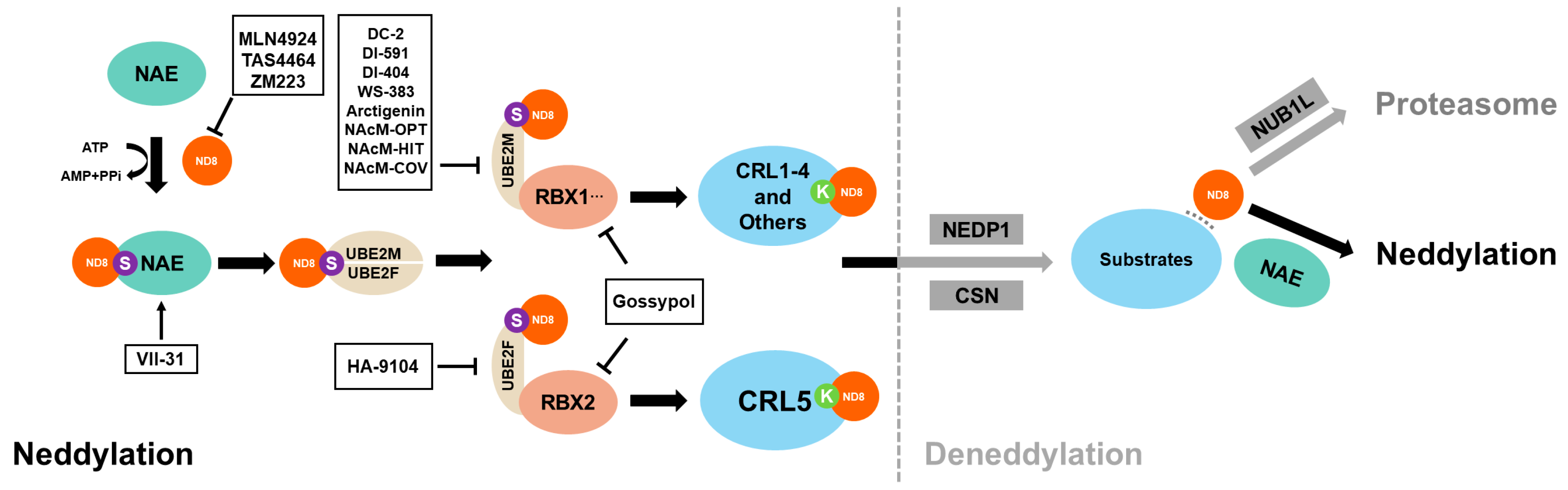
| Neddylation Inhibitor | Target | Clinical Status | References |
|---|---|---|---|
| MLN4924 | NAE | Phase I/II/III | [16] |
| TAS4464 | NAE | Phase I | [79] |
| ZM223 | NAE | N/A | [96] |
| DC-2 | UBE2M | Phase I/II | [97] |
| DI-591 | UBE2M | N/A | [97] |
| DI-404 | UBE2M | N/A | [97] |
| DI-1859 | UBE2M | N/A | [98] |
| WS-383 | UBE2M | N/A | [97] |
| Arctigenin | UBE2M | Phase I | [99] |
| NAcM-OPT | UBE2M | N/A | [43] |
| NAcM-HIT | UBE2M | N/A | [97] |
| NAcM-COV | UBE2M | N/A | [97] |
| HA-9104 | UBE2F | N/A | [100] |
| Gossypol | RBX1/RBX2 | Phase II/III/IV | [5] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.; Pang, X.; Li, S.; Tang, L. The Double-Edged Effects of MLN4924: Rethinking Anti-Cancer Drugs Targeting the Neddylation Pathway. Biomolecules 2024, 14, 738. https://doi.org/10.3390/biom14070738
Tang H, Pang X, Li S, Tang L. The Double-Edged Effects of MLN4924: Rethinking Anti-Cancer Drugs Targeting the Neddylation Pathway. Biomolecules. 2024; 14(7):738. https://doi.org/10.3390/biom14070738
Chicago/Turabian StyleTang, Haoming, Xin Pang, Shun Li, and Liling Tang. 2024. "The Double-Edged Effects of MLN4924: Rethinking Anti-Cancer Drugs Targeting the Neddylation Pathway" Biomolecules 14, no. 7: 738. https://doi.org/10.3390/biom14070738