
Therapeutic Potential of Hydrogen Sulfide in Ischemia and Reperfusion Injury
Abstract
:1. Introduction
2. Generation of Endogenous H2S
3. H2S Donors in I/R injury
4. H2S and I/R Injury
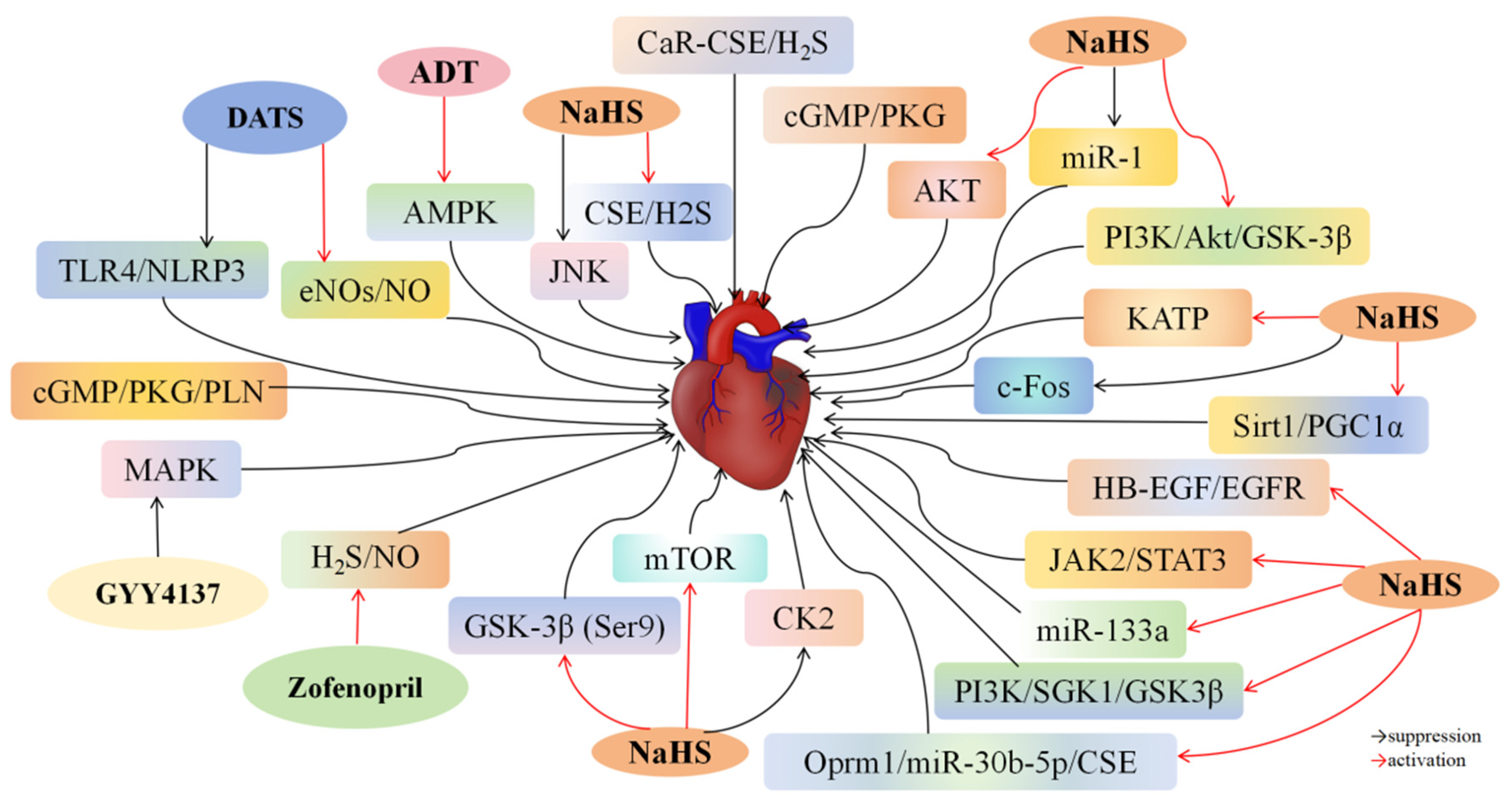
4.1. H2S and Myocardial I/R Injury
4.2. H2S and Cerebral I/R Injury
4.3. H2S and Hepatic I/R Injury
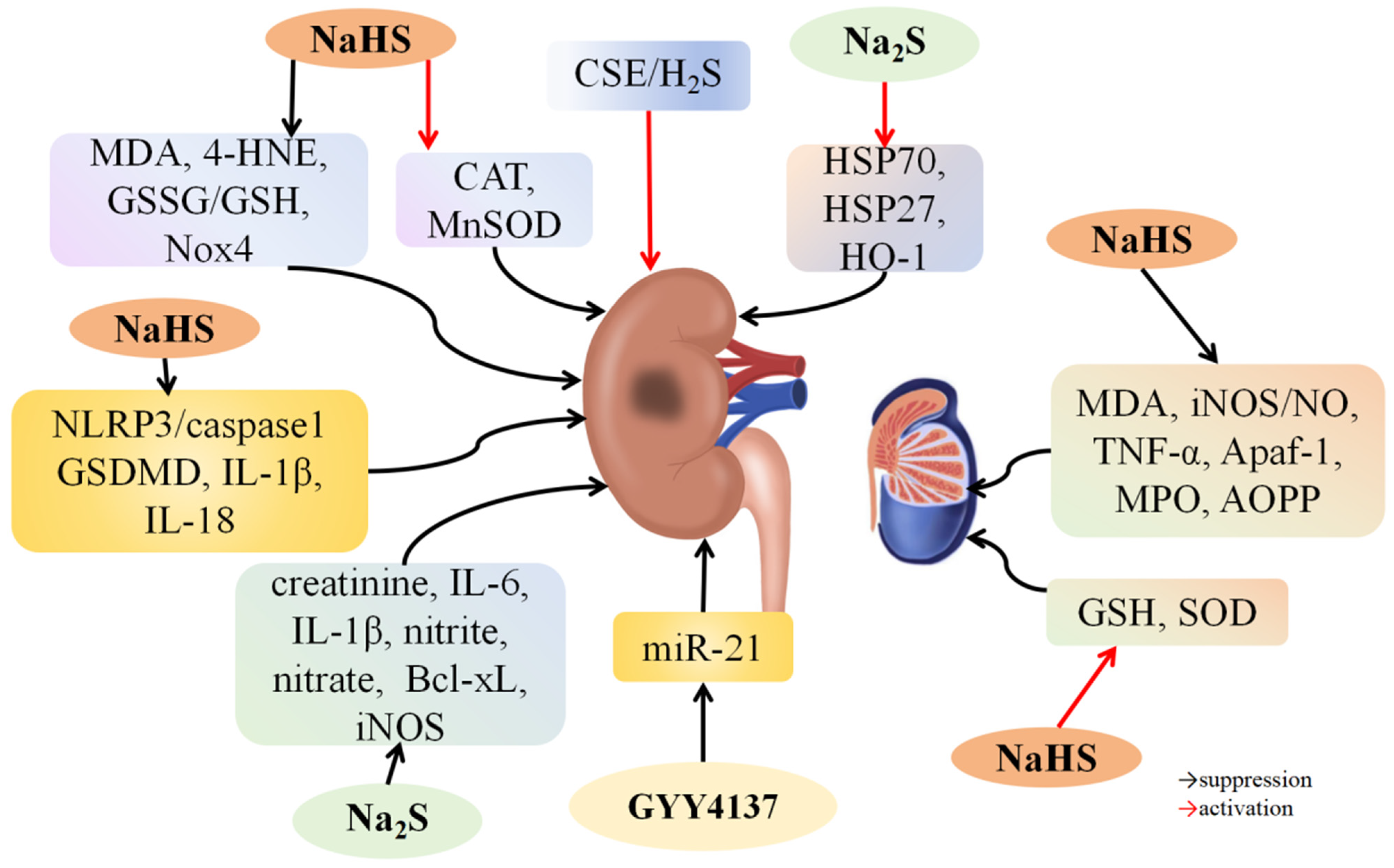
4.4. H2S and Renal I/R Injury
4.5. H2S and Testicular I/R Injury
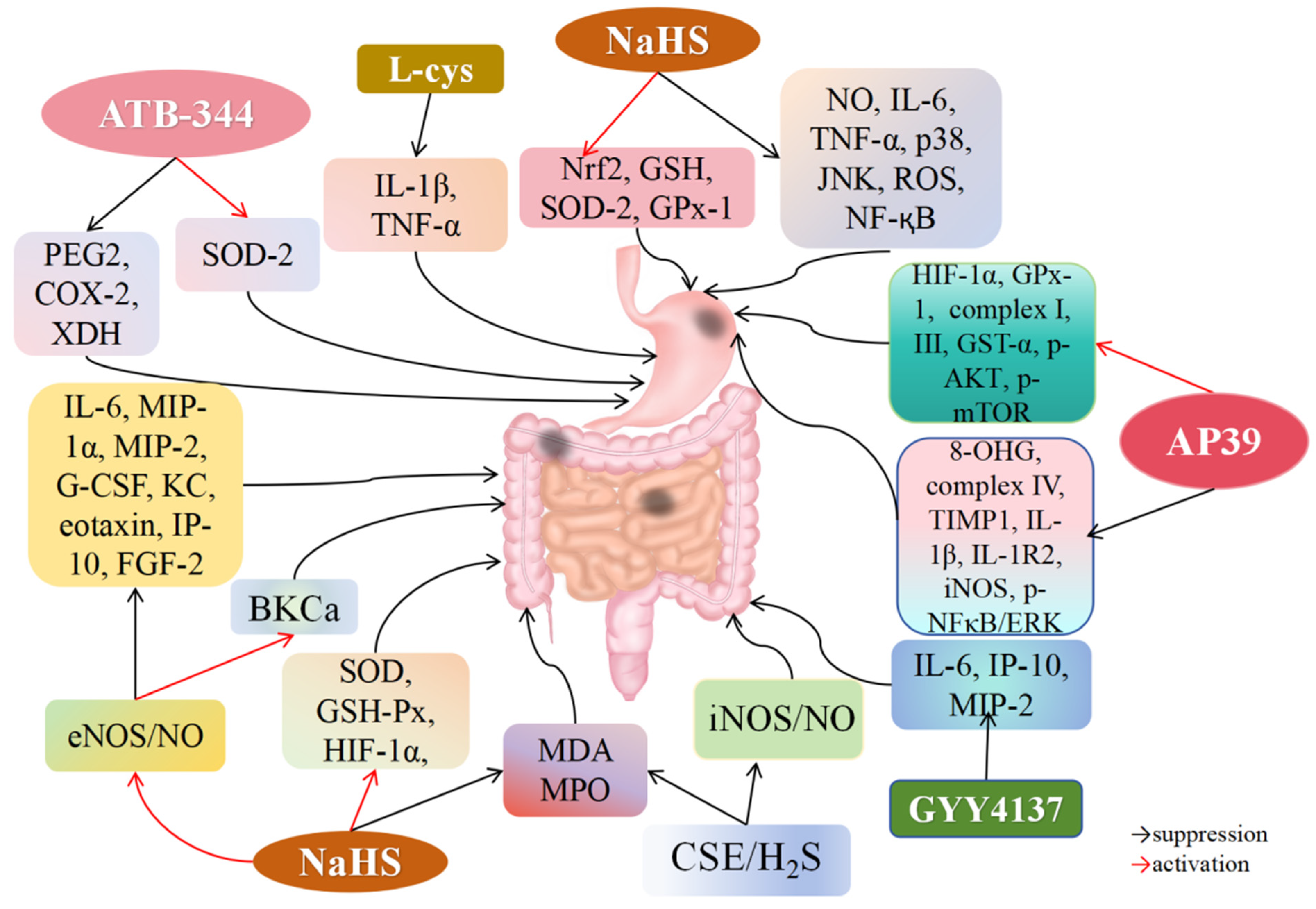
4.6. H2S and Gastric I/R Injury
4.7. H2S and Intestinal I/R Injury
4.8. H2S and Lung I/R Injury
4.9. H2S and Spinal Cord I/R Injury
4.10. H2S and Retinal I/R Injury
5. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Yapca, O.E.; Borekci, B.; Suleyman, H. Ischemia-reperfusion damage. Eurasian J. Med. 2013, 45, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yu, Y.; Wang, P.; Shen, H.; Ling, X.; Xue, X.; Yang, Q.; Zhang, Y.; Xiao, J.; Wang, Z. Role of Hydrogen Sulfide in Myocardial Ischemia-Reperfusion Injury. J. Cardiovasc. Pharmacol. 2021, 77, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Sodha, N.R.; Sellke, F.W. Attenuation of inflammatory responses by hydrogen sulfide (H2S) in ischemia/reperfusion injury. Methods Enzymol. 2015, 555, 127–144. [Google Scholar] [PubMed]
- Lv, S.; Wang, Z.; Wang, J.; Wang, H. Exogenous Hydrogen Sulfide Plays an Important Role Through Regulating Autophagy in Ischemia/Reperfusion Injury. Front. Mol. Biosci. 2021, 8, 681676. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yiang, G.; Liao, W.; Tsai, A.P.; Cheng, Y.; Cheng, P.; Li, C.; Li, C. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Pacheco, A.; Xian, M. Medicinal Chemistry: Insights into the Development of Novel H2S Donors. Handb. Exp. Pharmacol. 2015, 230, 365–388. [Google Scholar] [PubMed]
- Kimura, H. The physiological role of hydrogen sulfide and beyond. Nitric Oxide Biol. Chem. 2014, 41, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Bailey, S.M. Redox Biology of Hydrogen Sulfide: Implications for Physiology, Pathophysiology, and Pharmacology. Redox Biol. 2013, 1, 32–39. [Google Scholar] [CrossRef]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef]
- Andreadou, I.; Iliodromitis, E.K.; Rassaf, T.; Schulz, R.; Papapetropoulos, A.; Ferdinandy, P. The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br. J. Pharmacol. 2015, 172, 1587–1606. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, C.K.; Calvert, J.W. Hydrogen sulfide and ischemia-reperfusion injury. Pharmacol. Res. 2010, 62, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, W.; Dai, J.; Jin, S.; Huang, J.; Guo, C.; Wang, C.; Pang, L.; Wang, Y. A Long-Term and Slow-Releasing Hydrogen Sulfide Donor Protects against Myocardial Ischemia/Reperfusion Injury. Sci. Rep. 2017, 7, 3541. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Bornbaum, J.; Boengler, K.; Torregrossa, R.; Whiteman, M.; Wood, M.E.; Schulz, R.; Baxter, G.F. AP39, a mitochondria-targeting hydrogen sulfide (H(2)S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br. J. Pharmacol. 2017, 174, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, E.; Ali, M.J.; Rushing, A.M.; Scarborough, A.L.; Bradley, J.M.; Organ, C.L.; Islam, K.N.; Polhemus, D.J.; Evangelista, S.; Cirino, G.; et al. Zofenopril Protects Against Myocardial Ischemia-Reperfusion Injury by Increasing Nitric Oxide and Hydrogen Sulfide Bioavailability. J. Am. Heart Assoc. 2016, 5, e003531. [Google Scholar] [CrossRef] [PubMed]
- Głowacka, U.; Magierowski, M.; Śliwowski, Z.; Cieszkowski, J.; Szetela, M.; Wójcik-Grzybek, D.; Chmura, A.; Brzozowski, T.; Wallace, J.L.; Magierowska, K. Hydrogen Sulfide-Releasing Indomethacin-Derivative (ATB-344) Prevents the Development of Oxidative Gastric Mucosal Injuries. Antioxidants 2023, 12, 1545. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.J.; Lefer, D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014, 114, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Chen, S.; Tang, C.; Jin, H.; Du, J.; Huang, Y. Hydrogen sulfide and vascular regulation—An update. J. Adv. Res. 2021, 27, 85–97. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef]
- Kamoun, P. Endogenous production of hydrogen sulfide in mammals. Amino Acids 2004, 26, 243–254. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Xie, P.; Banerjee, R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 2011, 15, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen sulfide: Its production, release and functionsc. Amino Acids 2011, 41, 113–121. [Google Scholar] [CrossRef]
- Olson, K.R.; Whitfield, N.L.; Bearden, S.E.; St Leger, J.; Nilson, E.; Gao, Y.; Madden, J.A. Hypoxic pulmonary vasodilation: A paradigm shift with a hydrogen sulfide mechanism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R51–R60. [Google Scholar] [CrossRef]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H(2)S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Amagase, H. Clarifying the real bioactive constituents of garlic. J. Nutr. 2006, 136, 716S–725S. [Google Scholar] [CrossRef]
- Sun, X.; Wang, W.; Dai, J.; Huang, J.; Shi, M.; Chu, X.; Wang, F.; Guo, C.; Wang, C.; Pang, L.; et al. Donor heart preservation with a novel long-term and slow-releasing hydrogen sulfide system. Nitric Oxide Biol. Chem. 2018, 81, 1–10. [Google Scholar] [CrossRef]
- Marwah, M.K.; Shokr, H.; Sanchez-Aranguren, L.; Badhan, R.K.S.; Wang, K.; Ahmad, S. Transdermal Delivery of a Hydrogen Sulphide Donor, ADT-OH Using Aqueous Gel Formulations for the Treatment of Impaired Vascular Function: An Ex Vivo Study. Pharm. Res. 2022, 39, 341–352. [Google Scholar] [CrossRef]
- Cai, F.; Li, D.; Xie, Y.; Wang, X.; Ma, H.; Xu, H.; Cheng, J.; Zhuang, H.; Hua, Z. Sulfide:quinone oxidoreductase alleviates ferroptosis in acute kidney injury via ameliorating mitochondrial dysfunction of renal tubular epithelial cells. Redox Biol. 2024, 69, 102973. [Google Scholar] [CrossRef]
- Duan, S.; Zhang, M.; Dong, Q.; Yang, B.; Liu, W.; Zhang, X.; Yu, H.; Zhang, S.; Khan, N.H.; Wu, D.; et al. A Water-Soluble Hydrogen Sulfide Donor Suppresses the Growth of Hepatocellular Carcinoma via Inhibiting the AKT/GSK-3β/β-Catenin and TGF-β/Smad2/3 Signaling Pathways. J. Oncol. 2023, 2023, 8456852. [Google Scholar] [CrossRef]
- Bi, Z.; Chen, J.; Chang, X.; Li, D.; Yao, Y.; Cai, F.; Xu, H.; Cheng, J.; Hua, Z.; Zhuang, H. ADT-OH improves intestinal barrier function and remodels the gut microbiota in DSS-induced colitis. Front. Med. 2023, 17, 972–992. [Google Scholar] [CrossRef]
