

The Role of Hyperuricemia in Cardiac Diseases: Evidence, Controversies, and Therapeutic Strategies
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Current Status of Clinical Research on HUA and Myocardial Injury-Related Diseases
2.1. HUA Is Involved in MI Development and Prognosis
2.2. HUA Increases the Risk of Atrial Fibrillation (AF)
2.3. HUA Is Associated with the Onset and Outcome of HF
2.4. Controversies
3. Potential Mechanisms of HUA in Myocardial Injury
3.1. Direct Effects of UA on the Myocardium
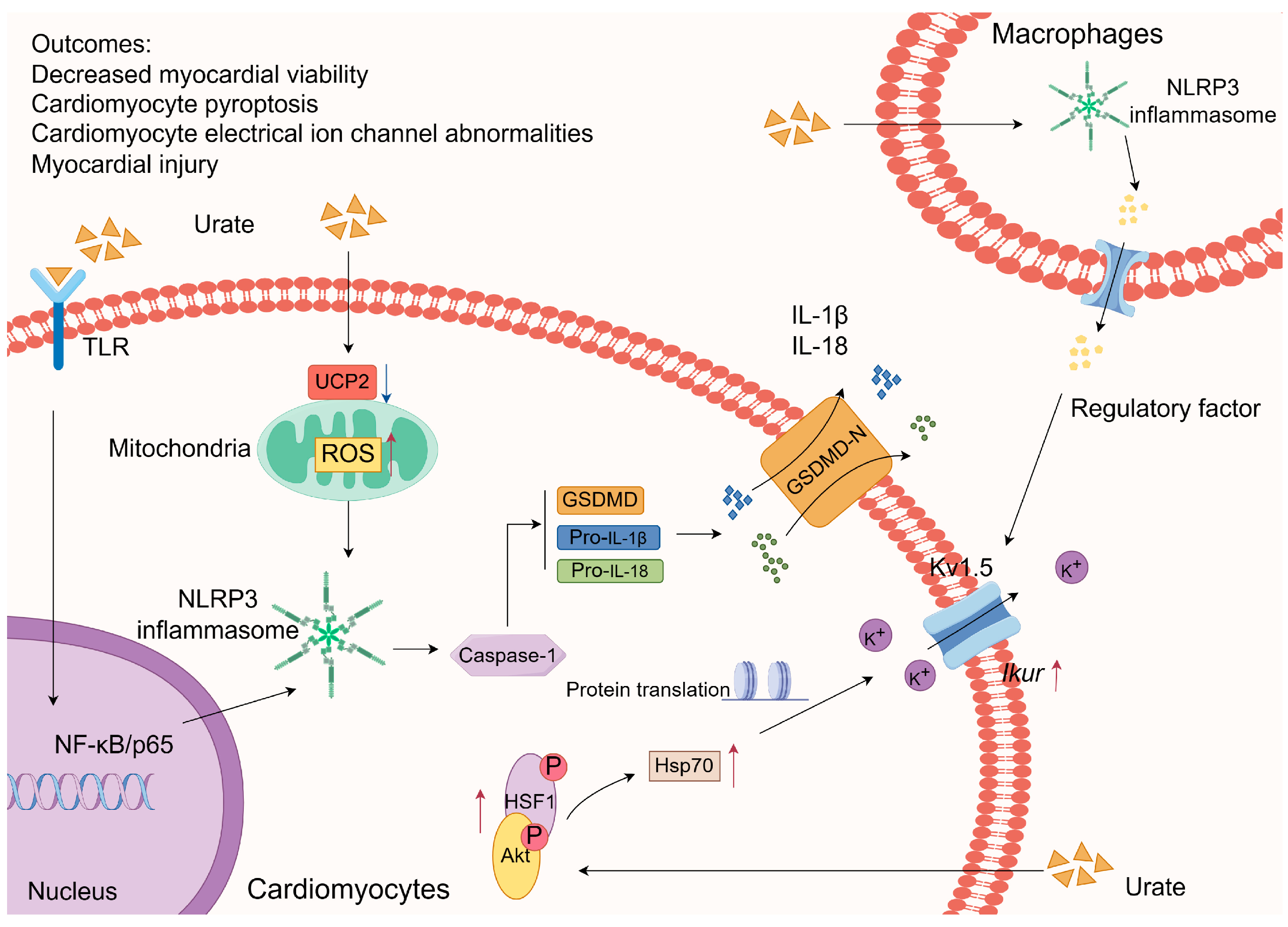
3.1.1. Inflammasome Activation
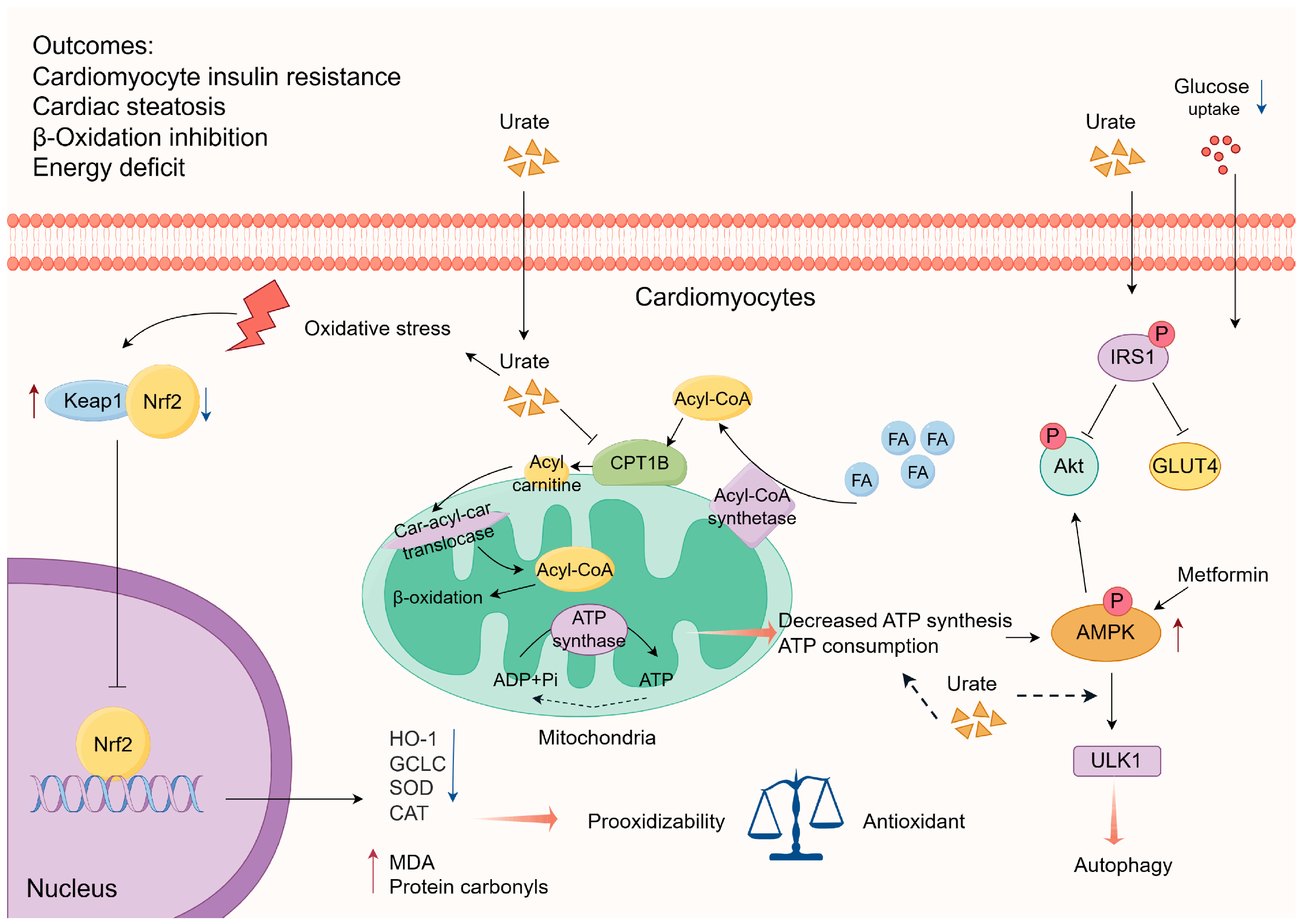
3.1.2. Interference with Energy Metabolism
3.1.3. Weakened Antioxidative Capacity
3.2. Myocardial Ischemia-Reperfusion Injury (MIRI) Model
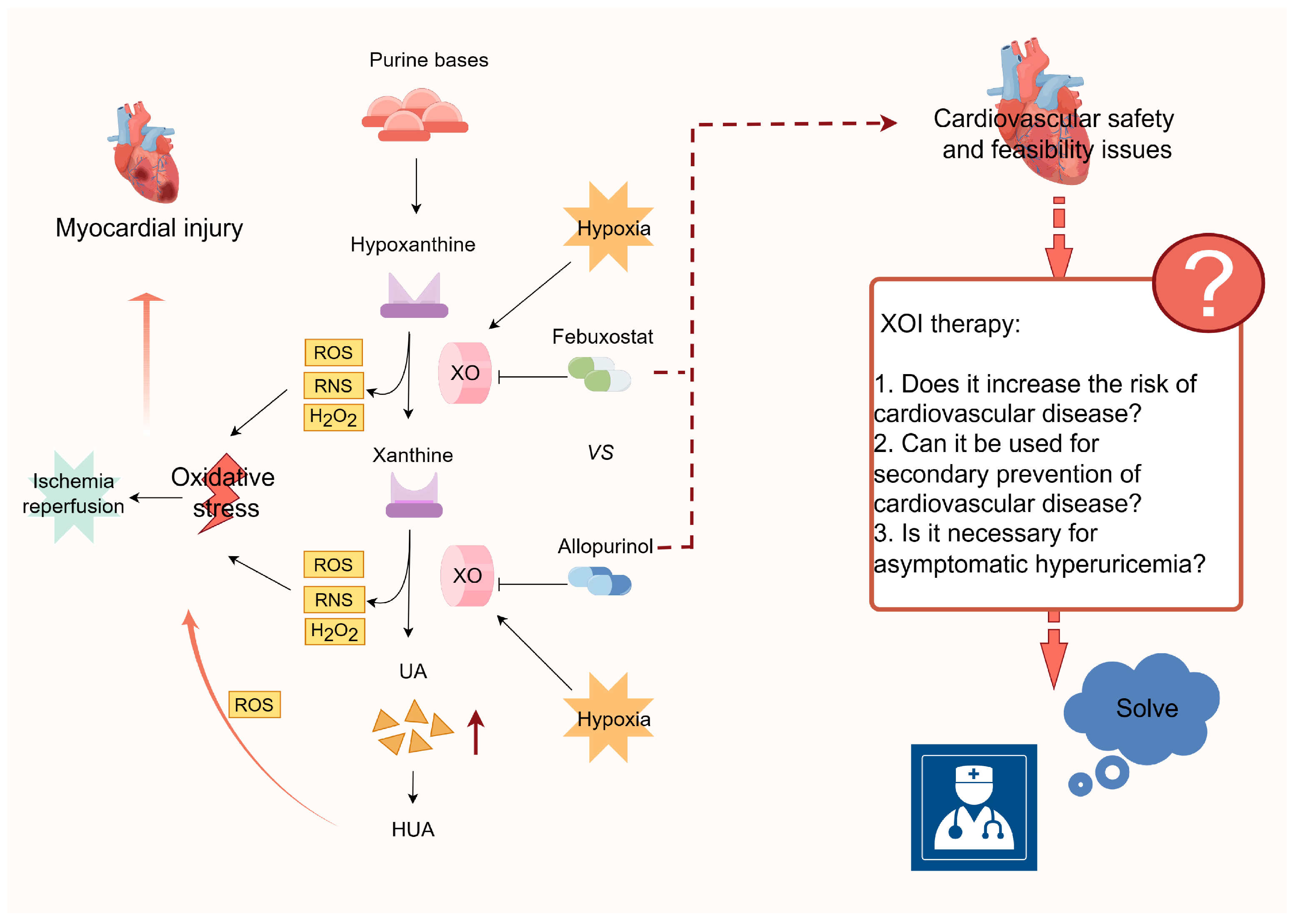
3.2.1. Xanthine Oxidase (XO)-Mediated ROS Production
3.2.2. ROS-Mediated Activation of the NLRP3 Inflammasome
4. Uric acid-Lowering Interventions
5. Summary and Outlook
6. Limitations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and Mitochondrial Integrity in Cardiac Ischemia-Reperfusion Injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Hirschhauser, C.; Bornbaum, J.; Sydykov, A.; Dempfle, A.; Schneider, A.; Braun, T.; Schluter, K.D.; Schulz, R. Cardiomyocytes-Specific Deletion of Monoamine Oxidase B Reduces Irreversible Myocardial Ischemia/Reperfusion Injury. Free Radic. Biol. Med. 2021, 165, 14–23. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, H.; Wang, T.; Liu, Y.; Meng, C. Mechanisms of Myocardial Damage Due to Hyperlipidemia: A Review of Recent Studies. Med. Sci. Monit. 2022, 28, e937051. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Andreadou, I.; Aragno, M.; Beauloye, C.; Bertrand, L.; Lazou, A.; Falcao-Pires, I.; Bell, R.; Zuurbier, C.J.; Pagliaro, P.; et al. Effect of Hyperglycaemia and Diabetes on Acute Myocardial Ischaemia-Reperfusion Injury and Cardioprotection by Ischaemic Conditioning Protocols. Br. J. Pharmacol. 2020, 177, 5312–5335. [Google Scholar] [CrossRef]
- Jin, L.; Deng, Z.; Zhang, J.; Yang, C.; Liu, J.; Han, W.; Ye, P.; Si, Y.; Chen, G. Mesenchymal Stem Cells Promote Type 2 Macrophage Polarization to Ameliorate the Myocardial Injury Caused by Diabetic Cardiomyopathy. J. Transl. Med. 2019, 17, 251. [Google Scholar] [CrossRef]
- Li, B.; Cao, X.; Ai, G.; Liu, Y.; Lv, C.; Jin, L.; Xu, R.; Zhao, G.; Yuan, H. Interleukin-37 Alleviates Myocardial Injury Induced by Coxsackievirus B3 Via Inhibiting Neutrophil Extracellular Traps Formation. Int. Immunopharmacol. 2022, 113, 109343. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, Z.; Zheng, Y.; Yang, S.; Huang, K.; Li, H. Roles of Inflammasomes in Viral Myocarditis. Front. Cell. Infect. Microbiol. 2023, 13, 1149911. [Google Scholar] [CrossRef]
- Wang, X.; Hou, Y.; Wang, X.; Li, Z.; Wang, X.; Li, H.; Shang, L.; Zhou, J.; Zhang, Y.; Ren, M.; et al. Relationship between Serum Uric Acid Levels and Different Types of Atrial Fibrillation: An Updated Meta-Analysis. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Liu, J.; Sun, N.; Huo, Y.; The Success Investigation Group. Hyperuricemia Is Associated with More Cardiometabolic Risk Factors in Hypertensive Younger Chinese Adults Than in Elderly. Front. Cardiovasc. Med. 2023, 10, 1133724. [Google Scholar] [CrossRef]
- Kim, J.H.; Kwon, M.J.; Choi, H.G.; Lee, S.J.; Kim, S.W.; Kim, J.H.; Kwon, B.C.; Lee, J.W. The Association between Hyperuricemia and Cardiovascular Disease History: A Cross-Sectional Study Using Koges Hexa Data. Medicine 2022, 101, e32338. [Google Scholar] [CrossRef]
- Kuwabara, M.; Fukuuchi, T.; Aoki, Y.; Mizuta, E.; Ouchi, M.; Kurajoh, M.; Maruhashi, T.; Tanaka, A.; Morikawa, N.; Nishimiya, K.; et al. Exploring the Multifaceted Nexus of Uric Acid and Health: A Review of Recent Studies on Diverse Diseases. Biomolecules 2023, 13, 1519. [Google Scholar] [CrossRef]
