
Expression Proteomics and Histone Analysis Reveal Extensive Chromatin Network Changes and a Role for Histone Tail Trimming during Cellular Differentiation


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Differentiation of NTERA Cells
2.2. Real-Time Quantitative PCR
2.3. Cell Viability Assay
2.4. Immunoblotting
2.5. Cell Fractionation
2.6. Sample Preparation for Mass Spectrometry
2.7. Mass Spectrometry Analysis
2.8. MS Proteome Data Analysis
2.9. Quantitative Analysis
2.10. Histone PTM Analysis
2.11. Histone Data Analysis
2.12. NanoLCMS/MS for Histone Peptide Analysis
2.13. Histone Peptide Pull-Down Assays
3. Results
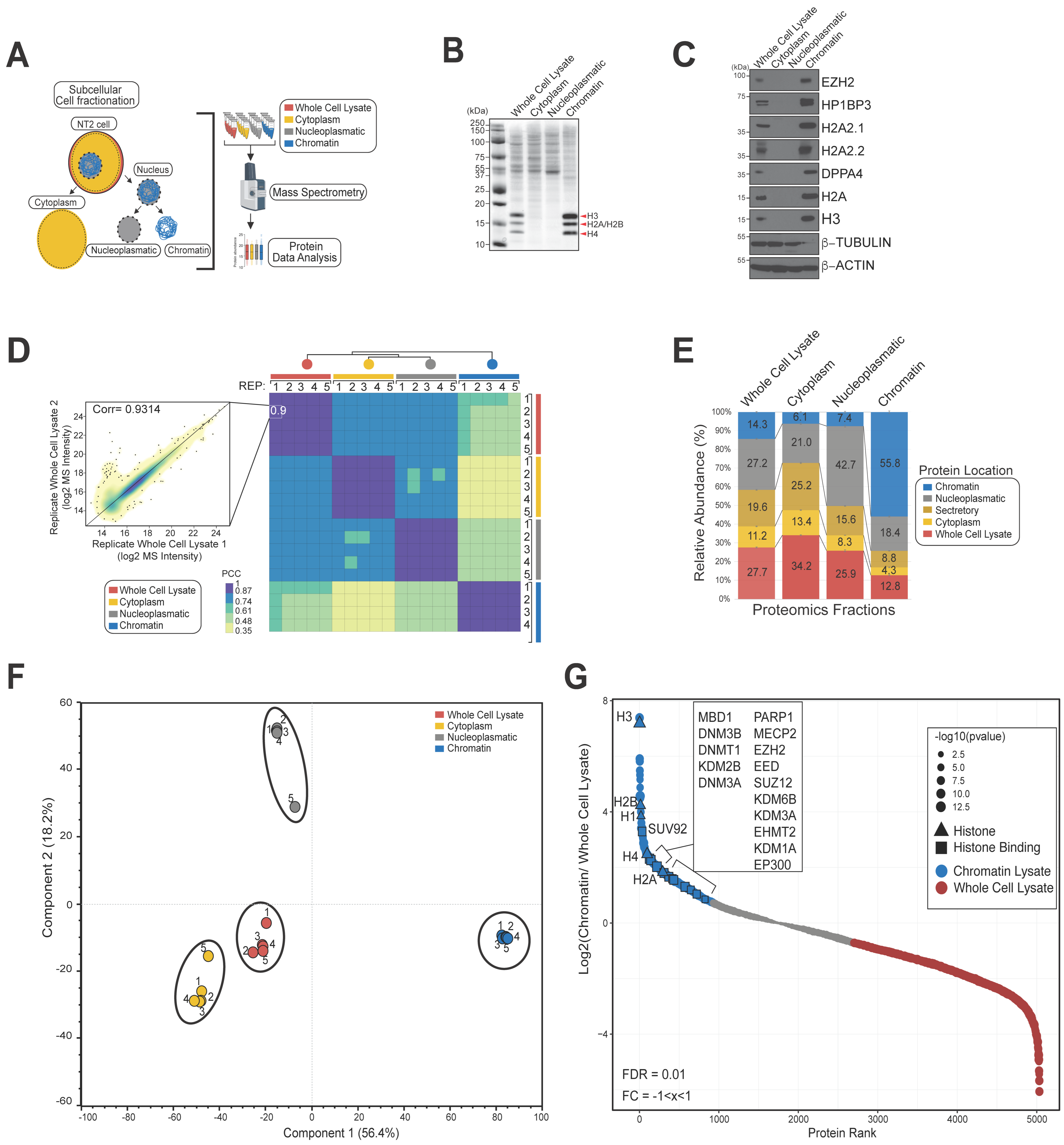
3.1. Expression Proteomics of Biochemically Separated Subcellular Fractions from a Model of Pluripotent Cell Differentiation
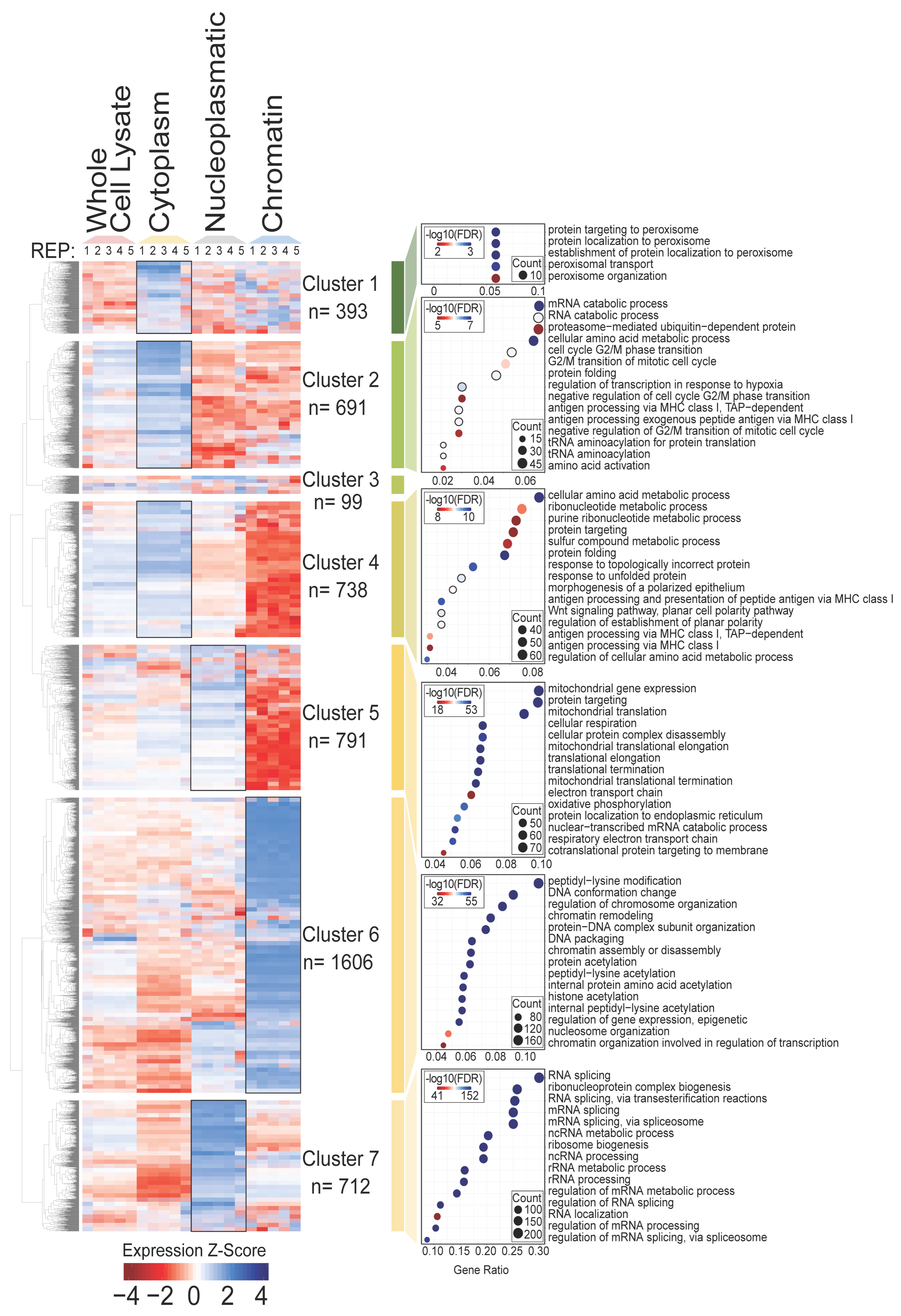
3.2. Molecular Functions Associated with Distinct Subcellular Fractions
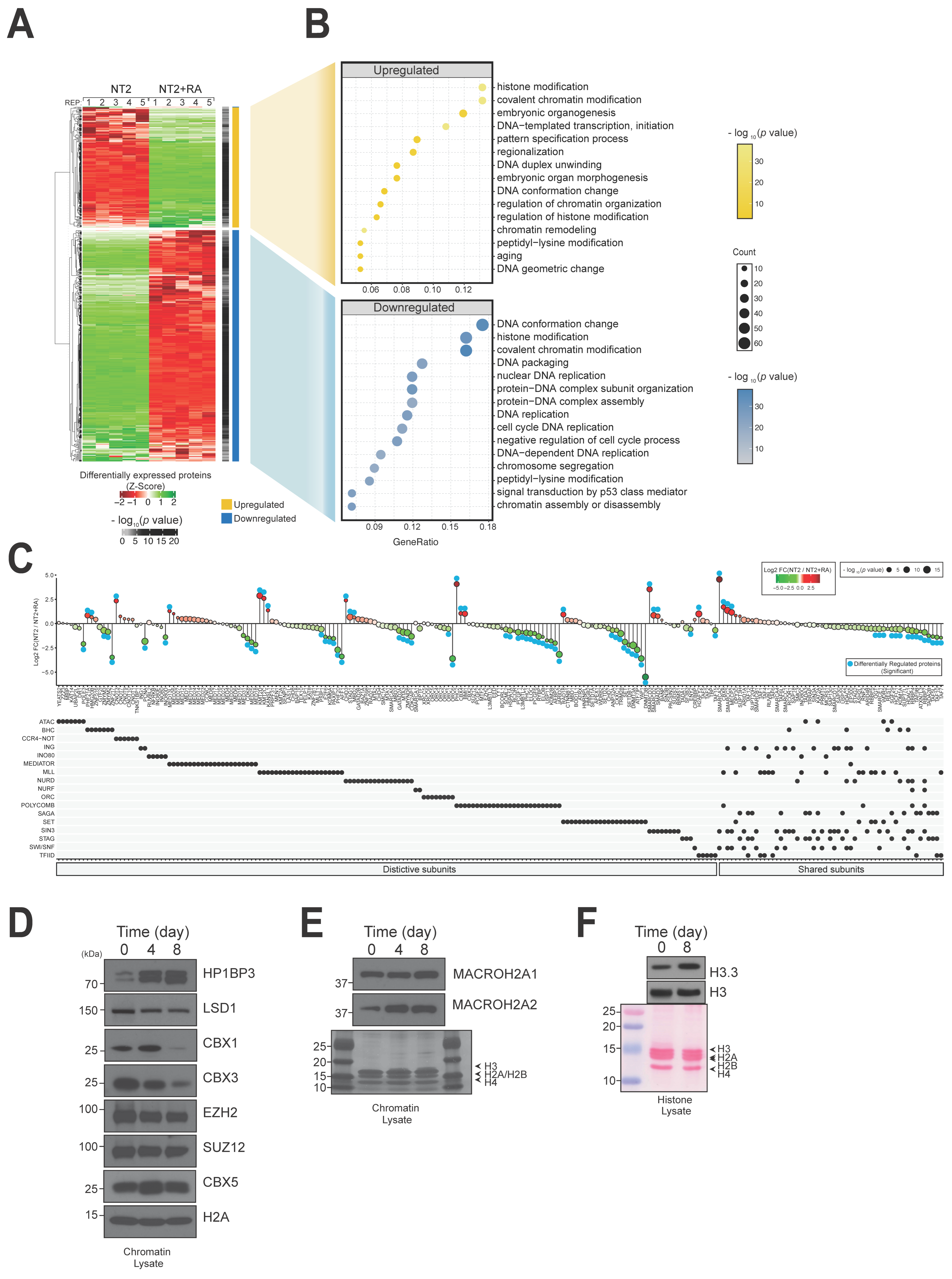
3.3. Chromatin Proteome Changes Induced by Differentiation
3.4. The NT2 Cell Differentiation Programme Centres on Processes Involving Chromatin Modification Pathways
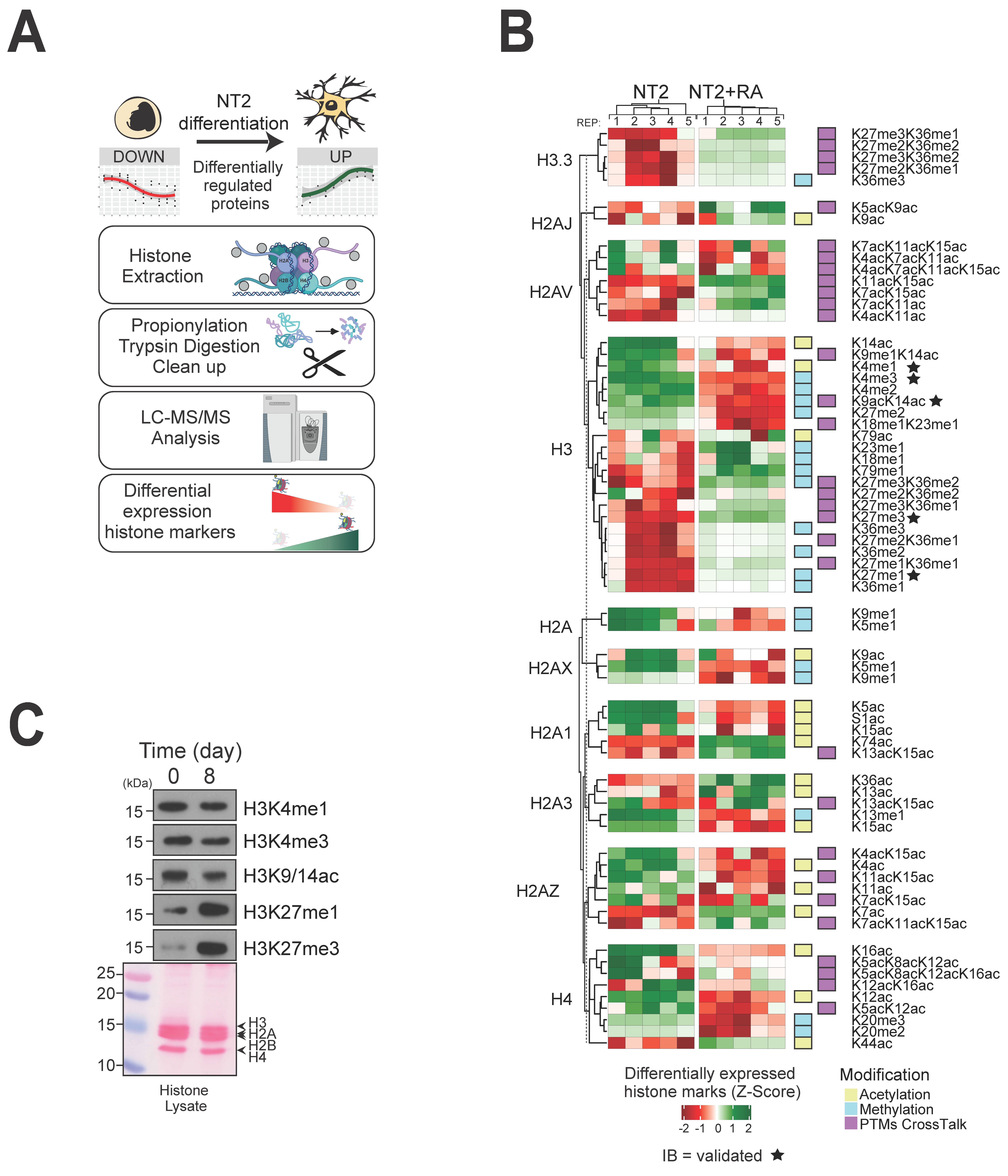
3.5. Analysis of Histone Post-Translational Modification Changes during Differentiation
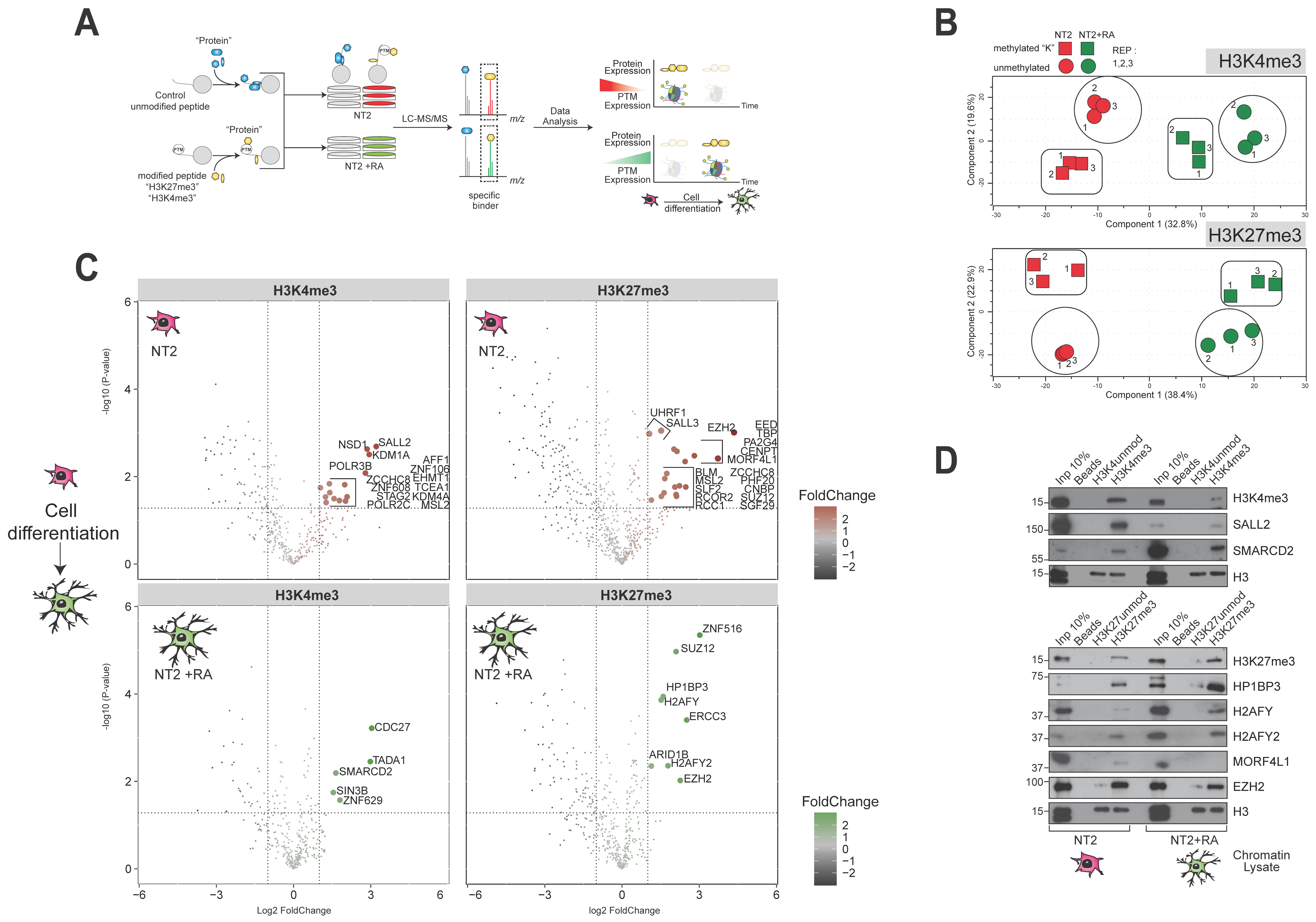
3.6. Histone Post-Translational Marks Associate with Distinct Protein Subsets during the Pluripotent and Differentiated States
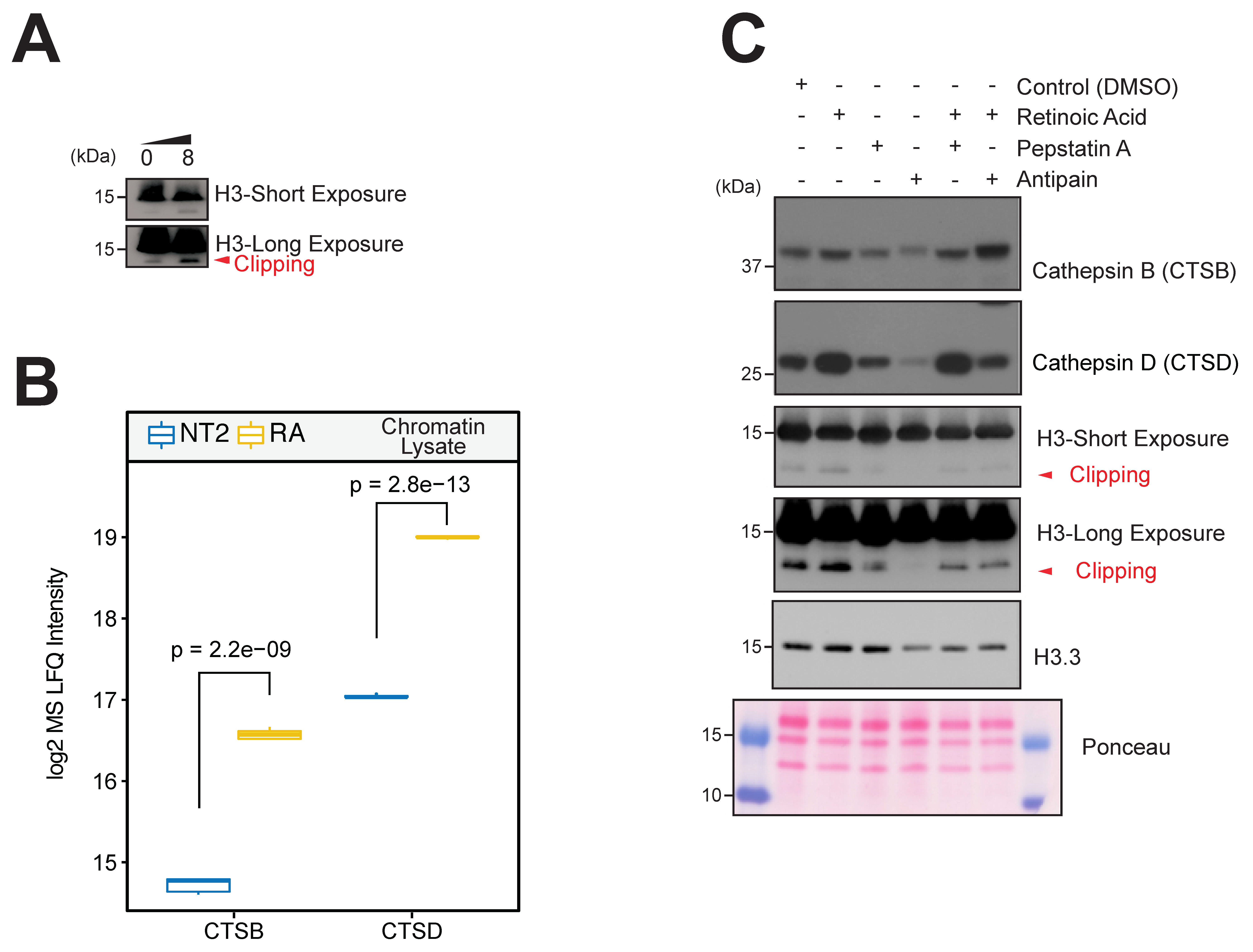
3.7. Histone Variant and Histone Proteolysis Changes Induced by Differentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, S.A. Cell differentiation: What have we learned in 50 years? J. Theor. Biol. 2020, 485, 110031. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef] [PubMed]