- Zhu, C.; Su, Y.; Juriasingani, S.; Zheng, H.; Veramkovich, V.; Jiang, J.; Sener, A.; Whiteman, M.; Lacefield, J.; Nagpal, D.; et al. Supplementing preservation solution with mitochondria-targeted H(2) S donor AP39 protects cardiac grafts from prolonged cold ischemia-reperfusion injury in heart transplantation. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transplant. Surg. 2019, 19, 3139–3148. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.; Hochholzer, W.; Siepe, M. ESC/EACTS guidelines on myocardial revascularization 2018: The most important innovations. Herz 2018, 43, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, L.; Xie, L.; Zhao, H.; Lu, Y.; Wu, B.; Wu, Z.; Zhang, Z.; Hao, Y.; Ou, W.; et al. Activation of the CaR-CSE/H2S pathway confers cardioprotection against ischemia-reperfusion injury. Exp. Cell Res. 2021, 398, 112389. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.N.; Sturz, G.R.; Yin, C.; Rehman, S.; Hoke, N.N.; Kukreja, R.C.; Xi, L. Beetroot juice reduces infarct size and improves cardiac function following ischemia-reperfusion injury: Possible involvement of endogenous H2S. Exp. Biol. Med. 2015, 240, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Liu, C. The cystathionine γ-lyase/hydrogen sulfide pathway mediates the trimetazidine-induced protection of H9c2 cells against hypoxia/reoxygenation-induced apoptosis and oxidative stress. Anat. J. Cardiol. 2019, 22, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Samidurai, A.; Hoke, N.N.; Kukreja, R.C.; Salloum, F.N. Hydrogen sulfide mediates the cardioprotective effects of gene therapy with PKG-Iα. Basic Res. Cardiol. 2015, 110, 42. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.N.; Das, A.; Samidurai, A.; Hoke, N.N.; Chau, V.Q.; Ockaili, R.A.; Stasch, J.; Kukreja, R.C. Cinaciguat, a novel activator of soluble guanylate cyclase, protects against ischemia/reperfusion injury: Role of hydrogen sulfide. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1347–H1354. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, B.; Wu, P.; Lang, Y.; Li, T. LncRNA Oprm1 overexpression attenuates myocardial ischemia/reperfusion injury by increasing endogenous hydrogen sulfide via Oprm1/miR-30b-5p/CSE axis. Life Sci. 2020, 254, 117699. [Google Scholar] [CrossRef] [PubMed]
- Ustunova, S.; Takir, S.; Yilmazer, N.; Bulut, H.; Altindirek, D.; Ng, O.H.; Tansel, C.D.; Dogan, B.S.U.; Ozbek, U.; Armutak, E.I.; et al. Hydrogen Sulphide and Nitric Oxide Cooperate in Cardioprotection Against Ischemia/Reperfusion Injury in Isolated Rat Heart. In Vivo 2020, 34, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, A.; Collino, M.; Yasin, M.; Benetti, E.; Gallicchio, M.; Mazzon, E.; Cuzzocrea, S.; Fantozzi, R.; Thiemermann, C. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 2009, 31, 267–274. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, H.; Liu, P.; Tang, C.; Wang, J. Hydrogen sulfide contributes to cardioprotection during ischemia-reperfusion injury by opening K ATP channels. Can. J. Physiol. Pharmacol. 2007, 85, 1248–1253. [Google Scholar] [CrossRef]
- Li, C.; Hu, M.; Wang, Y.; Lu, H.; Deng, J.; Yan, X. Hydrogen sulfide preconditioning protects against myocardial ischemia/reperfusion injury in rats through inhibition of endo/sarcoplasmic reticulum stress. Int. J. Clin. Exp. Pathol. 2015, 8, 7740–7751. [Google Scholar]
- Xie, H.; Xu, Q.; Jia, J.; Ao, G.; Sun, Y.; Hu, L.; Alkayed, N.J.; Wang, C.; Cheng, J. Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem. Biophys. Res. Commun. 2015, 458, 632–638. [Google Scholar] [CrossRef]
- Kar, S.; Bhandar, B.; Kavdia, M. Impact of SOD in eNOS uncoupling: A two-edged sword between hydrogen peroxide and peroxynitrite. Free Radic Res. 2012, 46, 1496–1513. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Bir, S.C.; Yuan, S.; Shen, X.; Pardue, S.; Wang, R.; Kevil, C.G. Cystathionine γ-lyase regulates arteriogenesis through NO-dependent monocyte recruitment. Cardiovasc. Res. 2015, 107, 590–600. [Google Scholar] [CrossRef]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef]
- King, A.L.; Polhemus, D.J.; Bhushan, S.; Otsuka, H.; Kondo, K.; Nicholson, C.K.; Bradley, J.M.; Islam, K.N.; Calvert, J.W.; Tao, Y.; et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 3182–3187. [Google Scholar] [CrossRef]
- Bibli, S.; Andreadou, I.; Chatzianastasiou, A.; Tzimas, C.; Sanoudou, D.; Kranias, E.; Brouckaert, P.; Coletta, C.; Szabo, C.; Kremastinos, D.T.; et al. Cardioprotection by H2S engages a cGMP-dependent protein kinase G/phospholamban pathway. Cardiovasc. Res. 2015, 106, 432–442. [Google Scholar] [CrossRef]
- Zhu, X.; Yan, X.; Chen, S. H(2)S protects myocardium against ischemia/reperfusion injury and its effect on c-Fos protein expression in rats. Sheng Li Xue Bao Acta Physiol. Sin. 2008, 60, 221–227. [Google Scholar]
- Ren, L.; Wang, Q.; Chen, Y.; Ma, Y.; Wang, D. Involvement of MicroRNA-133a in the Protective Effect of Hydrogen Sulfide against Ischemia/Reperfusion-Induced Endoplasmic Reticulum Stress and Cardiomyocyte Apoptosis. Pharmacology 2019, 103, 1–9. [Google Scholar] [CrossRef]
- Sodha, N.R.; Clements, R.T.; Feng, J.; Liu, Y.; Bianchi, C.; Horvath, E.M.; Szabo, C.; Sellke, F.W. The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia-reperfusion injury. Eur. J. Cardio Thorac. Surg. Off. J. Eur. Assoc. Cardio Thorac. Surg. 2008, 33, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Hong, J.; Xiao, J.; Zhu, X.; Ni, X.; Zhang, Y.; He, B.; Wang, Z. Involvement of miR-1 in the protective effect of hydrogen sulfide against cardiomyocyte apoptosis induced by ischemia/reperfusion. Mol. Biol. Rep. 2014, 41, 6845–6853. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, B.; Mao, H.; Sheng, Q.; Du, B.; Chen, J.; Pang, Q.; Ji, Y. Exogenous Hydrogen Sulfide Postconditioning Protects Isolated Rat Hearts from Ischemia/Reperfusion Injury Through Sirt1/PGC-1α Signaling Pathway. Int. Heart J. 2016, 57, 477–482. [Google Scholar] [CrossRef]
- Luan, H.; Zhao, Z.; Zhao, Q.; Zhu, P.; Xiu, M.; Ji, Y. Hydrogen sulfide postconditioning protects isolated rat hearts against ischemia and reperfusion injury mediated by the JAK2/STAT3 survival pathway. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Med. Biol. 2012, 45, 898–905. [Google Scholar] [CrossRef]
- Meng, G.; Wang, J.; Xiao, Y.; Bai, W.; Xie, L.; Shan, L.; Moore, P.K.; Ji, Y. GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J. Biomed. Res. 2015, 29, 203–213. [Google Scholar] [PubMed]
- Li, H.; Xiao, F. Effect of hydrogen sulfide on cardiomyocyte apoptosis in rats with myocardial ischemia-reperfusion injury via the JNK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2054–2061. [Google Scholar]
- Banu, S.A.; Ravindran, S.; Kurian, G.A. Hydrogen sulfide post-conditioning preserves interfibrillar mitochondria of rat heart during ischemia reperfusion injury. Cell Stress Chaperones 2016, 21, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef]
- Sun, W.; Liu, F.; Chen, Y.; Zhu, Y. Hydrogen sulfide decreases the levels of ROS by inhibiting mitochondrial complex IV and increasing SOD activities in cardiomyocytes under ischemia/reperfusion. Biochem. Biophys. Res. Commun. 2012, 421, 164–169. [Google Scholar] [CrossRef]