- Joosten, L.A.B.; Crisan, T.O.; Bjornstad, P.; Johnson, R.J. Asymptomatic Hyperuricaemia: A Silent Activator of the Innate Immune System. Nat. Rev. Rheumatol. 2020, 16, 75–86. [Google Scholar] [CrossRef]
- Elfishawi, M.M.; Zleik, N.; Kvrgic, Z.; Michet, C.J., Jr.; Crowson, C.S.; Matteson, E.L.; Bongartz, T. The Rising Incidence of Gout and the Increasing Burden of Comorbidities: A Population-Based Study over 20 Years. J. Rheumatol. 2018, 45, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhu, X.; Wu, J.; Huang, Z.; Zhao, Z.; Zhang, X.; Xue, Y.; Wan, W.; Li, C.; Zhang, W.; et al. Prevalence of Hyperuricemia among Chinese Adults: Findings from Two Nationally Representative Cross-Sectional Surveys in 2015-16 and 2018-19. Front. Immunol. 2021, 12, 791983. [Google Scholar] [CrossRef]
- Chen, F.; Yuan, L.; Xu, T.; Liu, J.; Han, S. Association of Hyperuricemia with 10-Year Atherosclerotic Cardiovascular Disease Risk among Chinese Adults and Elders. Int. J. Environ. Res. Public Health 2022, 19, 6713. [Google Scholar] [CrossRef] [PubMed]
- Casiglia, E.; Tikhonoff, V.; Virdis, A.; Masi, S.; Barbagallo, C.M.; Bombelli, M.; Bruno, B.; Cicero, A.F.G.; Cirillo, M.; Cirillo, P.; et al. Serum Uric Acid and Fatal Myocardial Infarction: Detection of Prognostic Cut-Off Values: The Urrah (Uric Acid Right for Heart Health) Study. J. Hypertens. 2020, 38, 412–419. [Google Scholar] [CrossRef]
- Muiesan, M.L.; Salvetti, M.; Virdis, A.; Masi, S.; Casiglia, E.; Tikhonoff, V.; Barbagallo, C.M.; Bombelli, M.; Cicero, A.F.G.; Cirillo, M.; et al. Serum Uric Acid, Predicts Heart Failure in a Large Italian Cohort: Search for a Cut-Off Value the Uric Acid Right for Heart Health Study. J. Hypertens. 2021, 39, 62–69. [Google Scholar] [CrossRef]
- Mengozzi, A.; Pugliese, N.R.; Desideri, G.; Masi, S.; Angeli, F.; Barbagallo, C.M.; Bombelli, M.; Cappelli, F.; Casiglia, E.; Cianci, R.; et al. Serum Uric Acid Predicts All-Cause and Cardiovascular Mortality Independently of Hypertriglyceridemia in Cardiometabolic Patients without Established Cv Disease: A Sub-Analysis of the Uric Acid Right for Heart Health (Urrah) Study. Metabolites 2023, 13, 244. [Google Scholar] [CrossRef]
- Lu, J.; Sun, W.; Cui, L.; Li, X.; He, Y.; Liu, Z.; Li, H.; Han, L.; Ji, A.; Wang, C.; et al. A Cross-Sectional Study on Uric Acid Levels among Chinese Adolescents. Pediatr. Nephrol. 2020, 35, 441–446. [Google Scholar] [CrossRef]
- von Lueder, T.G.; Girerd, N.; Atar, D.; Agewall, S.; Lamiral, Z.; Kanbay, M.; Pitt, B.; Dickstein, K.; Zannad, F.; Investigators High-Risk Myocardial Infarction Database Initiative; et al. Serum Uric Acid Is Associated with Mortality and Heart Failure Hospitalizations in Patients with Complicated Myocardial Infarction: Findings from the High-Risk Myocardial Infarction Database Initiative. Eur. J. Heart Fail. 2015, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Wang, A.; Wu, S.; Zuo, Y.; Chen, S.; Zhang, L.; Mo, D.; Luo, Y. Cumulative Serum Uric Acid and Its Time Course Are Associated with Risk of Myocardial Infarction and All-Cause Mortality. J. Am. Heart Assoc. 2021, 10, e020180. [Google Scholar] [CrossRef] [PubMed]
- Gertler, M.M.; Garn, S.M.; Levine, S.A. Serum Uric Acid in Relation to Age and Physique in Health and in Coronary Heart Disease. Ann. Intern. Med. 1951, 34, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Ranjith, N.; Myeni, N.N.; Sartorius, B.; Mayise, C. Association between Hyperuricemia and Major Adverse Cardiac Events in Patients with Acute Myocardial Infarction. Metab. Syndr. Relat. Disord. 2017, 15, 18–25. [Google Scholar] [CrossRef]
