
Helicobacter pylori-Induced Decrease in Membrane Expression of Na,K-ATPase Leads to Gastric Injury

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Cultured Cells
2.3. Gastric Organoids
2.4. Gastric Organoid Cells Grown as 2D Monolayers
2.5. Silencing of Na,K-ATPase α1 Subunit
2.6. Infection of Gastric Cells and 2D Organoids with H. pylori
2.7. Paracellular Permeability
2.8. Trans-Epithelial Electrical Resistance (TEER) of Cell Monolayers
2.9. TEER of Gerbil Gastric Tissue
2.10. Immunofluorescence and Confocal Microscopy
2.11. Cell Lysis
2.12. Western Blotting
3. Results
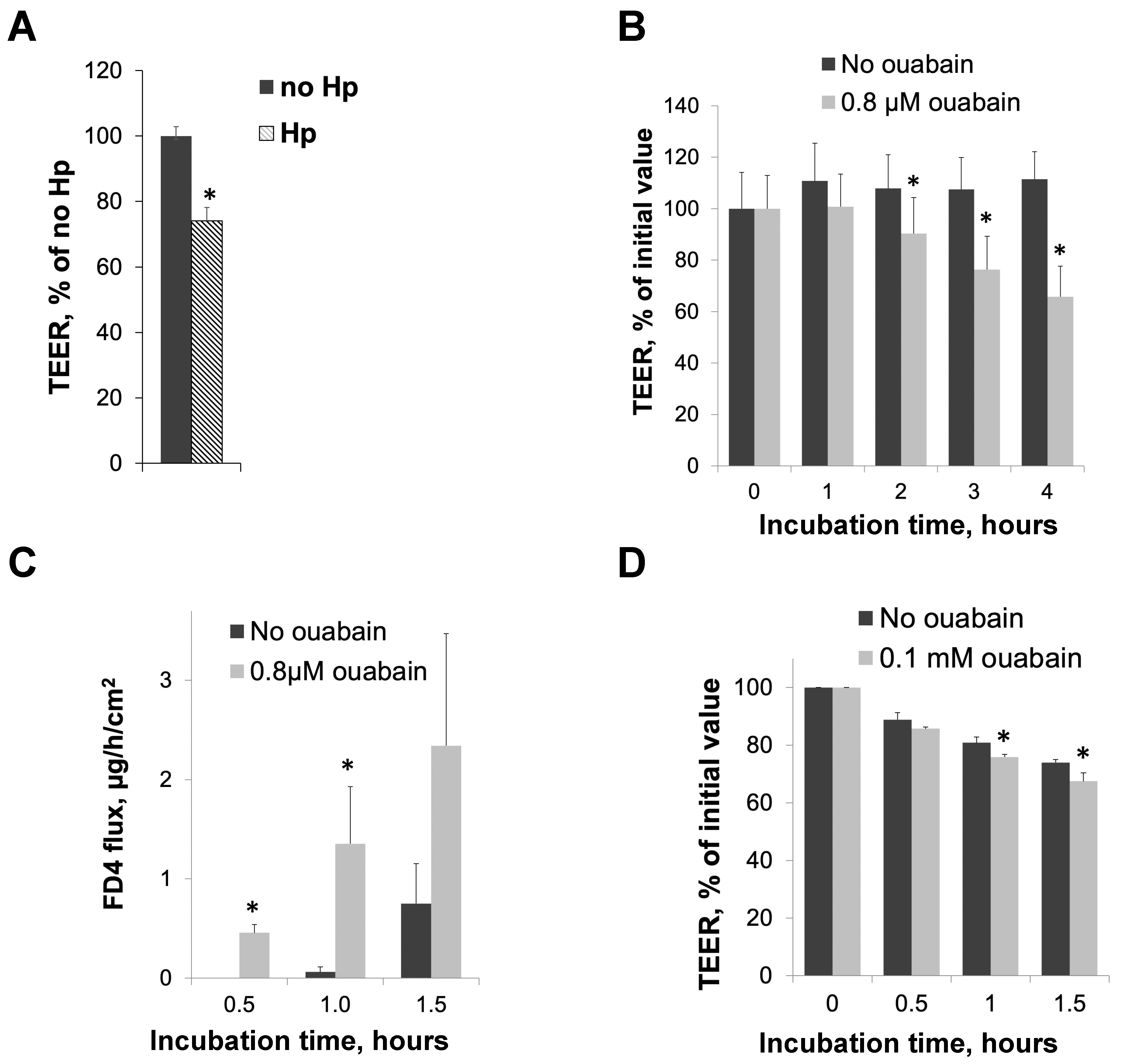
3.1. H. pylori Infection and Inhibition of Na,K-ATPase Activity with Ouabain Impair Intracellular Junctions in Cultured Gastric Epithelial Cells and Gerbil Gastric Mucosa
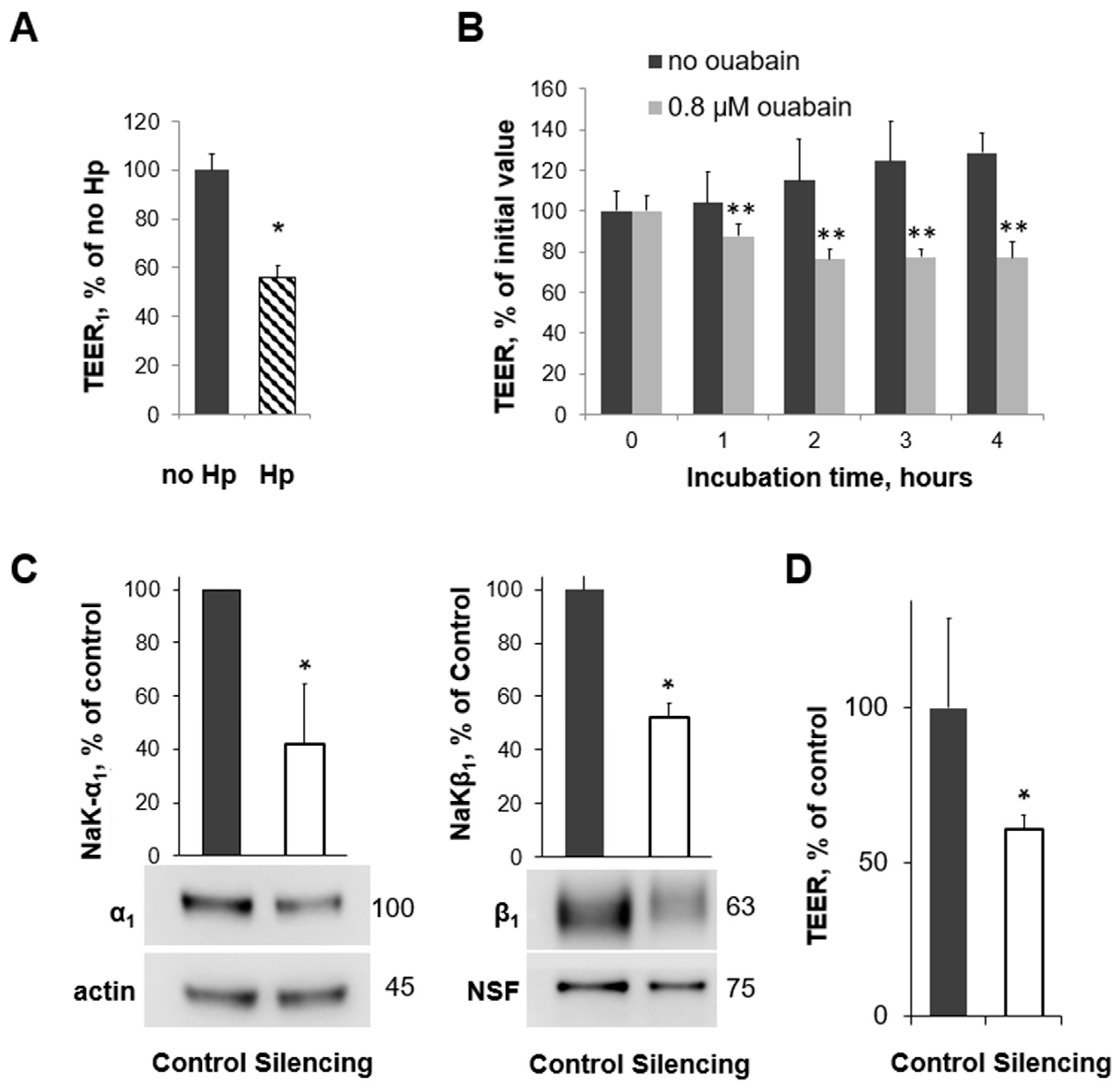
3.2. Human Gastric Organoids Express Na,K-ATPase and Demonstrate Reduction in Transporter in the Membrane in the Presence of H. pylori
3.3. H. pylori Infection, Na,K-ATPase Silencing, and Inhibition of Na,K-ATPase Activity All Decrease TEER in Human Gastric Organoids
3.4. Reduction of Na,K-ATPase Activity with Ouabain or Downregulation with H. pylori Disrupts the Organization and Integrity of Adherens Junctions in 2D Human Gastric Organoids
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blaser, M.J. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology 1992, 102, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Stemmermann, G.N.; Chyou, P.H.; Kato, I.; Perez-Perez, G.I.; Blaser, M.J. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 1991, 325, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J. Gastric adenocarcinoma and Helicobacter pylori infection. West. J. Med. 1994, 161, 60. [Google Scholar] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Correa, P.; Haenszel, W.; Cuello, C.; Tannenbaum, S.; Archer, M. A model for gastric cancer epidemiology. Lancet 1975, 2, 58–60. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Infection with Helicobacter pylori. In IARC Monographs on the Evaluation of the Carcinogenic Risk to Humans-Schistosomes, Liver Flukes, and Helicobacter pylori; International Agency for Research on Cancer: Lyon, France, 1994; Volume 61, pp. 177–240. [Google Scholar]
- Tytgat, G.N. Etiopathogenetic principles and peptic ulcer disease classification. Dig. Dis. 2011, 29, 454–458. [Google Scholar] [CrossRef]
- Ford, A.C.; Delaney, B.C.; Forman, D.; Moayyedi, P. Eradication therapy for peptic ulcer disease in Helicobacter pylori positive patients. Cochrane Database Syst. Rev. 2006, 2, CD003840. [Google Scholar]