- Yao, X.; Tan, G.; He, C.; Gao, Y.; Pan, S.; Jiang, H.; Zhang, Y.; Sun, X. Hydrogen sulfide protects cardiomyocytes from myocardial ischemia-reperfusion injury by enhancing phosphorylation of apoptosis repressor with caspase recruitment domain. Tohoku J. Exp. Med. 2012, 226, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Huang, X.; Wang, Y.; Cao, Y.; Zhang, C.; Zhu, Y. Hydrogen sulfide protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by preventing GSK-3beta-dependent opening of mPTP. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1310–H1319. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, D.; Gao, X.; Lew, K.; Richards, A.M.; Wang, P. mTORC2 phosphorylation of Akt1: A possible mechanism for hydrogen sulfide-induced cardioprotection. PLoS ONE 2014, 9, e99665. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhu, X.; Kang, B.; Xu, J.; Wu, L.; Hong, J.; Zhang, Y.; Ni, X.; Wang, Z. Hydrogen Sulfide Attenuates Myocardial Hypoxia-Reoxygenation Injury by Inhibiting Autophagy via mTOR Activation. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 37, 2444–2453. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xiao, J.; Kang, B.; Zhu, X.; Xin, N.; Wang, Z. PI3K/SGK1/GSK3β signaling pathway is involved in inhibition of autophagy in neonatal rat cardiomyocytes exposed to hypoxia/reoxygenation by hydrogen sulfide. Exp. Cell Res. 2016, 345, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Predmore, B.L.; Kondo, K.; Bhushan, S.; Zlatopolsky, M.A.; King, A.L.; Aragon, J.P.; Grinsfelder, D.B.; Condit, M.E.; Lefer, D.J. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2410–H2418. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Quindry, J.; Hamilton, K. Aging, exercise, and cardioprotection. Ann. N. Y. Acad. Sci. 2004, 1019, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. The Influence of Obese Insulin-Resistance on the Outcome of the Ischemia/Reperfusion Insult to the Heart. Curr. Med. Chem. 2018, 25, 1501–1509. [Google Scholar] [CrossRef]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Ravani, S.; Chatzianastasiou, A.; Papapetropoulos, A. Using mechanism-based combinations of H(2)S-donors to maximize the cardioprotective action of H(2)S. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024, 397, 1853–1864. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Wei, C.; Bai, S.; Zhao, Y.; Li, H.; Wu, B.; Wang, R.; Wu, L.; Xu, C. Mediation of exogenous hydrogen sulfide in recovery of ischemic post-conditioning-induced cardioprotection via down-regulating oxidative stress and up-regulating PI3K/Akt/GSK-3β pathway in isolated aging rat hearts. Cell Biosci. 2015, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, J.; Sun, W.; Li, L.; Wang, Y.; Bai, S.; Li, X.; Wang, R.; Wu, L.; Li, H.; et al. Involvement of exogenous H2S in recovery of cardioprotection from ischemic post-conditioning via increase of autophagy in the aged hearts. Int. J. Cardiol. 2016, 220, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, J.; Sun, W.; Wen, X.; Xi, Y.; Wang, Y.; Wei, C.; Xu, C.; Li, H. H(2)S restores the cardioprotective effects of ischemic post-conditioning by upregulating HB-EGF/EGFR signaling. Aging 2019, 11, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Gheibi, S.; Aboutaleb, N.; Khaksari, M.; Kalalian-Moghaddam, H.; Vakili, A.; Asadi, Y.; Mehrjerdi, F.Z.; Gheibi, A. Hydrogen sulfide protects the brain against ischemic reperfusion injury in a transient model of focal cerebral ischemia. J. Mol. Neurosci. MN 2014, 54, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Li, W.; Liu, Y.; Wei, Y.; Ren, Y.; Mai, C.; Zhang, S.; Zuo, Y.; Sun, Z.; Li, D.; et al. Hydrogen Sulfide Attenuates Neuroinflammation by Inhibiting the NLRP3/Caspase-1/GSDMD Pathway in Retina or Brain Neuron following Rat Ischemia/Reperfusion. Brain Sci. 2022, 12, 1245. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Xue, L.; Cheng, J.; Bai, Y. Preconditioning of H2S inhalation protects against cerebral ischemia/reperfusion injury by induction of HSP70 through PI3K/Akt/Nrf2 pathway. Brain Res. Bull. 2016, 121, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Mao, Z.; Wang, S.; Xin, Y.; Li, P.; Maharjan, S.; Zhang, B. GYY4137 protects against MCAO via p38 MAPK mediated anti-apoptotic signaling pathways in rats. Brain Res. Bull. 2020, 158, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, K.; Wang, X.; Ding, Y.; Ren, Z.; Fang, J.; Sun, T.; Guo, Y.; Chen, Z.; Wen, J. CSE-Derived H(2)S Inhibits Reactive Astrocytes Proliferation and Promotes Neural Functional Recovery after Cerebral Ischemia/Reperfusion Injury in Mice Via Inhibition of RhoA/ROCK(2) Pathway. ACS Chem. Neurosci. 2021, 12, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef]
- Wen, J.; Wang, M.; Li, Y.; Jiang, H.; Sun, X.; Chen, Z. Vascular Protection of Hydrogen Sulfide on Cerebral Ischemia/Reperfusion Injury in Rats. Front. Neurol. 2018, 9, 779. [Google Scholar] [CrossRef]
- McCarter, K.D.; Li, C.; Li, J.; Xu, G.; Sun, H. Influence of low-dose alcohol consumption on post-ischemic inflammation: Role of cystathionine γ-lyase. Alcohol 2019, 76, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Lu, Z.; Tao, L.; Yang, L.; Guo, Y.; Yang, Y.; Sun, X.; Ding, Q. ROS-Dependent Neuroprotective Effects of NaHS in Ischemia Brain Injury Involves the PARP/AIF Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Hansagi, H.; Romelsjo, A.; de Verdier, M.G.; Andreasson, S.; Leifman, A. Alcohol consumption and stroke mortality. Stroke 1995, 26, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Shui, M.; Liu, X.; Zhu, Y.; Wang, Y. Exogenous hydrogen sulfide attenuates cerebral ischemia-reperfusion injury by inhibiting autophagy in mice. Can. J. Physiol. Pharmacol. 2016, 94, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shui, M.; Liu, X.; Hu, W.; Wang, Y. Increased autophagic degradation contributes to the neuroprotection of hydrogen sulfide against cerebral ischemia/reperfusion injury. Metab. Brain Dis. 2017, 32, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Huang, B.; Han, Y.; Deng, L.; Wu, L. Sodium hydrosulfide attenuates cerebral ischemia/reperfusion injury by suppressing overactivated autophagy in rats. FEBS Open Bio 2017, 7, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Guo, F.; Liu, X.; Xi, J.; Xue, M.; Guo, Y.; Wen, J.; Dong, L.; Chen, Z. Roles of the RhoA-ROCK Signaling Pathway in the Endothelial H2S Production and Vasodilation in Rat Cerebral Arteries. ACS Omega 2022, 7, 18498–18508. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Gao, S.; Chen, F.; Chen, S.; Wang, M.; Chen, Z. Role of CSE-Produced H(2)S on Cerebrovascular Relaxation via RhoA-ROCK Inhibition and Cerebral Ischemia-Reperfusion Injury in Mice. ACS Chem. Neurosci. 2019, 10, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhang, B.; Cheng, L.; Chi, M.; Deng, L.; Pan, H.; Yao, X.; Wang, G. Hydrogen sulfide induces neuroprotection against experimental stroke in rats by down-regulation of AQP4 via activating PKC. Brain Res. 2015, 1622, 292–299. [Google Scholar] [CrossRef]
- Hu, Y.; Li, R.; Yang, H.; Luo, H.; Chen, Z. Sirtuin 6 is essential for sodium sulfide-mediated cytoprotective effect in ischemia/reperfusion-stimulated brain endothelial cells. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2015, 24, 601–609. [Google Scholar] [CrossRef]