- Tian, X.; Zuo, Y.; Chen, S.; Wu, S.; Wang, A.; Luo, Y. High Serum Uric Acid Trajectories Are Associated with Risk of Myocardial Infarction and All-Cause Mortality in General Chinese Population. Arthritis Res. Ther. 2022, 24, 149. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Daimon, M.; Yoshida, Y.; Ishiwata, J.; Sawada, N.; Hirokawa, M.; Kaneko, H.; Nakao, T.; Mizuno, Y.; Morita, H.; et al. Serum Uric Acid Level and Subclinical Left Ventricular Dysfunction: A Community-Based Cohort Study. ESC Heart Fail. 2020, 7, 1031–1038. [Google Scholar] [CrossRef]
- Kobayashi, N.; Hata, N.; Tsurumi, M.; Shibata, Y.; Okazaki, H.; Shirakabe, A.; Takano, M.; Seino, Y.; Shimizu, W. Relation of Coronary Culprit Lesion Morphology Determined by Optical Coherence Tomography and Cardiac Outcomes to Serum Uric Acid Levels in Patients with Acute Coronary Syndrome. Am. J. Cardiol. 2018, 122, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Mandurino-Mirizzi, A.; Crimi, G.; Raineri, C.; Pica, S.; Ruffinazzi, M.; Gianni, U.; Repetto, A.; Ferlini, M.; Marinoni, B.; Leonardi, S.; et al. Elevated Serum Uric Acid Affects Myocardial Reperfusion and Infarct Size in Patients with St-Segment Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. J. Cardiovasc. Med. 2018, 19, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.F.; Song, K.Y.; Zhou, W.N.; Wei, Y.J. Association between Uric Acid and in-Hospital Heart Failure in Patients with Acute Myocardial Infarction Undergoing Percutaneous Coronary Intervention. Dis. Markers 2021, 2021, 7883723. [Google Scholar] [CrossRef] [PubMed]
- Nakahashi, T.; Sakata, K.; Masuda, J.; Kumagai, N.; Higuma, T.; Ogimoto, A.; Tanigawa, T.; Hanada, H.; Nakamura, M.; Takamura, M.; et al. Impact of Hyperuricemia on Coronary Blood Flow and in-Hospital Mortality in Patients with Acute Myocardial Infarction Undergoing Percutaneous Coronary Intervention. J. Cardiol. 2022, 80, 268–274. [Google Scholar] [CrossRef]
- Guo, W.; Yang, D.; Wu, D.; Liu, H.; Chen, S.; Liu, J.; Lei, L.; Liu, Y.; Rao, L.; Zhang, L.; et al. Hyperuricemia and Long-Term Mortality in Patients with Acute Myocardial Infarction Undergoing Percutaneous Coronary Intervention. Ann. Transl. Med. 2019, 7, 636. [Google Scholar] [CrossRef]
- Ma, W.; Gao, S.; Huang, S.; Yuan, J.; Yu, M. Hyperuricemia as a Prognostic Marker for Long-Term Outcomes in Patients with Myocardial Infarction with Nonobstructive Coronary Arteries. Nutr. Metab. 2021, 18, 107. [Google Scholar] [CrossRef] [PubMed]
- Mandurino-Mirizzi, A.; Cornara, S.; Somaschini, A.; Demarchi, A.; Galazzi, M.; Puccio, S.; Montalto, C.; Crimi, G.; Ferlini, M.; Camporotondo, R.; et al. Elevated Serum Uric Acid Is Associated with a Greater Inflammatory Response and with Short- and Long-Term Mortality in Patients with St-Segment Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Canga, Y.; Emre, A.; Karatas, M.B.; Calik, A.N.; Yelgec, N.S.; Inan, D.; Yuksel, G.; Terzi, S. Prognostic Value of Serum Uric Acid Levels in Patients with Non-Stemi Undergoing Percutaneous Coronary Intervention. Herz 2020, 45, 389–396. [Google Scholar] [CrossRef]
- Li, L.; Ma, Y.; Shang, X.M.; Hong, Y.; Wang, J.H.; Tan, Z.; Wang, Y.J.; Geng, X.B. Hyperuricemia Is Associated with Short-Term Outcomes in Elderly Patients with Acute Myocardial Infarction. Aging Clin. Exp. Res. 2018, 30, 1211–1215. [Google Scholar] [CrossRef]