- Gisbert, J.P.; Khorrami, S.; Carballo, F.; Calvet, X.; Gene, E.; Dominguez-Munoz, J.E. H. pylori eradication therapy vs. antisecretory non-eradication therapy (with or without long-term maintenance antisecretory therapy) for the prevention of recurrent bleeding from peptic ulcer. Cochrane Database Syst. Rev. 2003, 2, CD004062. [Google Scholar]
- Leodolter, A.; Kulig, M.; Brasch, H.; Meyer-Sabellek, W.; Willich, S.N.; Malfertheiner, P. A meta-analysis comparing eradication, healing and relapse rates in patients with Helicobacter pylori-associated gastric or duodenal ulcer. Aliment. Pharmacol. Ther. 2001, 15, 1949–1958. [Google Scholar] [CrossRef]
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Saenz, J.B.; Mills, J.C. Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Chronic gastritis: A clinico-pathological classification. Am. J. Gastroenterol. 1988, 83, 504–509. [Google Scholar]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H.; Correa, P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol 1996, 20, 1161–1181. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Svanes, C.; Stangeland, L.; Kvinnsland, S.; Svanes, K. Epithelial restitution and cellular proliferation after gastric mucosal damage caused by hypertonic NaCl in rats. Virchows Arch. A Pathol. Anat. Histopathol. 1988, 413, 445–455. [Google Scholar] [CrossRef]
- Malfertheiner, P. The intriguing relationship of Helicobacter pylori infection and acid secretion in peptic ulcer disease and gastric cancer. Dig. Dis. 2011, 29, 459–464. [Google Scholar] [CrossRef]
- Krueger, S.; Hundertmark, T.; Kuester, D.; Kalinski, T.; Peitz, U.; Roessner, A. Helicobacter pylori alters the distribution of ZO-1 and p120ctn in primary human gastric epithelial cells. Pathol. Res. Pract. 2007, 203, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M., Jr.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef]
- Oliveira, M.J.; Costa, A.M.; Costa, A.C.; Ferreira, R.M.; Sampaio, P.; Machado, J.C.; Seruca, R.; Mareel, M.; Figueiredo, C. CagA associates with c-Met, E-cadherin, and p120-catenin in a multiproteic complex that suppresses Helicobacter pylori-induced cell-invasive phenotype. J. Infect. Dis. 2009, 200, 745–755. [Google Scholar] [CrossRef]
