
De Novo Design of Inhibitors of DNA Methyltransferase 1: A Critical Comparison of Ligand- and Structure-Based Approaches

 , , , , and
, , , , and 
Abstract
:
1. Introduction
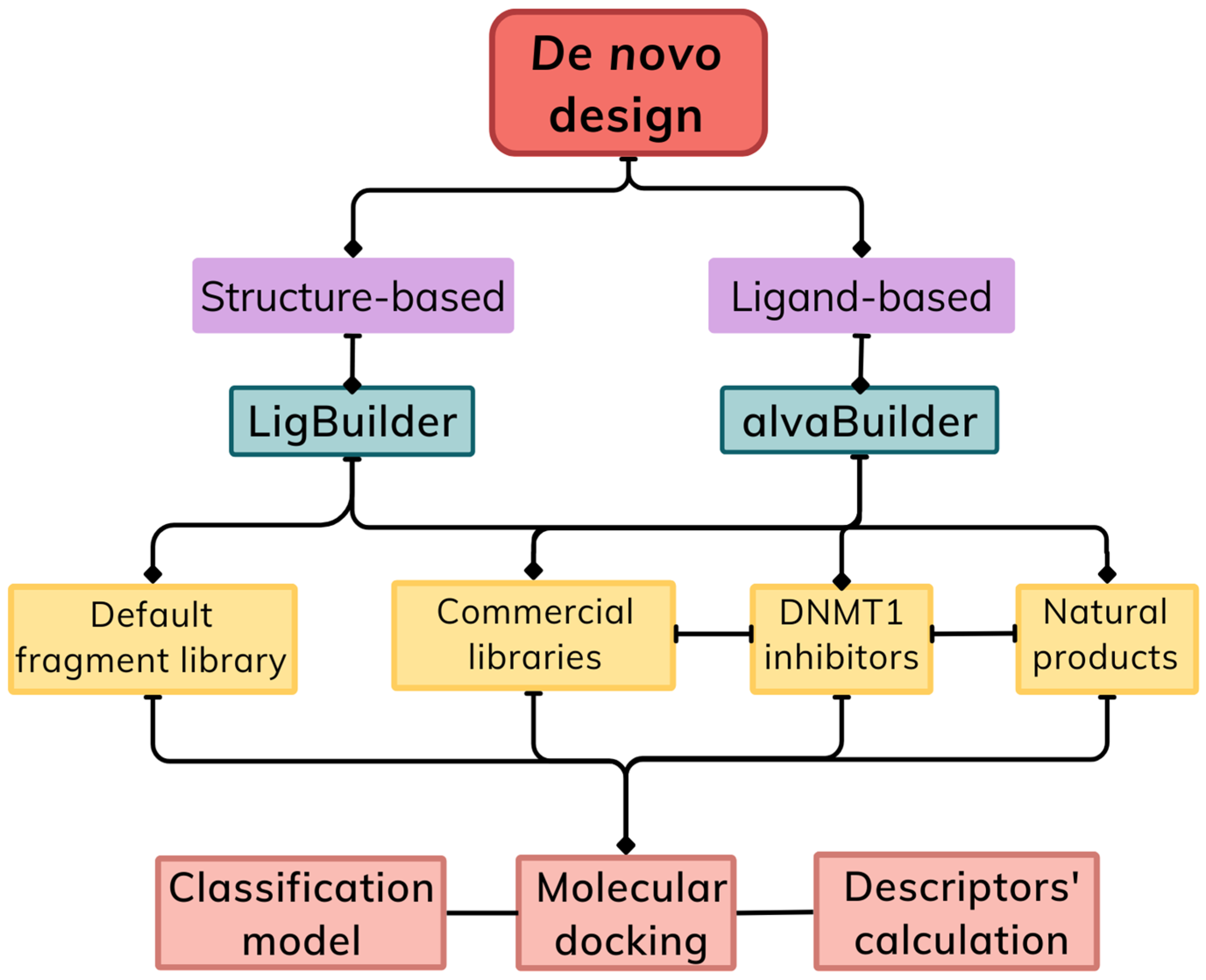
2. Methods
2.1. Data Curation
2.2. Ligand-Based De Novo Design
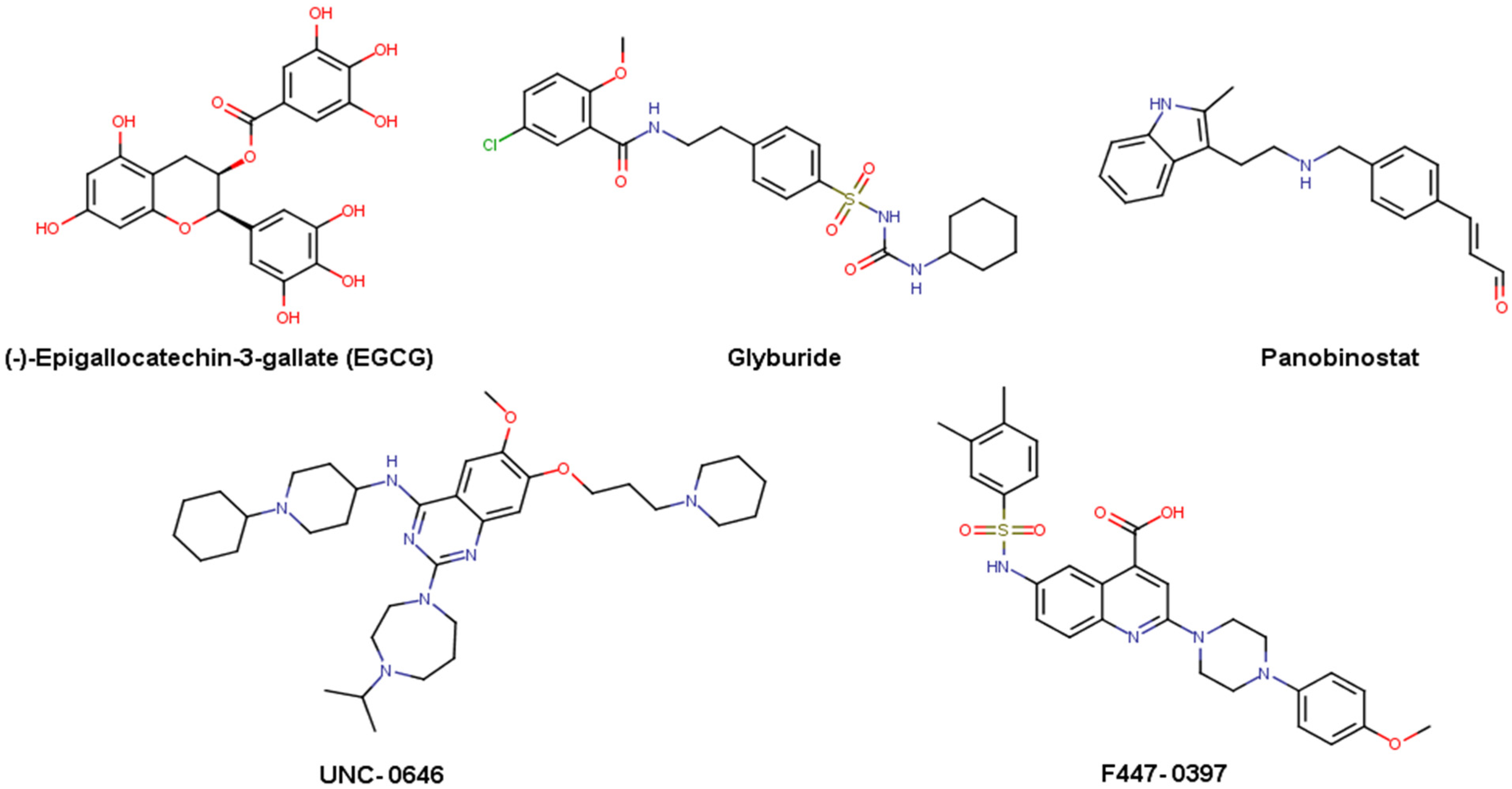