- Virdis, A.; Masi, S.; Casiglia, E.; Tikhonoff, V.; Cicero, A.F.G.; Ungar, A.; Rivasi, G.; Salvetti, M.; Barbagallo, C.M.; Bombelli, M.; et al. Identification of the Uric Acid Thresholds Predicting an Increased Total and Cardiovascular Mortality over 20 Years. Hypertension 2020, 75, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Donniacuo, M.; De Angelis, A.; Telesca, M.; Bellocchio, G.; Riemma, M.A.; Paolisso, P.; Scisciola, L.; Cianflone, E.; Torella, D.; Castaldo, G.; et al. Atrial Fibrillation: Epigenetic Aspects and Role of Sodium-Glucose Cotransporter 2 Inhibitors. Pharmacol. Res. 2023, 188, 106591. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Park, J.W.; Yang, P.S.; Hwang, I.; Kim, T.H.; Yu, H.T.; Uhm, J.S.; Joung, B.; Lee, M.H.; Jee, S.H.; et al. A Mendelian Randomization Analysis: The Causal Association between Serum Uric Acid and Atrial Fibrillation. Eur. J. Clin. Investig. 2020, 50, e13300. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Shao, W.; Yu, P.; Ma, J.; Liu, M.; Huang, S.; Liu, X.; Mei, K. Hyperuricemia Is Associated with the Risk of Atrial Fibrillation Independent of Sex: A Dose-Response Meta-Analysis. Front. Cardiovasc. Med. 2022, 9, 865036. [Google Scholar] [CrossRef]
- Kuwabara, M.; Niwa, K.; Nishihara, S.; Nishi, Y.; Takahashi, O.; Kario, K.; Yamamoto, K.; Yamashita, T.; Hisatome, I. Hyperuricemia Is an Independent Competing Risk Factor for Atrial Fibrillation. Int. J. Cardiol. 2017, 231, 137–142. [Google Scholar] [CrossRef]
- Mantovani, A.; Rigolon, R.; Civettini, A.; Bolzan, B.; Morani, G.; Bonapace, S.; Dugo, C.; Zoppini, G.; Bonora, E.; Targher, G. Hyperuricemia Is Associated with an Increased Prevalence of Paroxysmal Atrial Fibrillation in Patients with Type 2 Diabetes Referred for Clinically Indicated 24-H Holter Monitoring. J. Endocrinol. Investig. 2018, 41, 223–231. [Google Scholar] [CrossRef]
- Li, S.; Cheng, J.; Cui, L.; Gurol, M.E.; Bhatt, D.L.; Fonarow, G.C.; Benjamin, E.J.; Xing, A.; Xia, Y.; Wu, S.; et al. Cohort Study of Repeated Measurements of Serum Urate and Risk of Incident Atrial Fibrillation. J. Am. Heart Assoc. 2019, 8, e012020. [Google Scholar] [CrossRef]
- Zhong, X.; Jiao, H.; Zhao, D.; Yang, M.; Teng, J. Association between Serum Uric Acid Levels and Atrial Fibrillation in Different Fasting Glucose Patterns: A Case-Control Study. Front. Endocrinol. 2023, 14, 1021267. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Scisciola, L.; D’Onofrio, N.; Maiello, C.; Trotta, M.C.; Sardu, C.; Panarese, I.; Ferraraccio, F.; Capuano, A.; Barbieri, M.; et al. Sodium-Glucose Cotransporter-2 (Sglt2) Expression in Diabetic and Non-Diabetic Failing Human Cardiomyocytes. Pharmacol. Res. 2022, 184, 106448. [Google Scholar] [CrossRef]
- Vlad-Sabin, I.; Roxana, B.; Adina-Flavia, C.M.; Paul, C.; Melania, A.; Daniel, G.; Ilie, R.C.; Romulus, T.; Daniel, L. The Relationship between Serum Uric Acid and Ejection Fraction of the Left Ventricle. J. Clin. Med. 2021, 10, 4026. [Google Scholar] [CrossRef]
- Deng, X.L.; Yi, H.W.; Xiao, J.; Zhang, X.F.; Zhao, J.; Sun, M.; Wen, X.S.; Liu, Z.Q.; Gao, L.; Li, Z.Y.; et al. Serum Uric Acid: A Risk Factor for Right Ventricular Dysfunction and Prognosis in Heart Failure with Preserved Ejection Fraction. Front. Endocrinol. 2023, 14, 1143458. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.M.; Hsu, P.F.; Cheng, H.M.; Lu, D.Y.; Cheng, Y.L.; Guo, C.Y.; Sung, S.H.; Yu, W.C.; Chen, C.H. Determinants and Prognostic Impact of Hyperuricemia in Hospitalization for Acute Heart Failure. Circ. J. 2016, 80, 404–410. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Salvioni, E.; Corra, U.; Doni, F.; Bonomi, A.; La Gioia, R.; Limongelli, G.; Paolillo, S.; Sinagra, G.; Scardovi, A.B.; et al. Increased Serum Uric Acid Level Predicts Poor Prognosis in Mildly Severe Chronic Heart Failure with Reduced Ejection Fraction. An Analysis from the Mecki Score Research Group. Eur. J. Intern. Med. 2020, 72, 47–52. [Google Scholar] [CrossRef]