- Huang, X.; Gao, Y.; Qin, J.; Lu, S. The role of miR-34a in the hepatoprotective effect of hydrogen sulfide on ischemia/reperfusion injury in young and old rats. PLoS ONE 2014, 9, e113305. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Dai, W.; Wang, F.; Lu, J.; Shen, M.; Chen, K.; Li, J.; Zhang, Y.; Wang, C.; Yang, J.; et al. Ethyl pyruvate inhibits proliferation and induces apoptosis of hepatocellular carcinoma via regulation of the HMGB1-RAGE and AKT pathways. Biochem. Biophys. Res. Commun. 2014, 443, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Jiang, X.; Tong, L.; Zhang, F.; Ma, L.; Dong, X.; Sun, X. MicroRNA-21-Regulated Activation of the Akt Pathway Participates in the Protective Effects of H2S against Liver Ischemia-Reperfusion Injury. Biol. Pharm. Bull. 2018, 41, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Sameri, M.J.; Savari, F.; Hoseinynejad, K.; Danyaei, A.; Mard, S.A. The hepato-protective effect of H2S-modified and non-modified mesenchymal stem cell exosomes on liver ischemia-reperfusion injury in mice: The role of MALAT1. Biochem. Biophys. Res. Commun. 2022, 635, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ma, K.; Fan, H.; Wang, X.; Cao, T. Exogenous hydrogen sulfide protects against hepatic ischemia/reperfusion injury by inhibiting endoplasmic reticulum stress and cell apoptosis. Exp. Ther. Med. 2021, 22, 799. [Google Scholar] [CrossRef]
- Chen, L.; Lin, B.; Yang, J.; Zhong, L.; Xiong, X.; Wang, X. Hydrogen sulfide alleviates ischemia induced liver injury by repressing the SPHK1/S1P pathway. Ann. Transl. Med. 2023, 11, 73. [Google Scholar] [CrossRef]
- Zhang, Q.; Fu, H.; Zhang, H.; Xu, F.; Zou, Z.; Liu, M.; Wang, Q.; Miao, M.; Shi, X. Hydrogen sulfide preconditioning protects rat liver against ischemia/reperfusion injury by activating Akt-GSK-3β signaling and inhibiting mitochondrial permeability transition. PLoS ONE 2013, 8, e74422. [Google Scholar] [CrossRef]
- Pushpakumar, S.; Kundu, S.; Weber, G.; Sen, U. Exogenous hydrogen sulfide and miR-21 antagonism attenuates macrophage-mediated inflammation in ischemia reperfusion injury of the aged kidney. Geroscience 2021, 43, 1349–1367. [Google Scholar] [CrossRef]
- Han, S.J.; Kim, J.I.; Park, J.W.; Park, K.M. Hydrogen sulfide accelerates the recovery of kidney tubules after renal ischemia/reperfusion injury. Nephrol. Dial. Transplant. 2015, 30, 1497–1506. [Google Scholar] [CrossRef]
- Jiang, J.; Wen, C.; Li, Y.; Liu, G.; Chen, Z.; Zheng, D. IFC-305 attenuates renal ischemia-reperfusion injury by promoting the production of hydrogen sulfide (H2S) via suppressing the promoter methylation of cystathionine γ-lyase (CSE). Bioengineered 2022, 13, 12045–12054. [Google Scholar] [CrossRef]
- Niu, Y.; Du, C.; Cui, C.; Zhang, H.; Deng, Y.; Cai, J.; Chen, Z.; Geng, B. Norswertianolin Promotes Cystathionine γ-Lyase Activity and Attenuates Renal Ischemia/Reperfusion Injury and Hypertension. Front. Pharmacol. 2021, 12, 677212. [Google Scholar] [CrossRef]
- Simon, F.; Scheuerle, A.; Gröger, M.; Stahl, B.; Wachter, U.; Vogt, J.; Speit, G.; Hauser, B.; Möller, P.; Calzia, E.; et al. Effects of intravenous sulfide during porcine aortic occlusion-induced kidney ischemia/reperfusion injury. Shock 2011, 35, 156–163. [Google Scholar] [CrossRef]
- Du, Y.; Liu, X.H.; Zhu, H.C.; Wang, L.; Wang, Z.S.; Ning, J.Z.; Xiao, C.C. Hydrogen sulfide treatment protects against renal ischemia-reperfusion injury via induction of heat shock proteins in rats. Iran. J. Basic Med. Sci. 2019, 22, 99–105. [Google Scholar]
- Ni, J.; Jiang, L.; Shen, G.; Xia, Z.; Zhang, L.; Xu, J.; Feng, Q.; Qu, H.; Xu, F.; Li, X. Hydrogen sulfide reduces pyroptosis and alleviates ischemia-reperfusion-induced acute kidney injury by inhibiting NLRP3 inflammasome. Life Sci. 2021, 284, 119466. [Google Scholar] [CrossRef]
- Mard, S.A.; Neisi, N.; Solgi, G.; Hassanpour, M.; Darbor, M.; Maleki, M. Gastroprotective effect of NaHS against mucosal lesions induced by ischemia-reperfusion injury in rat. Dig. Dis. Sci. 2012, 57, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liang, F.; Shah Masood, W.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s-sulfhydration, MAPK dependent anti-apoptosis and NF-κB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Magierowski, M.; Magierowska, K.; Hubalewska-Mazgaj, M.; Sliwowski, Z.; Pajdo, R.; Ginter, G.; Kwiecien, S.; Brzozowski, T. Exogenous and Endogenous Hydrogen Sulfide Protects Gastric Mucosa against the Formation and Time-Dependent Development of Ischemia/Reperfusion-Induced Acute Lesions Progressing into Deeper Ulcerations. Molecules 2017, 22, 295. [Google Scholar] [CrossRef] [PubMed]
- Magierowska, K.; Korbut, E.; Wójcik-Grzybek, D.; Bakalarz, D.; Sliwowski, Z.; Cieszkowski, J.; Szetela, M.; Torregrossa, R.; Whiteman, M.; Magierowski, M. Mitochondria-targeted hydrogen sulfide donors versus acute oxidative gastric mucosal injury. J. Control. Release 2022, 348, 321–334. [Google Scholar] [CrossRef]
- Liu, H.; Bai, X.B.; Shi, S.; Cao, Y.X. Hydrogen sulfide protects from intestinal ischaemia-reperfusion injury in rats. J. Pharm. Pharmacol. 2009, 61, 207–212. [Google Scholar] [CrossRef]
- Jensen, A.R.; Drucker, N.A.; Khaneki, S.; Ferkowicz, M.J.; Markel, T.A. Hydrogen sulfide improves intestinal recovery following ischemia by endothelial nitric oxide-dependent mechanisms. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G450–G456. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, Y.; Wang, Y.; Tong, F. The interaction between CSE/H2S and the iNOS/NO-mediated resveratrol/poly(ethylene glycol)-poly(phenylalanine) complex alleviates intestinal ischemia/reperfusion injuries in diabetic rats. Biomed. Pharmacother. 2019, 112, 108736. [Google Scholar] [CrossRef] [PubMed]
- Zuidema, M.Y.; Yang, Y.; Wang, M.; Kalogeris, T.; Liu, Y.; Meininger, C.J.; Hill, M.A.; Davis, M.J.; Korthuis, R.J. Antecedent hydrogen sulfide elicits an anti-inflammatory phenotype in postischemic murine small intestine: Role of BK channels. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1554–H1567. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Chen, D.; Liu, B.; Xie, X.; Zhang, J.; Yang, G. Effects of sodium hydrosulfide on intestinal mucosal injury in a rat model of cardiac arrest and cardiopulmonary resuscitation. Life Sci. 2013, 93, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Liu, X.; Geng, B.; Fang, L.; Tang, C. Hydrogen sulfide protects rat lung from ischemia-reperfusion injury. Life Sci. 2008, 82, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.Y.; Chen, W.; Li, X.L.; Wang, Y.W.; Xie, X.H. H2S protecting against lung injury following limb ischemia-reperfusion by alleviating inflammation and water transport abnormality in rats. Biomed. Environ. Sci. 2014, 27, 410–418. [Google Scholar] [PubMed]
- Li, L.; Jiang, H.K.; Li, Y.P.; Guo, Y.P. Hydrogen sulfide protects spinal cord and induces autophagy via miR-30c in a rat model of spinal cord ischemia-reperfusion injury. J. Biomed. Sci. 2015, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Biermann, J.; Lagrèze, W.A.; Schallner, N.; Schwer, C.I.; Goebel, U. Inhalative preconditioning with hydrogen sulfide attenuated apoptosis after retinal ischemia/reperfusion injury. Mol. Vis. 2011, 17, 1275–1286. [Google Scholar] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e467–e492. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, S.; Iso, H.; Toyoshima, H.; Date, C.; Yamamoto, A.; Kikuchi, S. Alcohol consumption and mortality from stroke and coronary heart disease among Japanese men and women: The Japan collaborative cohort study. Stroke 2008, 39, 2936–2942. [Google Scholar] [CrossRef]