Training Chemical Libraries
2.3. Structure-Based De Novo Design
2.3.1. Binding Site Detection
2.3.2. Molecule Design
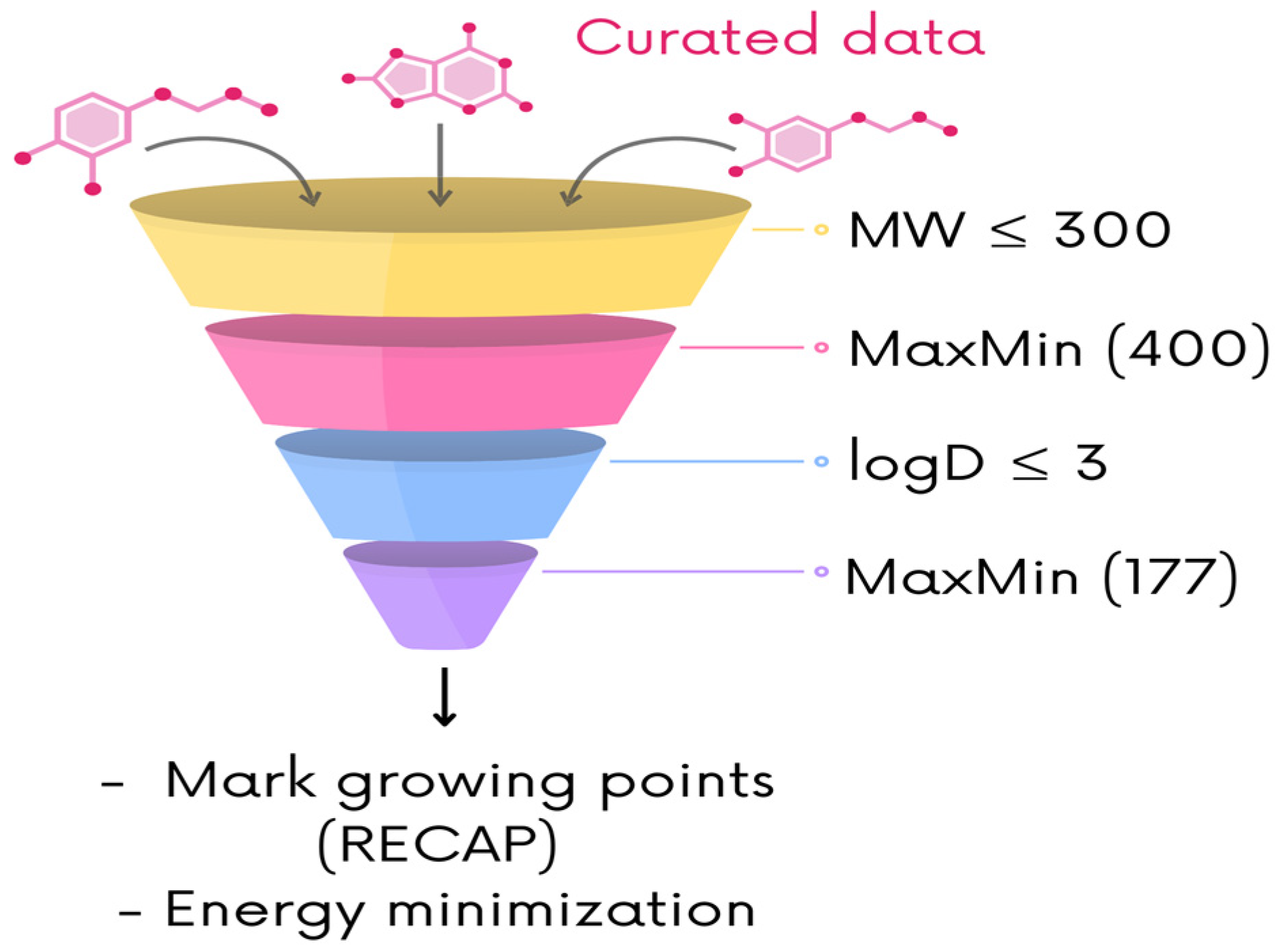
2.3.3. Fragment Libraries

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Description | Number of Fragments |
|---|---|---|