- Carnicelli, A.P.; Sun, J.L.; Alhanti, B.; Bjursell, M.; Perl, S.; Lytle, B.; Roe, M.T.; Mentz, R.J. Elevated Uric Acid Prevalence and Clinical Outcomes in Patients with Heart Failure with Preserved Ejection Fraction: Insights from Relax. Am. J. Med. 2020, 133, e716–e721. [Google Scholar] [CrossRef]
- Wu, X.; Jian, G.; Tang, Y.; Cheng, H.; Wang, N.; Wu, J. Asymptomatic Hyperuricemia and Incident Congestive Heart Failure in Elderly Patients without Comorbidities. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.H.; Chuang, S.Y.; Liu, W.L.; Cheng, H.M.; Hsu, P.F.; Pan, W.H. Hyperuricemia and Pulse Pressure Are Predictive of Incident Heart Failure in an Elderly Population. Int. J. Cardiol. 2020, 300, 178–183. [Google Scholar] [CrossRef]
- Yilmaz Oztekin, G.M.; Genc, A.; Cagirci, G.; Arslan, S. Prognostic Value of the Combination of Uric Acid and Nt-Probnp in Patients with Chronic Heart Failure. Hellenic J. Cardiol. 2022, 65, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Egami, Y.; Kawanami, S.; Sugae, H.; Ukita, K.; Kawamura, A.; Nakamura, H.; Matsuhiro, Y.; Yasumoto, K.; Osaka CardioVascular Conference-Heart Failure Investigators; et al. Lowering Uric Acid May Improve Prognosis in Patients with Hyperuricemia and Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2022, 11, e026301. [Google Scholar] [CrossRef] [PubMed]
- Naganuma, J.; Sakuma, M.; Kitahara, K.; Kato, T.; Yokomachi, J.; Yamauchi, F.; Inoue, R.; Iida, K.; Kohno, Y.; Inoue, K.; et al. Study Investigators Excited; et al. Optimal Uric Acid Reduction to Improve Vascular Endothelial Function in Patients with Chronic Heart Failure Complicated by Hyperuricemia. Hypertens. Res. 2023, 46, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Plata, G.; Nakafero, G.; Chakravorty, M.; Morgan, K.; Abhishek, A. Association between Serum Urate, Gout and Comorbidities: A Case-Control Study Using Data from the Uk Biobank. Rheumatology 2021, 60, 3243–3251. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; McCormick, N.; Yokose, C. Excess Comorbidities in Gout: The Causal Paradigm and Pleiotropic Approaches to Care. Nat. Rev. Rheumatol. 2022, 18, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Feuchtner, G.M.; Plank, F.; Beyer, C.; Schwabl, C.; Held, J.; Bellmann-Weiler, R.; Weiss, G.; Gruber, J.; Widmann, G.; Klauser, A.S. Monosodium Urate Crystal Deposition in Coronary Artery Plaque by 128-Slice Dual-Energy Computed Tomography: An Ex Vivo Phantom and in Vivo Study. J. Comput. Assist. Tomogr. 2021, 45, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Barazani, S.H.; Chi, W.W.; Pyzik, R.; Chang, H.; Jacobi, A.; O’Donnell, T.; Fayad, Z.A.; Ali, Y.; Mani, V. Quantification of Uric Acid in Vasculature of Patients with Gout Using Dual-Energy Computed Tomography. World J. Radiol. 2020, 12, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Klauser, A.S.; Halpern, E.J.; Strobl, S.; Gruber, J.; Feuchtner, G.; Bellmann-Weiler, R.; Weiss, G.; Stofferin, H.; Jaschke, W. Dual-Energy Computed Tomography Detection of Cardiovascular Monosodium Urate Deposits in Patients with Gout. JAMA Cardiol. 2019, 4, 1019–1028. [Google Scholar] [CrossRef]
- Saygin, M.; Asci, H.; Cankara, F.N.; Bayram, D.; Yesilot, S.; Candan, I.A.; Alp, H.H. The Impact of High Fructose on Cardiovascular System: Role of Alpha-Lipoic Acid. Hum. Exp. Toxicol. 2016, 35, 194–204. [Google Scholar] [CrossRef]