- Ronksley, P.E.; Brien, S.E.; Turner, B.J.; Mukamal, K.J.; Ghali, W.A. Association of alcohol consumption with selected cardiovascular disease outcomes: A systematic review and meta-analysis. BMJ 2011, 342, d671. [Google Scholar] [CrossRef]
- Hrelia, S.; Di Renzo, L.; Bavaresco, L.; Bernardi, E.; Malaguti, M.; Giacosa, A. Moderate Wine Consumption and Health: A Narrative Review. Nutrients 2022, 15, 175. [Google Scholar] [CrossRef] [PubMed]
- McCarter, K.D.; Li, C.; Jiang, Z.; Lu, W.; Smith, H.C.; Xu, G.; Mayhan, W.G.; Sun, H. Effect of Low-Dose Alcohol Consumption on Inflammation Following Transient Focal Cerebral Ischemia in Rats. Sci. Rep. 2017, 7, 12547. [Google Scholar] [CrossRef] [PubMed]
- Berrevoet, F.; Schäfer, T.; Vollmar, B.; Menger, M.D. Ischemic preconditioning: Enough evidence to support clinical application in liver surgery and transplantation? Acta Chir. Belg. 2003, 103, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Siempis, T.; Theodorakou, E.; Tsoulfas, G. Hepatic ischemia and reperfusion injury and trauma: Current concepts. Arch. Trauma Res. 2013, 2, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Woolbright, B.L. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant. Rev. 2012, 26, 103–114. [Google Scholar] [CrossRef]
- Miska, E.A. How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 2005, 15, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Muthusamy, S.; Liang, R.; Sarojini, H.; Wang, E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech. Ageing Dev. 2011, 132, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Takagi, Y.; Osanai, H.; Li, L.; Takeuchi, M.; Katoh, Y.; Kobayashi, M.; Yamamoto, M. Pi class glutathione S-transferase genes are regulated by Nrf 2 through an evolutionarily conserved regulatory element in zebrafish. Biochem. J. 2005, 388, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Sahin, K.; Orhan, C.; Tuzcu, M.; Sahin, N.; Ali, S.; Bahcecioglu, I.H.; Guler, O.; Ozercan, I.; Ilhan, N.; Kucuk, O. Orally administered lycopene attenuates diethylnitrosamine-induced hepatocarcinogenesis in rats by modulating Nrf-2/HO-1 and Akt/mTOR pathways. Nutr. Cancer 2014, 66, 590–598. [Google Scholar] [CrossRef]
- Zeng, T.; Zhang, C.; Song, F.; Zhao, X.; Yu, L.; Zhu, Z.; Xie, K. The activation of HO-1/Nrf-2 contributes to the protective effects of diallyl disulfide (DADS) against ethanol-induced oxidative stress. Biochim. Biophys. Acta 2013, 1830, 4848–4859. [Google Scholar] [CrossRef]
- Shen, M.; Lu, J.; Dai, W.; Wang, F.; Xu, L.; Chen, K.; He, L.; Cheng, P.; Zhang, Y.; Wang, C.; et al. Corrigendum to “Ethyl Pyruvate Ameliorates Hepatic Ischemia-Reperfusion Injury by Inhibiting Intrinsic Pathway of Apoptosis and Autophagy”. Mediat. Inflamm. 2016, 2016, 5434275. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, S.M. Ferulic acid attenuates ischemia/reperfusion-induced hepatocyte apoptosis via inhibition of JNK activation. Eur. J. Pharm. Sci. 2012, 45, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Hoffman, R.A.; Izuishi, K.; Critchlow, N.D.; Nakao, A.; Chan, M.H.; Lotze, M.T.; Geller, D.A.; Billiar, T.R. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J. Immunol. 2005, 175, 7661–7668. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wang, F.; Chen, K.; Shen, M.; Dai, W.; Xu, L.; Zhang, Y.; Wang, C.; Li, J.; Yang, J.; et al. Hydrogen sulfide ameliorates ischemia/reperfusion-induced hepatitis by inhibiting apoptosis and autophagy pathways. Mediat. Inflamm. 2014, 2014, 935251. [Google Scholar] [CrossRef] [PubMed]
- Spassov, S.G.; Donus, R.; Ihle, P.M.; Engelstaedter, H.; Hoetzel, A.; Faller, S. Hydrogen Sulfide Prevents Formation of Reactive Oxygen Species through PI3K/Akt Signaling and Limits Ventilator-Induced Lung Injury. Oxid. Med. Cell. Longev. 2017, 2017, 3715037. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ma, Y.; Li, Z.; Kang, K.; Sun, X.; Pan, S.; Wang, J.; Pan, H.; Liu, L.; Liang, D.; et al. The role of AKT1 and autophagy in the protective effect of hydrogen sulphide against hepatic ischemia/reperfusion injury in mice. Autophagy 2012, 8, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Suo, L.; Kang, K.; Wang, X.; Cao, Y.; Zhao, H.; Sun, X.; Tong, L.; Zhang, F. Carvacrol alleviates ischemia reperfusion injury by regulating the PI3K-Akt pathway in rats. PLoS ONE 2014, 9, e104043. [Google Scholar] [CrossRef]
- Bai, Y.; Yu, Z.; Luo, L.; Yi, J.; Xia, Q.; Zeng, Y. MicroRNA-21 accelerates hepatocyte proliferation in vitro via PI3K/Akt signaling by targeting PTEN. Biochem. Biophys. Res. Commun. 2014, 443, 802–807. [Google Scholar]
- Chen, X.; Song, M.; Chen, W.; Dimitrova-Shumkovska, J.; Zhao, Y.; Cao, Y.; Song, Y.; Yang, W.; Wang, F.; Xiang, Y.; et al. MicroRNA-21 Contributes to Liver Regeneration by Targeting PTEN. Med. Sci. Monit. 2016, 22, 83–91. [Google Scholar] [CrossRef]
- Juskeviciute, E.; Dippold, R.P.; Antony, A.N.; Swarup, A.; Vadigepalli, R.; Hoek, J.B. Inhibition of miR-21 rescues liver regeneration after partial hepatectomy in ethanol-fed rats. Am. J. Physiol. Gastrointest Liver Physiol. 2016, 311, G794–G806. [Google Scholar] [CrossRef]
- Jia, P.; Wu, X.; Dai, Y.; Teng, J.; Fang, Y.; Hu, J.; Zou, J.; Liang, M.; Ding, X. MicroRNA-21 Is Required for Local and Remote Ischemic Preconditioning in Multiple Organ Protection Against Sepsis. Crit. Care Med. 2017, 45, e703–e710. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Brown, J.; Gu, Z.; Garcia, C.A.; Liang, R.; Alard, P.; Beurel, E.; Jope, R.S.; Greenway, T.; Martin, M. Convergence of the mammalian target of rapamycin complex 1- and glycogen synthase kinase 3-β-signaling pathways regulates the innate inflammatory response. J. Immunol. 2011, 186, 5217–5226. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Srivastava, R.; Howell, S.H. Endoplasmic reticulum (ER) stress response and its physiological roles in plants. Int. J. Mol. Sci. 2013, 14, 8188–8212. [Google Scholar] [CrossRef] [PubMed]
- Emadali, A.; Nguyên, D.T.; Rochon, C.; Tzimas, G.N.; Metrakos, P.P.; Chevet, E. Distinct endoplasmic reticulum stress responses are triggered during human liver transplantation. J. Pathol. 2005, 207, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, L.; Chang, N.; Hou, L.; Zhou, X.; Dong, C.; Liu, F.; Yang, L.; Li, L. Neutrophil recruitment mediated by sphingosine 1-phosphate (S1P)/S1P receptors during chronic liver injury. Cell. Immunol. 2021, 359, 104243. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Wang, W.; Chen, J.; Dai, L.; Kaczorowski, D.; Gao, X.; Xia, P. Sphingosine Kinase 1 Protects Hepatocytes from Lipotoxicity via Down-regulation of IRE1α Protein Expression. J. Biol. Chem. 2015, 290, 23282–23290. [Google Scholar] [CrossRef]
- Kim, J.S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 463–470. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. New directions for protecting the heart against ischaemia-reperfusion injury: Targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc. Res. 2004, 61, 448–460. [Google Scholar] [CrossRef]
- Barry, S.P.; Townsend, P.A.; Latchman, D.S.; Stephanou, A. Role of the JAK-STAT pathway in myocardial injury. Trends Mol. Med. 2007, 13, 82–89. [Google Scholar] [CrossRef]