- Song, X.; Chen, H.X.; Wang, X.Y.; Deng, X.Y.; Xi, Y.X.; He, Q.; Peng, T.L.; Chen, J.; Chen, W.; Wong, B.C.; et al. H. pylori-encoded CagA disrupts tight junctions and induces invasiveness of AGS gastric carcinoma cells via Cdx2-dependent targeting of Claudin-2. Cell Immunol. 2013, 286, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.R.; Vogelmann, R.; Covacci, A.; Tompkins, L.S.; Nelson, W.J.; Falkow, S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003, 300, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Sharafutdinov, I.; Harrer, A.; Musken, M.; Rottner, K.; Sticht, H.; Tager, C.; Naumann, M.; Tegtmeyer, N.; Backert, S. Cortactin-dependent control of Par1b-regulated epithelial cell polarity in Helicobacter infection. Cell Insight 2024, 3, 100161. [Google Scholar] [CrossRef]
- Bernegger, S.; Hutterer, E.; Zarzecka, U.; Schmidt, T.P.; Huemer, M.; Widlroither, I.; Posselt, G.; Skorko-Glonek, J.; Wessler, S. E-Cadherin Orthologues as Substrates for the Serine Protease High Temperature Requirement A (HtrA). Biomolecules 2022, 12, 356. [Google Scholar] [CrossRef] [PubMed]
- Weydig, C.; Starzinski-Powitz, A.; Carra, G.; Lower, J.; Wessler, S. CagA-independent disruption of adherence junction complexes involves E-cadherin shedding and implies multiple steps in Helicobacter pylori pathogenicity. Exp. Cell Res. 2007, 313, 3459–3471. [Google Scholar] [CrossRef]
- Vagin, O.; Dada, L.A.; Tokhtaeva, E.; Sachs, G. The Na-K-ATPase alpha(1)beta(1) heterodimer as a cell adhesion molecule in epithelia. Am. J. Physiol. Cell Physiol. 2012, 302, C1271–C1281. [Google Scholar] [CrossRef]
- Ricci, V.; Sommi, P.; Cova, E.; Fiocca, R.; Romano, M.; Ivey, K.J.; Solcia, E.; Ventura, U. Na+,K(+)-ATPase of gastric cells. A target of Helicobacter pylori cytotoxic activity. FEBS Lett. 1993, 334, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Marcus, E.A.; Tokhtaeva, E.; Jimenez, J.L.; Wen, Y.; Naini, B.V.; Heard, A.N.; Kim, S.; Capri, J.; Cohn, W.; Whitelegge, J.P.; et al. Helicobacter pylori infection impairs chaperone-assisted maturation of Na-K-ATPase in gastric epithelium. Am. J. Physiol. 2020, 318, G931–G945. [Google Scholar] [CrossRef]
- Jorgensen, P.L.; Hakansson, K.O.; Karlish, S.J. Structure and mechanism of Na,K-ATPase: Functional sites and their interactions. Annu. Rev. Physiol. 2003, 65, 817–849. [Google Scholar] [CrossRef]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef]
- Contreras, R.G.; Flores-Maldonado, C.; Lazaro, A.; Shoshani, L.; Flores-Benitez, D.; Larre, I.; Cereijido, M. Ouabain binding to Na+,K+-ATPase relaxes cell attachment and sends a specific signal (NACos) to the nucleus. J. Membr. Biol. 2004, 198, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.G.; Shoshani, L.; Flores-Maldonado, C.; Lazaro, A.; Cereijido, M. Relationship between Na(+),K(+)-ATPase and cell attachment. J. Cell Sci. 1999, 112 Pt 23, 4223–4232. [Google Scholar] [CrossRef]