- Zhang, J.; Lin, X.; Xu, J.; Tang, F.; Tan, L. Ctrp3 Protects against Uric Acid-Induced Endothelial Injury by Inhibiting Inflammation and Oxidase Stress in Rats. Exp. Biol. Med. 2022, 247, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, L.; Zhang, G.; Liu, Y.; Liang, Z.; Zhao, J.; Yin, S.; Su, M.; Zhang, S.; Wei, Y.; et al. Increased Susceptibility of Atrial Fibrillation Induced by Hyperuricemia in Rats: Mechanisms and Implications. Cardiovasc. Toxicol. 2021, 21, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Liu, W.; Xie, D.; Wang, Q.; Xu, C.; Zhao, H.; Lv, J.; He, F.; Chen, B.; Yamamoto, T.; et al. High Level of Uric Acid Promotes Atherosclerosis by Targeting Nrf2-Mediated Autophagy Dysfunction and Ferroptosis. Oxid. Med. Cell. Longev. 2022, 2022, 9304383. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Zheng, Q.; Li, Y.; Wu, T.; Luo, J.; Jiang, Y.; Huang, Q.; Yan, C.; Zhang, L.; Zhang, W.; et al. Assessment of the Influence on Left Ventricle by Potassium Oxonate and Hypoxanthine-Induced Chronic Hyperuricemia. Exp. Biol. Med. 2023, 248, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Badejogbin, C.; Areola, D.E.; Olaniyi, K.S.; Adeyanju, O.A.; Adeosun, I.O. Sodium Butyrate Recovers High-Fat Diet-Fed Female Wistar Rats from Glucose Dysmetabolism and Uric Acid-Associated Cardiac Tissue Damage. Naunyn Schmiedeberg Arch. Pharmacol. 2019, 392, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Schreckenberg, R.; Schluter, K.D. Uric Acid Deteriorates Load-Free Cell Shortening of Cultured Adult Rat Ventricular Cardiomyocytes Via Stimulation of Arginine Turnover. Biology 2022, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Y.; Cao, R.; Wang, G.; Li, S.; Cao, Y.; Zhang, H.; Liu, M.; Liu, G.; Zhang, J.; et al. Soluble Uric Acid Induces Myocardial Damage through Activating the Nlrp3 Inflammasome. J. Cell. Mol. Med. 2020, 24, 8849–8861. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Dai, R.; Wang, M.; Chen, C. Uric Acid Promotes Myocardial Infarction Injury Via Activating Pyrin Domain-Containing 3 Inflammasome and Reactive Oxygen Species/Transient Receptor Potential Melastatin 2/Ca2+Pathway. BMC Cardiovasc. Disord. 2023, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Liu, K.; Zhu, L. Emerging Roles of Inflammasomes in Cardiovascular Diseases. Front. Immunol. 2022, 13, 834289. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kurata, Y.; Taufiq, F.; Kuwabara, M.; Ninomiya, H.; Higaki, K.; Tsuneto, M.; Shirayoshi, Y.; Lanaspa, M.A.; Hisatome, I. Kv1.5 Channel Mediates Monosodium Urate-Induced Activation of Nlrp3 Inflammasome in Macrophages and Arrhythmogenic Effects of Urate on Cardiomyocytes. Mol. Biol. Rep. 2022, 49, 5939–5952. [Google Scholar] [CrossRef]
- Taufiq, F.; Maharani, N.; Li, P.; Kurata, Y.; Ikeda, N.; Kuwabara, M.; Otani, N.; Miake, J.; Hasegawa, A.; Tsuneto, M.; et al. Uric Acid-Induced Enhancements of Kv1.5 Protein Expression and Channel Activity Via the Akt-Hsf1-Hsp70 Pathway in Hl-1 Atrial Myocytes. Circ. J. 2019, 83, 718–726. [Google Scholar] [CrossRef]
- So, A.; Thorens, B. Uric Acid Transport and Disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Lee, G.; Lee, G.S. Lower Temperatures Exacerbate Nlrp3 Inflammasome Activation by Promoting Monosodium Urate Crystallization, Causing Gout. Cells 2021, 10, 1919. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Guan, C.; Xu, L.; Wang, L.; Yang, C.; Zhao, L.; Zhou, B.; Luo, C.; Luan, H.; Jiang, W.; et al. Scutellarin Ameliorates Renal Injury Via Increasing Ccn1 Expression and Suppressing Nlrp3 Inflammasome Activation in Hyperuricemic Mice. Front. Pharmacol. 2020, 11, 584942. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Liu, S.; Tang, M.; Lu, Y.; Zhao, M.; Mao, R.; Wang, C.; Yuan, Y.; Li, L.; Chen, Y.; et al. Phloretin Ameliorates Hyperuricemia-Induced Chronic Renal Dysfunction through Inhibiting Nlrp3 Inflammasome and Uric Acid Reabsorption. Phytomedicine 2020, 66, 153111. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Xu, C.; Lin, Y.; Lu, C.; Li, D.; Sang, J.; He, H.; Liu, X.; Li, Y.; Yu, C. Uric Acid Regulates Hepatic Steatosis and Insulin Resistance through the Nlrp3 Inflammasome-Dependent Mechanism. J. Hepatol. 2016, 64, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Xu, D.; Ma, J.; Wang, Y.; Yang, X.; Zhao, P.; Ma, L.; Li, Z.; Yang, W.; Liu, X.; et al. Uric Acid Drives Intestinal Barrier Dysfunction through Tspo-Mediated Nlrp3 Inflammasome Activation. Inflamm. Res. 2021, 70, 127–137. [Google Scholar] [CrossRef]