- Pefanis, A.; Ierino, F.L.; Murphy, J.M.; Cowan, P.J. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. 2019, 96, 291–301. [Google Scholar] [CrossRef]
- Turner, T.T.; Bang, H.J.; Lysiak, J.L. The molecular pathology of experimental testicular torsion suggests adjunct therapy to surgical repair. J. Urol. 2004, 172 Pt 2, 2574–2578. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, S.; Erginel, B.; Bingul, I.; Ozluk, Y.; Karatay, H.; Aydın, F.; Keskin, E. The effect of hydrogen sulfide on ischemïa/reperfusion injury in an experimental testicular torsion model. J. Pediatr. Urol. 2022, 18, 16.e1–16.e7. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, M.; Degirmentepe, R.B.; Polat, E.C.; Yildirim, F.; Sonmez, K.; Cekmen, M.; Eraldemir, C.; Otunctemur, A. Protective effect of hydrogen sulfide on experimental testicular ischemia reperfusion in rats. J. Pediatr. Urol. 2020, 16, 40.e1–40.e8. [Google Scholar] [CrossRef] [PubMed]
- Oates, P.J.; Hakkinen, J.P. Studies on the mechanism of ethanol-induced gastric damage in rats. Gastroenterology 1988, 94, 10–21. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.F.; Zhang, Y.M.; Ma, X.B. The protective effect of genistein postconditioning on hypoxia/reoxygenation-induced injury in human gastric epithelial cells. Acta Pharmacol. Sin. 2009, 30, 576–581. [Google Scholar] [CrossRef]
- Piper, H.M.; Meuter, K.; Schäfer, C. Cellular mechanisms of ischemia-reperfusion injury. Ann. Thorac. Surg. 2003, 75, S644–S648. [Google Scholar] [CrossRef]
- Liu, M.J.; Fei, S.J.; Qiao, W.L.; Du, D.S.; Zhang, Y.M.; Li, Y.; Zhang, J.F. The protective effect of 17beta-estradiol postconditioning against hypoxia/reoxygenation injury in human gastric epithelial cells. Eur. J. Pharmacol. 2010, 645, 151–157. [Google Scholar] [CrossRef]
- Ju, Y.; Zhang, W.; Pei, Y.; Yang, G. H(2)S signaling in redox regulation of cellular functions. Can. J. Physiol. Pharmacol. 2013, 91, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rossoni, G.; Sparatore, A.; Lee, L.C.; Del Soldato, P.; Moore, P.K. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic. Biol. Med. 2007, 42, 706–719. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.M.; Qiao, W.L.; Zhang, J.F.; Wang, L. Role of mitogen-activated protein kinases in the regulation of paraventricular nucleus to gastric ischemia-reperfusion injuries. Chin. Med. J. 2007, 120, 1082–1087. [Google Scholar] [CrossRef]
- Lagoutte, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 2010, 1797, 1500–1511. [Google Scholar] [CrossRef]
- Yang, M.X.; Wu, Z.Z.; Liao, X.Y.; Zhang, B.L.; Chen, X.; Wu, Y.; Lin, J.D. Remifentanil reduces multiple organ and energy metabolism disturbances in a rat sepsis model. J. Physiol. Pharmacol. 2022, 73, 81–87. [Google Scholar]
- Polishchuk, S.; Tsekhmistrenko, S.; Polishchuk, V.; Tsekhmistrenko, O.; Zdorovtseva, L.; Kotula-Balak, M.; Tarasiuk, K.; Ievstafiieva, Y.; Hutsol, T. Status of prooxidant and antioxidant systems in the sperm and seminal plasma of breeding boars of large white breed and SS23 synthetic line. J. Physiol. Pharmacol. 2022, 73, 71–79. [Google Scholar]
- Li, Q.; Zhang, W.; Xiao, E. SOD2 overexpression in bone marrow-derived mesenchymal stem cells ameliorates hepatic ischemia/reperfusion injury. Mol. Med. Rep. 2021, 24, 671. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef]
- Palma, F.R.; He, C.; Danes, J.M.; Paviani, V.; Coelho, D.R.; Gantner, B.N.; Bonini, M.G. Mitochondrial Superoxide Dismutase: What the Established, the Intriguing, and the Novel Reveal About a Key Cellular Redox Switch. Antioxid. Redox Signal. 2020, 32, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Mallick, I.H.; Yang, W.; Winslet, M.C.; Seifalian, A.M. Ischemia-reperfusion injury of the intestine and protective strategies against injury. Dig. Dis. Sci. 2004, 49, 1359–1377. [Google Scholar] [CrossRef]
- Qu, K.; Lee, S.W.; Bian, J.S.; Low, C.M.; Wong, P.T. Hydrogen sulfide: Neurochemistry and neurobiology. Neurochem. Int. 2008, 52, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Jawa, R.S.; Quist, E.; Boyer, C.W.; Shostrom, V.K.; Mercer, D.W. Mesenteric ischemia-reperfusion injury up-regulates certain CC, CXC, and XC chemokines and results in multi-organ injury in a time-dependent manner. Eur. Cytokine Netw. 2013, 24, 148–156. [Google Scholar] [CrossRef]
- Jin, X.; Zimmers, T.A.; Zhang, Z.; Pierce, R.H.; Koniaris, L.G. Interleukin-6 is an important in vivo inhibitor of intestinal epithelial cell death in mice. Gut 2010, 59, 186–196. [Google Scholar] [CrossRef]
- Jensen, A.R.; Drucker, N.A.; Te Winkel, J.P.; Ferkowicz, M.J.; Markel, T.A. The route and timing of hydrogen sulfide therapy critically impacts intestinal recovery following ischemia and reperfusion injury. J. Pediatr. Surg. 2018, 53, 1111–1117. [Google Scholar] [CrossRef]
- Henderson, P.W.; Weinstein, A.L.; Sohn, A.M.; Jimenez, N.; Krijgh, D.D.; Spector, J.A. Hydrogen sulfide attenuates intestinal ischemia-reperfusion injury when delivered in the post-ischemic period. J. Gastroenterol. Hepatol. 2010, 25, 1642–1647. [Google Scholar] [CrossRef]
- Popov, S.V.; Mukhomedzyanov, A.V.; Voronkov, N.S.; Derkachev, I.A.; Boshchenko, A.A.; Fu, F.; Sufianova, G.Z.; Khlestkina, M.S.; Maslov, L.N. Regulation of autophagy of the heart in ischemia and reperfusion. Apoptosis 2023, 28, 55–80. [Google Scholar] [CrossRef]
- Chen, Y.H.; Yao, W.Z.; Geng, B.; Ding, Y.L.; Lu, M.; Zhao, M.W.; Tang, C.S. Endogenous hydrogen sulfide in patients with COPD. Chest 2005, 128, 3205–3211. [Google Scholar] [CrossRef]
- Zhang, H.; Zhi, L.; Moochhala, S.; Moore, P.K.; Bhatia, M. Hydrogen sulfide acts as an inflammatory mediator in cecal ligation and puncture-induced sepsis in mice by upregulating the production of cytokines and chemokines via NF-kappaB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L960–L971. [Google Scholar] [CrossRef]
- Fang, L.; Li, H.; Tang, C.; Geng, B.; Qi, Y.; Liu, X. Hydrogen sulfide attenuates the pathogenesis of pulmonary fibrosis induced by bleomycin in rats. Can. J. Physiol. Pharmacol. 2009, 87, 531–538. [Google Scholar] [CrossRef]
- Rong, Y.; Fan, J.; Ji, C.; Wang, Z.; Ge, X.; Wang, J.; Ye, W.; Yin, G.; Cai, W.; Liu, W. USP11 regulates autophagy-dependent ferroptosis after spinal cord ischemia-reperfusion injury by deubiquitinating Beclin 1. Cell Death Differ. 2022, 29, 1164–1175. [Google Scholar] [CrossRef]
- Wynn, M.M.; Acher, C.W. A modern theory of spinal cord ischemia/injury in thoracoabdominal aortic surgery and its implications for prevention of paralysis. J. Cardiothorac. Vasc. Anesth. 2014, 28, 1088–1099. [Google Scholar] [CrossRef]
- Bell, M.T.; Puskas, F.; Agoston, V.A.; Cleveland, J.C., Jr.; Freeman, K.A.; Gamboni, F.; Herson, P.S.; Meng, X.; Smith, P.D.; Weyant, M.J.; et al. Toll-like receptor 4-dependent microglial activation mediates spinal cord ischemia-reperfusion injury. Circulation 2013, 128 (Suppl. S1), S152–S156. [Google Scholar] [CrossRef]
- Ege, E.; Ilhan, A.; Gurel, A.; Akyol, O.; Ozen, S. Erdosteine ameliorates neurological outcome and oxidative stress due to ischemia/reperfusion injury in rabbit spinal cord. Eur. J. Vasc. Endovasc. Surg. 2004, 28, 379–386. [Google Scholar] [CrossRef]