- Richly, H.; Rocha-Viegas, L.; Ribeiro, J.D.; Demajo, S.; Gundem, G.; Lopez-Bigas, N.; Nakagawa, T.; Rospert, S.; Ito, T.; Di Croce, L. Transcriptional activation of polycomb-repressed genes by ZRF1. Nature 2010, 468, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, N.; Lerdrup, M.; Landt, E.; Agrawal-Singh, S.; Bak, M.; Tommerup, N.; Rappsilber, J.; Södersten, E.; Hansen, K. REST-mediated recruitment of polycomb repressor complexes in mammalian cells. PLoS Genet. 2012, 8, e1002494. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Andrews, P.W. Differentiation of NTERA-2 clonal human embryonal carcinoma cells into neurons involves the induction of all three neurofilament proteins. J. Neurosci. 1986, 6, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Tegenge, M.A.; Roloff, F.; Bicker, G. Rapid differentiation of human embryonal carcinoma stem cells (NT2) into neurons for neurite outgrowth analysis. Cell Mol. Neurobiol. 2011, 31, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Ghyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J. Retinoids induce stem cell differentiation via epigenetic changes. Semin. Cell Dev. Biol. 2013, 24, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- De, S.; Mitra, A.; Cheng, Y.; Pfeifer, K.; Kassis, J.A. Formation of a Polycomb-Domain in the Absence of Strong Polycomb Response Elements. PLoS Genet. 2016, 12, e1006200. [Google Scholar] [CrossRef]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2014, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.A.; Marcum, R.D.; He, Y. Structure and Function of Chromatin Remodelers. J. Mol. Biol. 2021, 433, 166929. [Google Scholar] [CrossRef] [PubMed]