- Jiao, Z.; Chen, Y.; Xie, Y.; Li, Y.; Li, Z. Metformin Protects against Insulin Resistance Induced by High Uric Acid in Cardiomyocytes Via Ampk Signalling Pathways in Vitro and in Vivo. J. Cell. Mol. Med. 2021, 25, 6733–6745. [Google Scholar] [CrossRef]
- Yu, W.; Xie, D.; Yamamoto, T.; Koyama, H.; Cheng, J. Mechanistic Insights of Soluble Uric Acid-Induced Insulin Resistance: Insulin Signaling and Beyond. Rev. Endocr. Metab. Disord. 2023, 24, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Ampk: A Key Regulator of Energy Balance in the Single Cell and the Whole Organism. Int. J. Obes. 2008, 32 (Suppl. S4), S7–S12. [Google Scholar] [CrossRef]
- Zhang, X.J.; Liu, D.M.; Sun, Y.; Li, Y.S.; Ma, L.L.; Kong, X.F.; Cui, X.M.; Chen, R.Y.; Zhang, Z.J.; Jiang, L.D. Potential Risk of Hyperuricemia: Leading Cardiomyocyte Hypertrophy by Inducing Autophagy. Am. J. Transl. Res. 2020, 12, 1894–1903. [Google Scholar]
- Yang, Y.; Lin, C.; Zheng, Q.; Zhang, L.; Li, Y.; Huang, Q.; Wu, T.; Zhao, Z.; Li, L.; Luo, J.; et al. L-Carnitine Attenuated Hyperuricemia-Associated Left Ventricular Remodeling through Ameliorating Cardiomyocytic Lipid Deposition. Front. Pharmacol. 2023, 14, 1016633. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Zhao, M.; Li, Y.; Zhang, G.; Ma, Y.; Shi, Y.; Su, P.; Chen, R.; Tang, Z.G.; Zhang, Y.; et al. The Protective Effects of Uric Acid against Myocardial Ischemia Via the Nrf2 Pathway. Eur. J. Pharmacol. 2023, 959, 176062. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiao, H.; Zhao, J.; Wang, X.; Lin, H. Unexpected Effect of Urate on Hydrogen Peroxide-Induced Oxidative Damage in Embryonic Chicken Cardiac Cells. Free Radic. Res. 2017, 51, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiao, H.; Zhao, J.; Wang, X.; Lin, H. Rule of Ua on Cardiac Myocytes Uric Acid Differently Influence the Oxidative Damage Induced by Acute Exposure of High Level of Glucose in Chicken Cardiac Myocytes. Front. Vet. Sci. 2020, 7, 602419. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Nrf2, Cellular Redox Regulation, and Neurologic Implications. Neurology 2017, 88, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, C.; Zhuang, W.; Wang, W.; Liu, W.; Zhao, H.; Lv, J.; Xie, D.; Wang, Q.; He, F.; et al. Silencing Txnip Ameliorates High Uric Acid-Induced Insulin Resistance Via the Irs2/Akt and Nrf2/Ho-1 Pathways in Macrophages. Free Radic. Biol. Med. 2022, 178, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Lu, L.Q.; Jiang, Y.Q.; Li, N.S.; Luo, X.J.; Peng, J.W.; Peng, J. Allopurinol Attenuates Oxidative Injury in Rat Hearts Suffered Ischemia/Reperfusion Via Suppressing the Xanthine Oxidase/Vascular Peroxidase 1 Pathway. Eur. J. Pharmacol. 2021, 908, 174368. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, E.; Clausen, M.H.; Laraia, L. Small-Molecule Inhibitors of Reactive Oxygen Species Production. J. Med. Chem. 2021, 64, 5252–5275. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Bortolotti, M.; Battelli, M.G.; Bolognesi, A. Xanthine Oxidoreductase: A Leading Actor in Cardiovascular Disease Drama. Redox Biol. 2021, 48, 102195. [Google Scholar] [CrossRef]
- Garattini, E.; Mendel, R.; Romao, M.J.; Wright, R.; Terao, M. Mammalian Molybdo-Flavoenzymes, an Expanding Family of Proteins: Structure, Genetics, Regulation, Function and Pathophysiology. Biochem. J. 2003, 372 Pt 1, 15–32. [Google Scholar] [CrossRef]