- Mellios, N.; Huang, H.S.; Grigorenko, A.; Rogaev, E.; Akbarian, S. A set of differentially expressed miRNAs, including miR-30a-5p, act as post-transcriptional inhibitors of BDNF in prefrontal cortex. Hum. Mol. Genet. 2008, 17, 3030–3042. [Google Scholar] [CrossRef]
- Wang, P.; Liang, J.; Li, Y.; Li, J.; Yang, X.; Zhang, X.; Han, S.; Li, S.; Li, J. Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem. Res. 2014, 39, 1279–1291. [Google Scholar] [CrossRef]
- Valente, G.; Morani, F.; Nicotra, G.; Fusco, N.; Peracchio, C.; Titone, R.; Alabiso, O.; Arisio, R.; Katsaros, D.; Benedetto, C.; et al. Expression and clinical significance of the autophagy proteins BECLIN 1 and LC3 in ovarian cancer. BioMed. Res. Int. 2014, 2014, 462658. [Google Scholar] [CrossRef]
- Hu, J.R.; Lv, G.H.; Yin, B.L. Altered microRNA expression in the ischemic-reperfusion spinal cord with atorvastatin therapy. J. Pharmacol. Sci. 2013, 121, 343–346. [Google Scholar] [CrossRef]
- Sheng, W.S.; Hu, S.; Ni, H.T.; Rowen, T.N.; Lokensgard, J.R.; Peterson, P.K. TNF-alpha-induced chemokine production and apoptosis in human neural precursor cells. J. Leukoc. Biol. 2005, 78, 1233–1241. [Google Scholar] [CrossRef]
- Talley, A.K.; Dewhurst, S.; Perry, S.W.; Dollard, S.C.; Gummuluru, S.; Fine, S.M.; New, D.; Epstein, L.G.; Gendelman, H.E.; Gelbard, H.A. Tumor necrosis factor alpha-induced apoptosis in human neuronal cells: Protection by the antioxidant N-acetylcysteine and the genes bcl-2 and crmA. Mol. Cell. Biol. 1995, 15, 2359–2366. [Google Scholar] [CrossRef]
- Yang, T.; Wu, L.; Wang, H.; Fang, J.; Yao, N.; Xu, Y. Inflammation Level after Decompression Surgery for a Rat Model of Chronic Severe Spinal Cord Compression and Effects on Ischemia-Reperfusion Injury. Neurol. Med. Chir. 2015, 55, 578–586. [Google Scholar] [CrossRef]
- Fang, B.; Wang, H.; Sun, X.J.; Li, X.Q.; Ai, C.Y.; Tan, W.F.; White, P.F.; Ma, H. Intrathecal transplantation of bone marrow stromal cells attenuates blood-spinal cord barrier disruption induced by spinal cord ischemia-reperfusion injury in rabbits. J. Vasc. Surg. 2013, 58, 1043–1052. [Google Scholar] [CrossRef]
- Vy Tran, A.H.; Hahm, S.H.; Han, S.H.; Chung, J.H.; Park, G.T.; Han, Y.S. Functional interaction between hMYH and hTRADD in the TNF-α-mediated survival and death pathways of HeLa cells. Mutat. Res. 2015, 777, 11–19. [Google Scholar] [CrossRef]
- Swarup, V.; Das, S.; Ghosh, S.; Basu, A. Tumor necrosis factor receptor-1-induced neuronal death by TRADD contributes to the pathogenesis of Japanese encephalitis. J. Neurochem. 2007, 103, 771–783. [Google Scholar] [CrossRef]
- Kokkinakis, D.M.; Liu, X.; Chada, S.; Ahmed, M.M.; Shareef, M.M.; Singha, U.K.; Yang, S.; Luo, J. Modulation of gene expression in human central nervous system tumors under methionine deprivation-induced stress. Cancer Res. 2004, 64, 7513–7525. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Jeong, M.S.; Jang, S.B. Death domain complex of the TNFR-1, TRADD, and RIP1 proteins for death-inducing signaling. Biochem. Biophys. Res. Commun. 2014, 443, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Z.; Zhang, D.; Li, H.; Sun, Z. Hydrogen sulfide protects against TNF-α induced neuronal cell apoptosis through miR-485-5p/TRADD signaling. Biochem. Biophys. Res. Commun. 2016, 478, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Nickells, R.W. Retinal ganglion cell death in glaucoma: The how, the why, and the maybe. J. Glaucoma 1996, 5, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.K.; Choi, J.S.; Ko, H.W.; Park, K.Y.; Sohn, S.; Chun, M.H.; Oh, Y.J.; Gwag, B.J. Necrosis and apoptosis after retinal ischemia: Involvement of NMDA-mediated excitotoxicity and p53. Investig. Ophthalmol. Vis. Sci. 1999, 40, 713–720. [Google Scholar]
- Tobalem, S.; Schutz, J.S.; Chronopoulos, A. Central retinal artery occlusion—Rethinking retinal survival time. BMC Ophthalmol. 2018, 18, 101. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, Y.; Machida, S.; Masuda, T.; Tazawa, Y. Correlation of retinal function with retinal histopathology following ischemia-reperfusion in rat eyes. Curr. Eye Res. 2004, 28, 381–389. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Y.; Ojwang, B.A.; Brantley, M.A., Jr.; Gidday, J.M. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: Role of HIF-1alpha and heme oxygenase-1. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1735–1743. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classify of I/R | H2S Donors (Concentration) | Effects | Mechanisms | Reference |
|---|---|---|---|---|
| Myocardial I/R | NaHS (400 μg/kg) | Cardiomyocyte apoptosis ↓ | CSE/H2S pathway ↑ | [33] |
| Oxidative damage and apoptosis in cardiomyocytes ↓ | CaR-CSE/H2S pathway ↑ | [33] | ||
| Myocardial infarction and ventricular dysfunction ↓ | CSE expression ↑ | [34] | ||
| Cell apoptosis and oxidative stress ↓ | CSE and H2S ↑ | [35] | ||
| Myocardial infarction ↓ | H2S levels ↑ | [36] | ||
| Cardiomyocyte apoptosis ↓ | H2S levels ↑ | [36] | ||
| Myocardial infarction area and restore left ventricular function ↓ | cGMP/PKG pathway and H2S levels ↑ | [37] | ||
| Myocardial injury and enhancing cardiac function in rats ↓ | Oprm1/miR-30b-5p/CSE/H2S axis ↑ | [38] | ||
| Cardiomyocyte apoptosis ↓ | Oprm1/miR-30b-5p/CSE/H2S axis ↑ | [38] | ||
| NaHS (40 μM) | Myocardial injury ↓ | CSE expression ↑ | [39] | |
| NaHS (3 mg/kg) | Cardiomyocyte apoptosis ↓ | KATP pathway ↑, MAPK and NF-κB pathway ↓ | [40] | |
| NaHS (40 μM) | Heart function ↑ | KATP pathway ↑ | [41] | |
| NaHS (1.4, 2.8, 14 μM/kg) | Myocardial infarction area and restore left ventricular function ↓ | LDH, CK, CK-MB ↓ | [42] | |
| NaHS (100–400 μM) | Cardiomyocyte apoptosis ↓ | GRP78, CHOP, eIF2α ↓ | [42] | |
| H2S (50–200 μM) | Cardiomyocyte apoptosis and ER/SR stress ↓ | miR-133a expression ↑ | [51] | |
| ADT (50 mg/kg) | Myocardial infarction area and restore autophagy ↓ | AMPK pathway ↑ | [43] | |
| Zofenopril (10 μg/kg) | Myocardial infarction area ↓ | H2S and NO ↑ | [15] | |
| DATS | Myocardial infarction area ↓ | eNOS/NO ↑ | [48] | |
| NaHS (100 μg/kg) | Myocardial infarction area ↓ | cGMP/PKG/PLN ↑ | [49] | |
| NaHS (14, 28 μM/kg) | Myocardial infarction and ventricular dysfunction ↓ | c-Fos expression ↓ | [50] | |
| Na2S (100 μg/kg) | Myocardial infarction and ventricular dysfunction ↓ | CK-MB and FABP expression ↓ | [52] | |
| NaHS (30 μM/kg) | Cardiomyocyte apoptosis ↓ | miR-1 expression ↓ | [53] | |
| NaHS (30 μM) | Cardiomyocyte apoptosis ↓ | miR-1 expression ↓ | [53] | |
| NaHS (10 μM) | Myocardial infarction area ↓ | Sirt1/PGC1α pathway ↑ | [54] | |
| NaHS (10 μM) | Myocardial infarction area and restore left ventricular function ↓ | JAK2/STAT3 pathway ↑ | [55] | |
| GYY4137 (12.5, 25, 50 mg/kg) | Oxidative damage and apoptosis in cardiomyocytes ↓ | MAPK pathway ↓ | [56] | |
| NaHS | Cardiomyocyte and mitochondrial membrane damage ↓ | JNK pathway ↓ | [57] | |
| NaHS (1, 10, 20 μM) | Myocardial infarction area and restore left ventricular function ↓ | LDH, CK ↓ | [58] | |
| Na2S (50 μg/kg) | Myocardial infarction area and restore left ventricular function ↓ | mitochondrial dysfunction ↓ | [59] | |