- Yakushiji-Kaminatsui, N.; Lopez-Delisle, L.; Bolt, C.C.; Andrey, G.; Beccari, L.; Duboule, D. Similarities and differences in the regulation of HoxD genes during chick and mouse limb development. PLoS Biol. 2018, 16, e3000004. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kumar, S.; Duester, G. Retinoic acid controls body axis extension by directly repressing Fgf8 transcription. Development 2014, 141, 2972–2977. [Google Scholar] [CrossRef] [PubMed]
- Rampalli, S.; Li, L.; Mak, E.; Ge, K.; Brand, M.; Tapscott, S.J.; Dilworth, F.J. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 2007, 14, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.K.; Stadler, M.B.; Wirbelauer, C.; Paro, R.; Schübeler, D.; Beisel, C. A chromatin-modifying function of JNK during stem cell differentiation. Nat. Genet. 2011, 44, 94–100. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Voss, T.C.; Hager, G.L. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 2014, 15, 69–81. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef]
- Streubel, G.; Watson, A.; Jammula, S.G.; Scelfo, A.; Fitzpatrick, D.J.; Oliviero, G.; McCole, R.; Conway, E.; Glancy, E.; Negri, G.L.; et al. The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol. Cell. 2018, 70, 371–379.e5. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Plazas-Mayorca, M.D.; Zee, B.M.; Young, N.L.; Fingerman, I.M.; LeRoy, G.; Briggs, S.D.; Garcia, B.A. One-pot shotgun quantitative mass spectrometry characterization of histones. J. Proteome Res. 2009, 8, 5367–5374. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yuan, Z.F.; Sidoli, S.; Marchione, D.M.; Simithy, J.; Janssen, K.A.; Szurgot, M.R.; Garcia, B.A. EpiProfile 2.0: A Computational Platform for Processing Epi-Proteomics Mass Spectrometry Data. J. Proteome Res. 2018, 17, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Gretarsson, K.H.; Hackett, J.A. Dppa2 and Dppa4 counteract de novo methylation to establish a permissive epigenome for development. Nat. Struct. Mol. Biol. 2020, 27, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.C.; Lemons, D.; McGinnis, W. Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 2005, 6, 893–904. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, M.J.; Erickson, S.L.; Kopp, D.; Wen, P.; Aghanoori, M.R.; Kedia, S.; Burns, K.M.L.; Vitobello, A.; Tran Mau-Them, F.; Thomas, Q.; et al. Nucleocytoplasmic transport of the RNA-binding protein CELF2 regulates neural stem cell fates. Cell Rep. 2021, 35, 109226. [Google Scholar] [CrossRef] [PubMed]
- de Lichtenberg, U.; Jensen, L.J.; Brunak, S.; Bork, P. Dynamic complex formation during the yeast cell cycle. Science 2005, 307, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Chalabi Hagkarim, N.; Grand, R.J. The Regulatory Properties of the Ccr4-Not Complex. Cells 2020, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pliatska, M.; Kapasa, M.; Kokkalis, A.; Polyzos, A.; Thanos, D. The Histone Variant MacroH2A Blocks Cellular Reprogramming by Inhibiting Mesenchymal-to-Epithelial Transition. Mol. Cell Biol. 2018, 38, e00669-17. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ha, M.; Kraushaar, D.C.; Zhao, K. Genome-wide analysis of H3.3 dissociation reveals high nucleosome turnover at distal regulatory regions of embryonic stem cells. Epigenet. Chromatin 2014, 7, 38. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Garcia, B.A.; Mollah, S.; Ueberheide, B.M.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Azad, G.K.; Swagatika, S.; Kumawat, M.; Kumawat, R.; Tomar, R.S. Modifying Chromatin by Histone Tail Clipping. J. Mol. Biol. 2018, 430, 3051–3067. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.M.; Muratore-Schroeder, T.L.; Cook, R.G.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Allis, C.D. Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 2008, 135, 284–294. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hurrell, T.; Segeritz, C.P.; Vallier, L.; Lilley, K.S.; Cromarty, A.D. A proteomic time course through the differentiation of human induced pluripotent stem cells into hepatocyte-like cells. Sci. Rep. 2019, 9, 3270. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Loo, L.S.W.; Vethe, H.; Soetedjo, A.A.P.; Paulo, J.A.; Jasmen, J.; Jackson, N.; Bjørlykke, Y.; Valdez, I.A.; Vaudel, M.; Barsnes, H.; et al. Dynamic proteome profiling of human pluripotent stem cell-derived pancreatic progenitors. Stem Cells 2020, 38, 542–555. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Varderidou-Minasian, S.; Verheijen, B.M.; Schätzle, P.; Hoogenraad, C.C.; Pasterkamp, R.J.; Altelaar, M. Deciphering the Proteome Dynamics during Development of Neurons Derived from Induced Pluripotent Stem Cells. J. Proteome Res. 2020, 19, 2391–2403. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sabatier, P.; Beusch, C.M.; Saei, A.A.; Aoun, M.; Moruzzi, N.; Coelho, A.; Leijten, N.; Nordenskjöld, M.; Micke, P.; Maltseva, D.; et al. An integrative proteomics method identifies a regulator of translation during stem cell maintenance and differentiation. Nat. Commun. 2021, 12, 6558. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.J.; et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J. Biol. Chem. 2017, 292, 14456–14472. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hermosilla, V.E.; Hepp, M.I.; Escobar, D.; Farkas, C.; Riffo, E.N.; Castro, A.F.; Pincheira, R. Developmental SALL2 transcription factor: A new player in cancer. Carcinogenesis 2017, 38, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cheng, K.; Shi, L.; Li, Z.; Negi, H.; Gao, G.; Kamle, S.; Li, D. Sal-like