- Rajasekaran, A.K.; Rajasekaran, S.A. Role of Na-K-ATPase in the assembly of tight junctions. Am. J. Physiol. Ren. Physiol. 2003, 285, F388–F396. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Hu, J.; Gopal, J.; Gallemore, R.; Ryazantsev, S.; Bok, D.; Rajasekaran, A.K. Na,K-ATPase inhibition alters tight junction structure and permeability in human retinal pigment epithelial cells. Am. J. Physiol. Cell Physiol. 2003, 284, C1497–C1507. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.A.; Palmer, L.G.; Moon, S.Y.; Peralta Soler, A.; Apodaca, G.L.; Harper, J.F.; Zheng, Y.; Rajasekaran, A.K. Na,K-ATPase activity is required for formation of tight junctions, desmosomes, and induction of polarity in epithelial cells. Mol. Biol. Cell 2001, 12, 3717–3732. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.A.; Palmer, L.G.; Quan, K.; Harper, J.F.; Ball, W.J., Jr.; Bander, N.H.; Peralta Soler, A.; Rajasekaran, A.K. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol. Biol. Cell 2001, 12, 279–295. [Google Scholar] [CrossRef]
- Shoshani, L.; Contreras, R.G.; Roldan, M.L.; Moreno, J.; Lazaro, A.; Balda, M.S.; Matter, K.; Cereijido, M. The polarized expression of Na+,K+-ATPase in epithelia depends on the association between beta-subunits located in neighboring cells. Mol. Biol. Cell 2005, 16, 1071–1081. [Google Scholar] [CrossRef]
- Vagin, O.; Tokhtaeva, E.; Sachs, G. The role of the beta1 subunit of the Na,K-ATPase and its glycosylation in cell-cell adhesion. J. Biol. Chem. 2006, 281, 39573–39587. [Google Scholar] [CrossRef]
- Tokhtaeva, E.; Sachs, G.; Souda, P.; Bassilian, S.; Whitelegge, J.P.; Shoshani, L.; Vagin, O. Epithelial junctions depend on intercellular trans-interactions between the Na,K-ATPase beta(1) subunits. J. Biol. Chem. 2011, 286, 25801–25812. [Google Scholar] [CrossRef]
- Vagin, O.; Tokhtaeva, E.; Yakubov, I.; Shevchenko, E.; Sachs, G. Inverse correlation between the extent of N-glycan branching and intercellular adhesion in epithelia. Contribution of the Na,K-ATPase beta1 subunit. J. Biol. Chem. 2008, 283, 2192–2202. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Rajasekaran, A.K. Na,K-ATPase and epithelial tight junctions. Front. Biosci. 2009, 14, 2130–2148. [Google Scholar] [CrossRef]