| NaHS (50 μM) | Oxidative damage ↓ | mitochondrial complex IV expression ↓ | [60] | |
| NaHS (0.122–250 μM) | Cardiomyocyte apoptosis ↓ | CK2 expression ↓ | [61] | |
| NaHS (30 μM/kg) | Cardiomyocyte apoptosis ↓ | GSK-3β (Ser9) expression ↑ | [62] | |
| NaHS (30 μM) | Cardiomyocyte apoptosis ↓ | GSK-3β (Ser9) expression ↑ | [62] | |
| NaHS (50 μM) | Myocardial infarction area ↓ | AKT pathway ↑ | [63] | |
| NaHS (50 μM) | Cardiomyocyte apoptosis ↓ | AKT pathway ↑ | [63] | |
| NaHS (30–100 μM/Kg) | Oxidative damage ↓ | mTOR pathway ↑ | [64] | |
| NaHS (10–100 μM) | Oxidative damage ↓ | mTOR pathway ↑ | [64] | |
| NaHS (30 μM) | Autophagy in cells ↓ | PI3K/SGK1/GSK3β pathway ↑ | [65] | |
| DATS (200 μg/kg) | Myocardial infarction area and restore left ventricular function ↓ | eNOS/NO ↑ | [66] | |
| DATS-MSN (2, 4 mg/kg) | Cardiomyocyte apoptosis and oxidative damage ↓ | [13] | ||
| DATS-MSN (2.2 μM) | Myocardial infarction area and restore left ventricular function ↓ | TLR4/NLRP3 pathway ↓ | [26] | |
| Na2S (1 μM/kg), GYY4137 (25 μM/kg), AP39 (0.25 μM/kg) | Myocardial infarction area and restore left ventricular function ↓ | H2S and NO levels ↑ | [70] | |
| NaHS (10 μM) | Myocardial tissue injury, cardiomyocyte apoptosis ↓ and cardiac function ↑ | PI3K/Akt/GSK-3β pathway ↑ | [71] | |
| NaHS (10 μM) | Myocardial tissue injury, cardiomyocyte apoptosis ↑ and cardiac function ↑ | AMPK/mTOR pathway ↑ | [72] | |
| NaHS (100 μM) | Cardiomyocyte apoptosis and oxidative damage ↓ | HB-EGF/EGFR ↑ | [73] | |
| Cerebral I/R | NaHS (1, 5 mg/kg) | Cerebral oedema and cell apoptosis ↑ | [74] | |
| NaHS (50 μM/kg) | Neuronal pyroptosis and inflammation ↓ | IL-1β, IL-18, NLRP3/caspase-1/GSDMD pathway ↓ | [75] | |
| H2S (40 ppm) | Cell apoptosis ↓ | HSP70 expression ↑, PI3K/Akt/Nrf2 pathway ↓ | [76] | |
| GYY4137 (5 mM) | Cerebral infarct size and cell apoptosis ↑ | p38 MAPK, ERK1/2 and JNK pathway ↓ | [77] | |
| NaHS (4.8 mg/kg) | Restore neural function | RhoA/ROCK2 pathway ↓ | [78] | |
| NaHS (0.2, 0.4 mM/kg) | Oxidative stress, inflammation and cell apoptosis ↓ | [79] | ||
| NaHS (0.1–10 mM/kg) | MCA relaxation ↓ | KATP pathway ↑ | [80] | |
| Post-ischemic inflammation ↓ | CSE/H2S pathway ↑ | [81] | ||
| NaHS (1.25, 2.5, 5 mg/kg) | Cerebral infarct size and cell apoptosis ↓ | MDA expression ↓, SOD and GSH-Px expression ↑ | [82] | |
| NaHS (0–250 μM) | Cell apoptosis ↓ | PARP-1/AIF pathway ↓ | [83] | |
| NaHS (1, 2, 4, 8, 16 mg/kg) | Infarct volume and neurological deficits ↓ | LC3-II expression ↓, p62 expression ↑ | [84] | |
| NaHS (1, 2, 4 mg/kg) | Infarct volume ↓ | LC3-II expression ↓, p62 expression ↑ | [85] | |
| NaHS (200 μM) | Autophagy ↓ | LC3-II expression ↓, p62 expression ↑ | [85] | |
| NaHS (5.6 mg/kg) | Infarct volume and neurological deficits ↓ | LDH, capase 3, LC3-II ↓, p62 ↑ | [86] | |
| NaHS (100 μM) | Cell apoptosis ↓ | LC3-II expression ↓, p62 expression ↑ | [86] | |
| NaHS (12.5–200 μM) | Diastolic blood vessels | CSE, 3-MST, H2S ↑, RhoA-ROCK ↓ | [87] | |
| NaHS (50–200 μM) | Diastolic blood vessels | RhoA-ROCK ↓ | [88] | |
| H2S (40, 80 ppm) | Infarct volume and neurological deficits ↓ | PKC expression ↑, AQP4 expression ↓ | [89] | |
| Na2S (25–100 mM) | Cell death ↓ | SIRT6 expression ↑ | [90] | |
| Hepatic I/R | NaHS (20 μM/kg) | Liver damage ↓ | Nrf2 expression ↑, NQO1, GST, HO-1, miR-34a expression ↓ | [91] |
| NaHS (14, 28 μM/kg) | Liver damage ↓ | JNK1 pathway ↓ | [92] | |
| NaHS (1, 3, 5, 7, 9 μM) | Cell apoptosis ↓ | BCl2 ↑, Bax, JNK1 pathway ↓ | [92] | |
| GYY4137 (133 μM/kg) | Liver damage and cell apoptosis ↓ | AKT pathway and miR-21 expression ↑ | [93] | |
| NaHS (20 μM) | Cell apoptosis ↓ | AKT pathway and miR-21 expression ↑ | [93] | |
| H2S-Exo | Liver function ↑ | [94] | ||
| NaHS (56 μM/kg) | Liver damage and cell apoptosis ↓ | ATF6, PERK, GRP78, TRAF2, CHOP ↓ | [95] | |
| NaHS (30 μM) | Liver damage and inflammation ↓ | SPHK1/S1P pathway ↓ | [96] | |
| NaHS (12.5, 25, 50 μM/kg) | The mitochondrial and hepatocellular injury ↓ | Akt-GSK-3β pathway ↑ | [97] | |
| Renal I/R | GYY4137 (0.25 mg/kg) | Kidney function ↑ | miR-21 expression ↓ | [98] |
| NaHS (500 μg/kg) | Kidney injury ↓ and renal function ↑ | [99] | ||
| Kidney injury ↓ | CSE promoter methylation ↓, H2S ↑ | [100] | ||
| Kidney injury ↓ and renal function ↑ | CSE/H2S pathway ↑ | [101] | ||
| Na2S | Kidney injury ↓ | IL-6, IL-1β, nitrite, nitrate ↓ | [102] | |
| NaHS (150 μM) | Liver damage and cell apoptosis ↓ | HSP70, HO-1, HSP27 expression ↑ | [103] | |
| NaHS (500 μg/kg) | Kidney injury ↓ | NLRP3/Caspase-1 pathway ↓ | [104] | |
| NaHS (100 μM) | Liver damage and cell apoptosis ↓ | NLRP3/Caspase-1 pathway ↓ | [104] | |
| Gastric I/R | NaHS (160, 320, 640 mg/kg) | Gastric mucosal damage ↓ | CSE expression ↑ | [105] |
| ATB-344 (7 mg/kg) | Gastric mucosal damage ↓ | Anti-inflammation | [16] | |
| NaHS (30–300 μM) | Gastric mucosal damage ↓ | Keap1 s-sulfhydration, MAPK pathway ↑, NF-KB pathway ↓ | [106] | |
| NaHS, GYY4137 | Gastric mucosal damage ↓ | SOD-2, GPx-1 expression ↑ | [107] | |
| AP39 | Gastric mucosal damage ↓ | HIF-1α, GPx-1, ATP production, mitochondria complex I, complex III, GST-α, p-AKT and p-mTOR ↑, 8-OHG, complex IV, TIMP1, IL-1β, IL-1R2, iNOS, p-NF-κB, p-ERK ↓ | [108] | |
| Intestinal I/R | NaHS (7, 14 μM/kg) | Intestine injury ↓ | SOD and GSH-Px ↑, MDA ↓ | [109] |
| NaHS (2 μM/kg) | Intestine injury ↓ | eNOS/NO ↑ | [110] | |
| Intestine oxidative injury ↓ | CSE/H2S ↑, iNOS/NO ↓ | [111] | ||
| NaHS (14 μM/kg) | Intestine oxidative injury ↓ | BK pathway ↓ | [112] | |
| GYY4137 (50 mg/kg) | Intestinal mucosal injury ↓ | IL-6, IP-10, MIP-2 ↓ | [110] | |
| NaHS (0.5 mg/kg) | Intestine oxidative injury ↓ | HIF-1α expression ↑ | [113] | |
| Lung I/R | H2S (50, 100 μM) | Lung oxidative injury ↓ | CSE/H2S pathway ↑ | [114] |
| NaHS (0.78 mg/kg) | Lung injury ↓ | AQP1/AQP5 ↑, TLR4/Myd88/NFκB ↓ | [115] | |
| Spinal cord I/R | NaHS (30 μM/kg) | Spinal cord infarct zone ↓ and hind motor function ↑ | miR-30c expression ↓ | [116] |
| NaSH (10, 100, 200 μM) | Cellular ischemic injury ↑ | Beclin-1 and LC3II ↑, miR-30c ↓ | [116] | |
| Testicular I/R | NaHS (75 μM/kg) | Testicular torsion ↓ | TNF-α, iNOS, MDA, NO, GSH, and SOD ↓ | [108] |
| NaHS (10 mg/kg) | Testicular torsion ↓ | GSH ↑, MPO and AOPP ↓ | [107] | |
| Retinal I/R | H2S (80 ppm) | Neuronal injuries ↓ | HSP-90α ↑, NF-κB, caspase-3, p-JNK ↓ | [117] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Wu, S.; Mao, C.; Qu, Y.; Xu, Z.; Xie, Y.; Jiang, D.; Song, Y. Therapeutic Potential of Hydrogen Sulfide in Ischemia and Reperfusion Injury. Biomolecules 2024, 14, 740. https://doi.org/10.3390/biom14070740
Sun X, Wu S, Mao C, Qu Y, Xu Z, Xie Y, Jiang D, Song Y. Therapeutic Potential of Hydrogen Sulfide in Ischemia and Reperfusion Injury. Biomolecules. 2024; 14(7):740. https://doi.org/10.3390/biom14070740
Chicago/Turabian StyleSun, Xutao, Siyu Wu, Caiyun Mao, Ying Qu, Zihang Xu, Ying Xie, Deyou Jiang, and Yunjia Song. 2024. "Therapeutic Potential of Hydrogen Sulfide in Ischemia and Reperfusion Injury" Biomolecules 14, no. 7: 740. https://doi.org/10.3390/biom14070740