protein 2 upregulates p16 expression through a proximal promoter element. Cancer Sci. 2015, 106, 253–261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, D.; Tian, Y.; Ma, Y.; Benjamin, T. p150(Sal2) is a p53-independent regulator of p21(WAF1/CIP). Mol. Cell. Biol. 2004, 24, 3885–3893. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Álvarez, C.; Quiroz, A.; Benítez-Riquelme, D.; Riffo, E.; Castro, A.F.; Pincheira, R. SALL Proteins; Common and Antagonistic Roles in Cancer. Cancers 2021, 13, 6292. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bainor, A.J.; Saini, S.; Calderon, A.; Casado-Polanco, R.; Giner-Ramirez, B.; Moncada, C.; Cantor, D.J.; Ernlund, A.; Litovchick, L.; David, G. The HDAC-Associated Sin3B Protein Represses DREAM Complex Targets and Cooperates with APC/C to Promote Quiescence. Cell Rep. 2018, 25, 2797–2807.e8. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Islam, A.; Turner, E.L.; Menzel, J.; Malo, M.E.; Harkness, T.A. Antagonistic Gcn5-Hda1 interactions revealed by mutations to the Anaphase Promoting Complex in yeast. Cell Div. 2011, 6, 13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salifou, K.; Ray, S.; Verrier, L.; Aguirrebengoa, M.; Trouche, D.; Panov, K.I.; Vandromme, M. The histone demethylase JMJD2A/KDM4A links ribosomal RNA transcription to nutrients and growth factors availability. Nat. Commun. 2016, 7, 10174. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, W.; Liu, W.; Jia, L.; Chen, D.; Chang, I.; Lake, M.; Bentolila, L.A.; Wang, C.Y. Targeting KDM4A epigenetically activates tumor-cell-intrinsic immunity by inducing DNA replication stress. Mol. Cell. 2021, 81, 2148–2165.e9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Han, X.; Gui, B.; Xiong, C.; Zhao, L.; Liang, J.; Sun, L.; Yang, X.; Yu, W.; Si, W.; Yan, R.; et