- Baltrus, D.A.; Amieva, M.R.; Covacci, A.; Lowe, T.M.; Merrell, D.S.; Ottemann, K.M.; Stein, M.; Salama, N.R.; Guillemin, K. The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 2009, 191, 447–448. [Google Scholar] [CrossRef]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petracca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N.; et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 5791–5795. [Google Scholar] [CrossRef]
- Chailler, P.; Menard, D. Establishment of human gastric epithelial (HGE) cell lines exhibiting barrier function, progenitor, and prezymogenic characteristics. J. Cell. Physiol. 2005, 202, 263–274. [Google Scholar] [CrossRef]
- Marcus, E.A.; Vagin, O.; Tokhtaeva, E.; Sachs, G.; Scott, D.R. Helicobacter pylori impedes acid-induced tightening of gastric epithelial junctions. Am. J. Physiol. 2013, 305, G731–G739. [Google Scholar] [CrossRef]
- Akera, T.; Larsen, F.S.; Brody, T.M. Correlation of cardiac sodium- and potassium-activated adenosine triphosphatase activity with ouabain-induced inotropic stimulation. J. Pharmacol. Exp. Ther. 1970, 173, 145–151. [Google Scholar]
- Smith, T.W.; Wagner, H., Jr.; Markis, J.E.; Young, M. Studies on the localization of the cardiac glycoside receptor. J. Clin. Investig. 1972, 51, 1777–1789. [Google Scholar] [CrossRef]
- Habeck, M.; Tokhtaeva, E.; Nadav, Y.; Ben Zeev, E.; Ferris, S.P.; Kaufman, R.J.; Bab-Dinitz, E.; Kaplan, J.H.; Dada, L.A.; Farfel, Z.; et al. Selective Assembly of Na,K-ATPase alpha2beta2 Heterodimers in the Heart: Distinct Functional Properties and Isoform-Selective Inhibitors. J. Biol. Chem. 2016, 291, 23159–23174. [Google Scholar] [CrossRef]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. 1998, 275, F633–F650. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B. Na,K-ATPase: Isoform structure, function, and expression. J. Bioenerg. Biomembr. 1992, 24, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Huang, L.; Xie, Z.; Huang, W.H.; Askari, A. Partial inhibition of Na+/K+-ATPase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J. Biol. Chem. 1996, 271, 10372–10378. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.; Grupp, G.; Wallick, E.; Grupp, I.L.; Ball, W.J., Jr. Role of the Na+K+-ATPase in the cardiotonic action of cardiac glycosides. Prog. Clin. Biol. Res. 1988, 268B, 321–338. [Google Scholar]
- Sweadner, K.J. Isozymes of the Na+/K+-ATPase. Biochim. Biophys. Acta 1989, 988, 185–220. [Google Scholar] [CrossRef]
- Sweadner, K.J. Overview: Subunit diversity in the Na,K-ATPase. Soc. Gen. Physiol. Ser. 1991, 46, 63–76. [Google Scholar] [PubMed]
- Xie, Z.J.; Wang, Y.H.; Ganjeizadeh, M.; McGee, R., Jr.; Askari, A. Determination of total (Na+ + K+)-ATPase activity of isolated or cultured cells. Anal. Biochem. 1989, 183, 215–219. [Google Scholar] [CrossRef]