- Shen, S.; He, F.; Cheng, C.; Xu, B.; Sheng, J. Uric Acid Aggravates Myocardial Ischemia-Reperfusion Injury Via Ros/Nlrp3 Pyroptosis Pathway. Biomed. Pharmacother. 2021, 133, 110990. [Google Scholar] [CrossRef]
- Tai, C.J.; Wu, C.C.; Lee, K.T.; Tseng, T.G.; Wang, H.C.; Chang, F.R.; Yang, Y.H. The Impact of Urate-Lowering Therapy in Post-Myocardial Infarction Patients: Insights from a Population-Based, Propensity Score-Matched Analysis. Clin. Pharmacol. Ther. 2022, 111, 655–663. [Google Scholar] [CrossRef]
- Rodriguez-Martin, S.; de Abajo, F.J.; Gil, M.; Gonzalez-Bermejo, D.; Rodriguez-Miguel, A.; Barreira-Hernandez, D.; Mazzucchelli, R.; Garcia-Lledo, A.; Garcia-Rodriguez, L.A. Risk of Acute Myocardial Infarction among New Users of Allopurinol According to Serum Urate Level: A Nested Case-Control Study. J. Clin. Med. 2019, 8, 2150. [Google Scholar] [CrossRef] [PubMed]
- Omizo, H.; Tamura, Y.; Morimoto, C.; Ueno, M.; Hayama, Y.; Kuribayashi-Okuma, E.; Uchida, S.; Shibata, S. Cardio-Renal Protective Effect of the Xanthine Oxidase Inhibitor Febuxostat in the 5/6 Nephrectomy Model with Hyperuricemia. Sci. Rep. 2020, 10, 9326. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nagoshi, T.; Yoshii, A.; Oi, Y.; Takahashi, H.; Kimura, H.; Ito, K.; Kashiwagi, Y.; Tanaka, T.D.; Yoshimura, M. Xanthine Oxidase Inhibition Attenuates Doxorubicin-Induced Cardiotoxicity in Mice. Free Radic. Biol. Med. 2021, 162, 298–308. [Google Scholar] [CrossRef]
- Mackenzie, I.S.; Hawkey, C.J.; Ford, I.; Greenlaw, N.; Pigazzani, F.; Rogers, A.; Struthers, A.D.; Begg, A.G.; Wei, L.; All-Heart Study Group; et al. Allopurinol Versus Usual Care in Uk Patients with Ischaemic Heart Disease (All-Heart): A Multicentre, Prospective, Randomised, Open-Label, Blinded-Endpoint Trial. Lancet 2022, 400, 1195–1205. [Google Scholar] [CrossRef]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L.; Cares Investigators. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.S.; Ford, I.; Nuki, G.; Hallas, J.; Hawkey, C.J.; Webster, J.; Ralston, S.H.; Walters, M.; Robertson, M.; Fast Study Group; et al. Long-Term Cardiovascular Safety of Febuxostat Compared with Allopurinol in Patients with Gout (Fast): A Multicentre, Prospective, Randomised, Open-Label, Non-Inferiority Trial. Lancet 2020, 396, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Care Res. 2020, 72, 744–760. [Google Scholar] [CrossRef] [PubMed]
- Talpur, A.S.; Fattah, A.; Hewadmal, H.; Hafizyar, F.; Farooq, J.; Shaik, T.A.; Qadar, L.T.; Zaidi, S.M.H.; Pirzada, S.; Bahar, A.R. Asymptomatic Hyperuricemia as an Independent Risk Factor for Myocardial Infarction in Adult Population: A Four-Year Follow-up Study. Cureus 2023, 15, e34614. [Google Scholar] [CrossRef]
- Chen, C.; Dong, J.; Lv, Q.; Liu, X.; Zhang, Q.; Du, X. Effect of Asymptomatic Hyperuricemia on Mortality of Elderly Patients after Elective Percutaneous Coronary Intervention. Front. Cardiovasc. Med. 2022, 9, 800414. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Chen, Z.; Yang, J.; Zheng, J.; Shui, X.; Yan, Y.; Huang, S.; Liang, Z.; Lei, W.; He, Y. The Role of Hyperuricemia in Cardiac Diseases: Evidence, Controversies, and Therapeutic Strategies. Biomolecules 2024, 14, 753. https://doi.org/10.3390/biom14070753
Zheng Y, Chen Z, Yang J, Zheng J, Shui X, Yan Y, Huang S, Liang Z, Lei W, He Y. The Role of Hyperuricemia in Cardiac Diseases: Evidence, Controversies, and Therapeutic Strategies. Biomolecules. 2024; 14(7):753. https://doi.org/10.3390/biom14070753
Chicago/Turabian StyleZheng, Yue, Zhirui Chen, Jinya Yang, Jing Zheng, Xiaorong Shui, Yiguang Yan, Shian Huang, Zheng Liang, Wei Lei, and Yuan He. 2024. "The Role of Hyperuricemia in Cardiac Diseases: Evidence, Controversies, and Therapeutic Strategies" Biomolecules 14, no. 7: 753. https://doi.org/10.3390/biom14070753