al. Destabilizing LSD1 by Jade-2 promotes neurogenesis: An antibraking system in neural development. Mol. Cell. 2014, 55, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Baksh, S.S.; Pratt, R.E.; Gomez, J.; Dzau, V.J.; Hodgkinson, C.P. A novel Cbx1, PurB, and Sp3 complex mediates long-term silencing of tissue- and lineage-specific genes. J. Biol. Chem. 2022, 298, 102053. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Yu, J.; Zhang, C.; Alejo, S.; Hoang, N.; Sun, H.; Lu, F.; Zhang, H. Methylated DNMT1 and E2F1 are targeted for proteolysis by L3MBTL3 and CRL4DCAF5 ubiquitin ligase. Nat. Commun. 2018, 9, 1641. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, T.; Park, S.S.; Giaimo, B.D.; Hall, D.; Ferrante, F.; Ho, D.M.; Hori, K.; Anhezini, L.; Ertl, I.; Bartkuhn, M.; et al. RBPJ/CBF1 interacts with L3MBTL3/MBT1 to promote repression of Notch signaling via histone demethylase KDM1A/LSD1. EMBO J. 2017, 36, 3232–3249. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mishra, S.; Van Rechem, C.; Pal, S.; Clarke, T.L.; Chakraborty, D.; Mahan, S.D.; Black, J.C.; Murphy, S.E.; Lawrence, M.S.; Daniels, D.L.; et al. Cross-Talk between Lysine-Modifying Enzymes Controls Site-Specific DNA Amplifications. Cell 2018, 174, 803–817.e16, Erratum in Cell 2018, 175, 1716. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sankar, A.; Lerdrup, M.; Manaf, A.; Johansen, J.V.; Gonzalez, J.M.; Borup, R.; Blanshard, R.; Klungland, A.; Hansen, K.; Andersen, C.Y.; et al. KDM4A regulates the maternal-to-zygotic transition by protecting broad H3K4me3 domains from H3K9me3 invasion in oocytes. Nat. Cell Biol. 2020, 22, 380–388. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fang, Y.; Tang, Y.; Zhang, Y.; Pan, Y.; Jia, J.; Sun, Z.; Zeng, W.; Chen, J.; Yuan, Y.; Fang, D. The H3K36me2 methyltransferase NSD1 modulates H3K27ac at active enhancers to safeguard gene expression. Nucleic Acids Res. 2021, 49, 6281–6295. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kunowska, N.; Rotival, M.; Yu, L.; Choudhary, J.; Dillon, N. Identification of protein complexes that bind to histone H3 combinatorial modifications using super-SILAC and weighted correlation network analysis. Nucleic Acids Res. 2015, 43, 1418–1432. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliviero, G.; Wynne, K.; Andrews, D.; Crean, J.; Kolch, W.; Cagney, G. Expression Proteomics and Histone Analysis Reveal Extensive Chromatin Network Changes and a Role for Histone Tail Trimming during Cellular Differentiation. Biomolecules 2024, 14, 747. https://doi.org/10.3390/biom14070747
Oliviero G, Wynne K, Andrews D, Crean J, Kolch W, Cagney G. Expression Proteomics and Histone Analysis Reveal Extensive Chromatin Network Changes and a Role for Histone Tail Trimming during Cellular Differentiation. Biomolecules. 2024; 14(7):747. https://doi.org/10.3390/biom14070747
Chicago/Turabian StyleOliviero, Giorgio, Kieran Wynne, Darrell Andrews, John Crean, Walter Kolch, and Gerard Cagney. 2024. "Expression Proteomics and Histone Analysis Reveal Extensive Chromatin Network Changes and a Role for Histone Tail Trimming during Cellular Differentiation" Biomolecules 14, no. 7: 747. https://doi.org/10.3390/biom14070747
APA StyleOliviero, G., Wynne, K., Andrews, D., Crean, J., Kolch, W., & Cagney, G. (2024). Expression Proteomics and Histone Analysis Reveal Extensive Chromatin Network Changes and a Role for Histone Tail Trimming during Cellular Differentiation. Biomolecules, 14(7), 747. https://doi.org/10.3390/biom14070747