- Sun, Y.Q.; Soderholm, J.D.; Petersson, F.; Borch, K. Long-standing gastric mucosal barrier dysfunction in Helicobacter pylori-induced gastritis in mongolian gerbils. Helicobacter 2004, 9, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Indra, I.; Choi, J.; Chen, C.S.; Troyanovsky, R.B.; Shapiro, L.; Honig, B.; Troyanovsky, S.M. Spatial and temporal organization of cadherin in punctate adherens junctions. Proc. Natl. Acad. Sci. USA 2018, 115, E4406–E4415. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; Contreras, R.G.; Shoshani, L.; Larre, I. The Na+-K+-ATPase as self-adhesion molecule and hormone receptor. Am. J. Physiol. Cell Physiol. 2012, 302, C473–C481. [Google Scholar] [CrossRef] [PubMed]
- Marcus, E.A.; Tokhtaeva, E.; Scott, D.R.; Naini, B.; Sachs, G.; Vagin, O. Helicobacter pylori infection decreases expression of the Na,K-ATPase in gastric epithelial cells, resulting in acid-induced gastric injury. Gastroenterology 2016, 150, S36. [Google Scholar] [CrossRef]
- Brody, T.M. Ouabain-induced inhibition of cardiac (Na+ plus K+)-ATPase and the positive inotropic response. Ann. N. Y. Acad. Sci. 1974, 242, 684–687. [Google Scholar] [CrossRef]
- Noto, J.M.; Romero-Gallo, J.; Piazuelo, M.B.; Peek, R.M. The Mongolian Gerbil: A Robust Model of Helicobacter pylori-Induced Gastric Inflammation and Cancer. Methods Mol. Biol. 2016, 1422, 263–280. [Google Scholar] [CrossRef]
- Capaldo, C.T.; Farkas, A.E.; Nusrat, A. Epithelial adhesive junctions. F1000Prime Rep. 2014, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Troyanovsky, S.M. Adherens junction: The ensemble of specialized cadherin clusters. Trends Cell Biol. 2023, 33, 374–387. [Google Scholar] [CrossRef]
- Laughery, M.D.; Todd, M.L.; Kaplan, J.H. Mutational analysis of alpha-beta subunit interactions in the delivery of Na,K-ATPase heterodimers to the plasma membrane. J. Biol. Chem. 2003, 278, 34794–34803. [Google Scholar] [CrossRef] [PubMed]
- Tamkun, M.M.; Fambrough, D.M. The (Na+ + K+)-ATPase of chick sensory neurons. Studies on biosynthesis and intracellular transport. J. Biol. Chem. 1986, 261, 1009–1019. [Google Scholar] [CrossRef]
- Beggah, A.T.; Jaunin, P.; Geering, K. Role of glycosylation and disulfide bond formation in the beta subunit in the folding and functional expression of Na,K-ATPase. J. Biol. Chem. 1997, 272, 10318–10326. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Noguchi, S.; Sugino, A.; Kawamura, M. Functional activity of oligosaccharide-deficient (Na,K)ATPase expressed in Xenopus oocytes. FEBS Lett. 1988, 238, 201–204. [Google Scholar] [CrossRef]
- Zamofing, D.; Rossier, B.C.; Geering, K. Inhibition of N-glycosylation affects transepithelial Na+ but not Na+-K+-ATPase transport. Am. J. Physiol. 1989, 256, C958–C966. [Google Scholar] [CrossRef]
- Dada, L.A.; Chandel, N.S.; Ridge, K.M.; Pedemonte, C.; Bertorello, A.M.; Sznajder, J.I. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J. Clin. Investig. 2003, 111, 1057–1064. [Google Scholar] [CrossRef]
- Zhou, G.; Dada, L.A.; Sznajder, J.I. Regulation of alveolar epithelial function by hypoxia. Eur. Respir. J. 2008, 31, 1107–1113. [Google Scholar] [CrossRef]
- Factor, P.; Dumasius, V.; Saldias, F.; Sznajder, J.I. Adenoviral-mediated overexpression of the NA,K-ATPase beta1 subunit gene increases lung edema clearance and improves survival during acute hyperoxic lung injury in rats. Chest 1999, 116, 24S–25S. [Google Scholar] [CrossRef] [PubMed]
- Factor, P.; Saldias, F.; Ridge, K.; Dumasius, V.; Zabner, J.; Jaffe, H.A.; Blanco, G.; Barnard, M.; Mercer, R.; Perrin, R.; et al. Augmentation of lung liquid clearance via adenovirus-mediated transfer of a Na,K-ATPase beta1 subunit gene. J. Clin. Investig. 1998, 102, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Machado-Aranda, D.; Adir, Y.; Young, J.L.; Briva, A.; Budinger, G.R.; Yeldandi, A.V.; Sznajder, J.I.; Dean, D.A. Gene transfer of the Na+,K+-ATPase beta1 subunit using electroporation increases lung liquid clearance. Am. J. Respir. Crit. Care Med. 2005, 171, 204–211. [Google Scholar] [CrossRef]
- Madan, P.; Rose, K.; Watson, A.J. Na/K-ATPase beta1 subunit expression is required for blastocyst formation and normal assembly of trophectoderm tight junction-associated proteins. J. Biol. Chem. 2007, 282, 12127–12134. [Google Scholar] [CrossRef] [PubMed]
- Violette, M.I.; Madan, P.; Watson, A.J. Na+/K+ -ATPase regulates tight junction formation and function during mouse preimplantation development. Dev. Biol. 2006, 289, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, T.; Beitel, G.J. Unexpected roles of the Na-K-ATPase and other ion transporters in cell junctions and tubulogenesis. Physiology 2009, 24, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Sive, H. Initial formation of zebrafish brain ventricles occurs independently of circulation and requires the nagie oko and snakehead/atp1a1a.1 gene products. Development 2005, 132, 2057–2067. [Google Scholar] [CrossRef]
- Shu, X.; Cheng, K.; Patel, N.; Chen, F.; Joseph, E.; Tsai, H.J.; Chen, J.N. Na,K-ATPase is essential for embryonic heart development in the zebrafish. Development 2003, 130, 6165–6173. [Google Scholar] [CrossRef]
- Yuan, S.; Joseph, E.M. The small heart mutation reveals novel roles of Na+/K+-ATPase in maintaining ventricular cardiomyocyte morphology and viability in zebrafish. Circ. Res. 2004, 95, 595–603. [Google Scholar] [CrossRef]
- Cibrian-Uhalte, E.; Langenbacher, A.; Shu, X.; Chen, J.N.; Abdelilah-Seyfried, S. Involvement of zebrafish Na+,K+ ATPase in myocardial cell junction maintenance. J. Cell Biol. 2007, 176, 223–230. [Google Scholar] [CrossRef]
- Fiocca, R.; Villani, L.; Turpini, F.; Turpini, R.; Solcia, E. High incidence of Campylobacter-like organisms in endoscopic biopsies from patients with gastritis, with or without peptic ulcer. Digestion 1987, 38, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Stremming, J.; Jansson, T.; Powell, T.L.; Rozance, P.J.; Brown, L.D. Reduced Na(+) K(+)-ATPase activity may reduce amino acid uptake in intrauterine growth restricted fetal sheep muscle despite unchanged ex vivo amino acid transporter activity. J. Physiol. 2020, 598, 1625–1639. [Google Scholar] [CrossRef]
- Palaniappan, B.; Arthur, S.; Sundaram, V.L.; Butts, M.; Sundaram, S.; Mani, K.; Singh, S.; Nepal, N.; Sundaram, U. Inhibition of intestinal villus cell Na/K-ATPase mediates altered glucose and NaCl absorption in obesity-associated diabetes and hypertension. FASEB J. 2019, 33, 9323–9333. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Zhang, J.; Chen, L.; Song, H.; Raina, H.; Kinsey, S.P.; Izuka, M.; Iwamoto, T.; Kotlikoff, M.I.; Lingrel, J.B.; et al. The pump, the exchanger, and endogenous ouabain: Signaling mechanisms that link salt retention to hypertension. Hypertension 2009, 53, 291–298. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vagin, O.; Tokhtaeva, E.; Larauche, M.; Davood, J.; Marcus, E.A. Helicobacter pylori-Induced Decrease in Membrane Expression of Na,K-ATPase Leads to Gastric Injury. Biomolecules 2024, 14, 772. https://doi.org/10.3390/biom14070772
Vagin O, Tokhtaeva E, Larauche M, Davood J, Marcus EA. Helicobacter pylori-Induced Decrease in Membrane Expression of Na,K-ATPase Leads to Gastric Injury. Biomolecules. 2024; 14(7):772. https://doi.org/10.3390/biom14070772
Chicago/Turabian StyleVagin, Olga, Elmira Tokhtaeva, Muriel Larauche, Joshua Davood, and Elizabeth A. Marcus. 2024. "Helicobacter pylori-Induced Decrease in Membrane Expression of Na,K-ATPase Leads to Gastric Injury" Biomolecules 14, no. 7: 772. https://doi.org/10.3390/biom14070772