| LigBuilder default [22] | Common chemical groups and ring frameworks observed in organic compounds. | 177 |
| ChemDiv Fragments Library [60] | Fragments with desirable properties, including diversity. | 11,269 |
| ChemDiv Epigenetics Fragments [61] | Privileged fragments focused on epigenetic regulators. | 9196 |
| ChEMBL actives [23,45] | Fragments from ChEMBL actives (Table 1) obtained with RECAP. | 1645 |
| FooDB [23,46] | Fragments from FooDB compounds up to 1500 Da obtained with RECAP. | 225,206 |
| Life Chemicals Soluble Fragments [62] | Diversity-oriented fragment library with experimental solubility data. | 1280 |
| Selleckchem [63] | A collection of fragments for Fragment-Based Drug Discovery. | 1015 |
| UNPD-A [23,49,50] | Fragments from UNPD-A up to 1500 Da (Table 1) obtained with RECAP. | 412,110 |
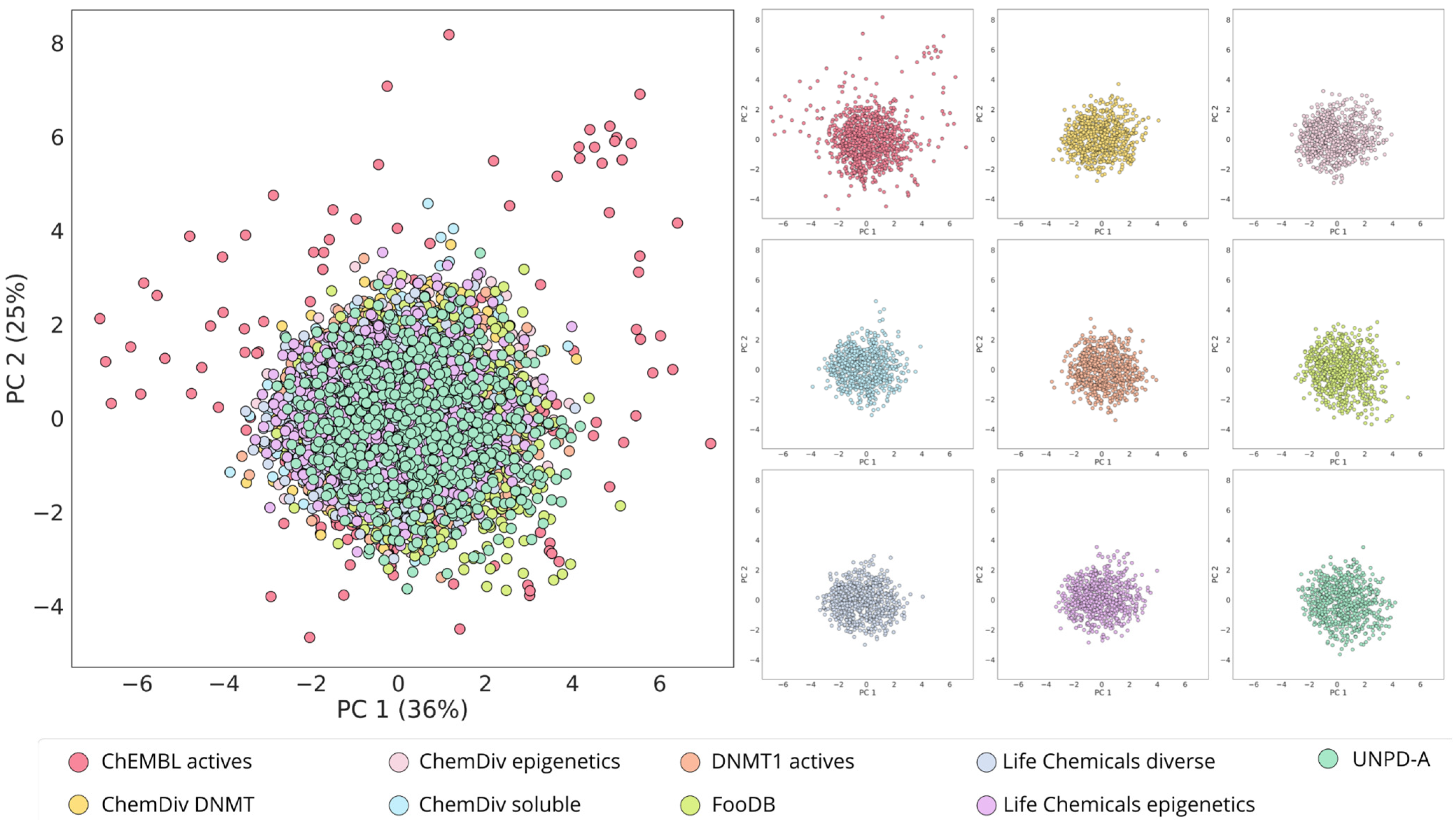
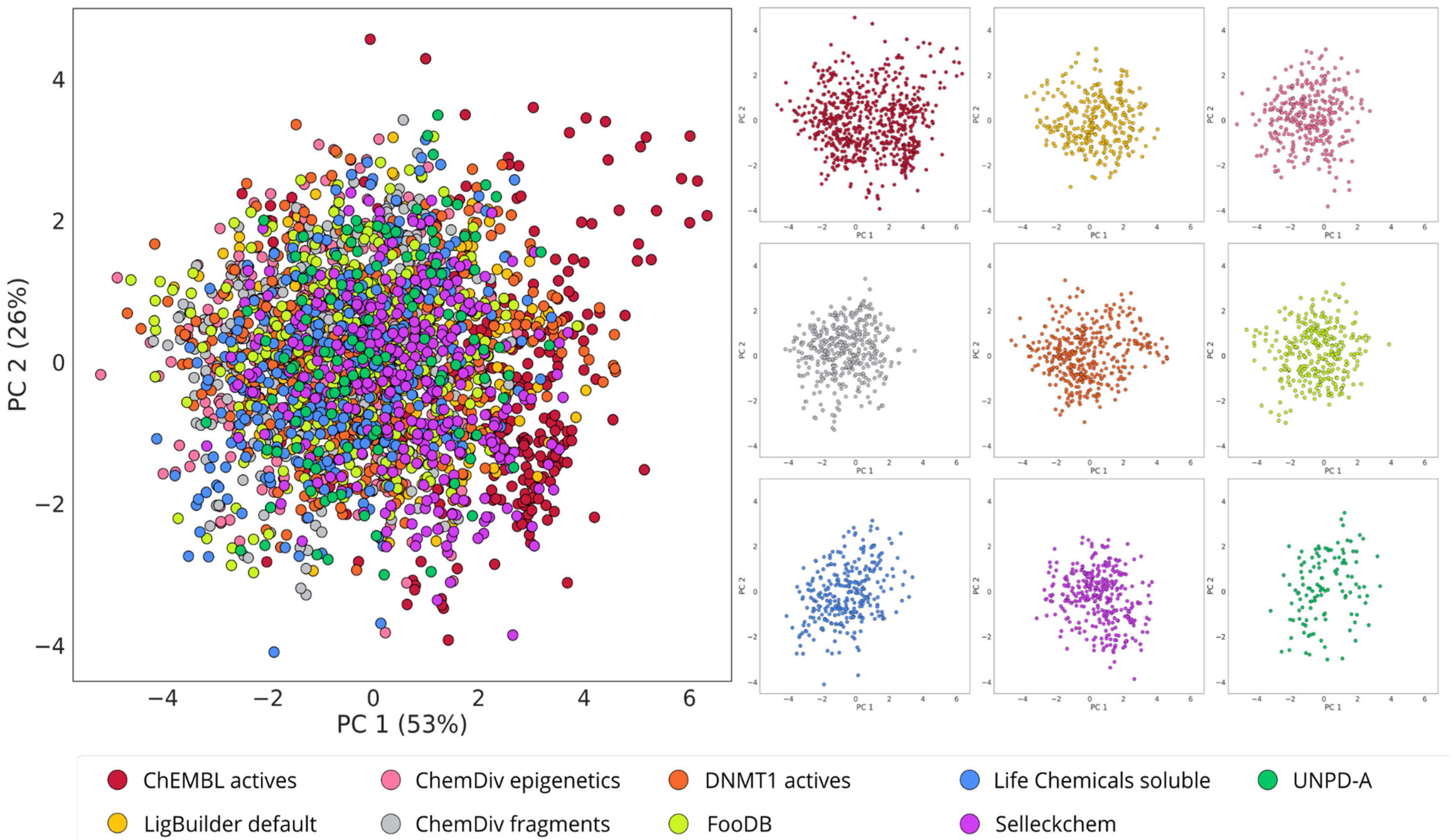
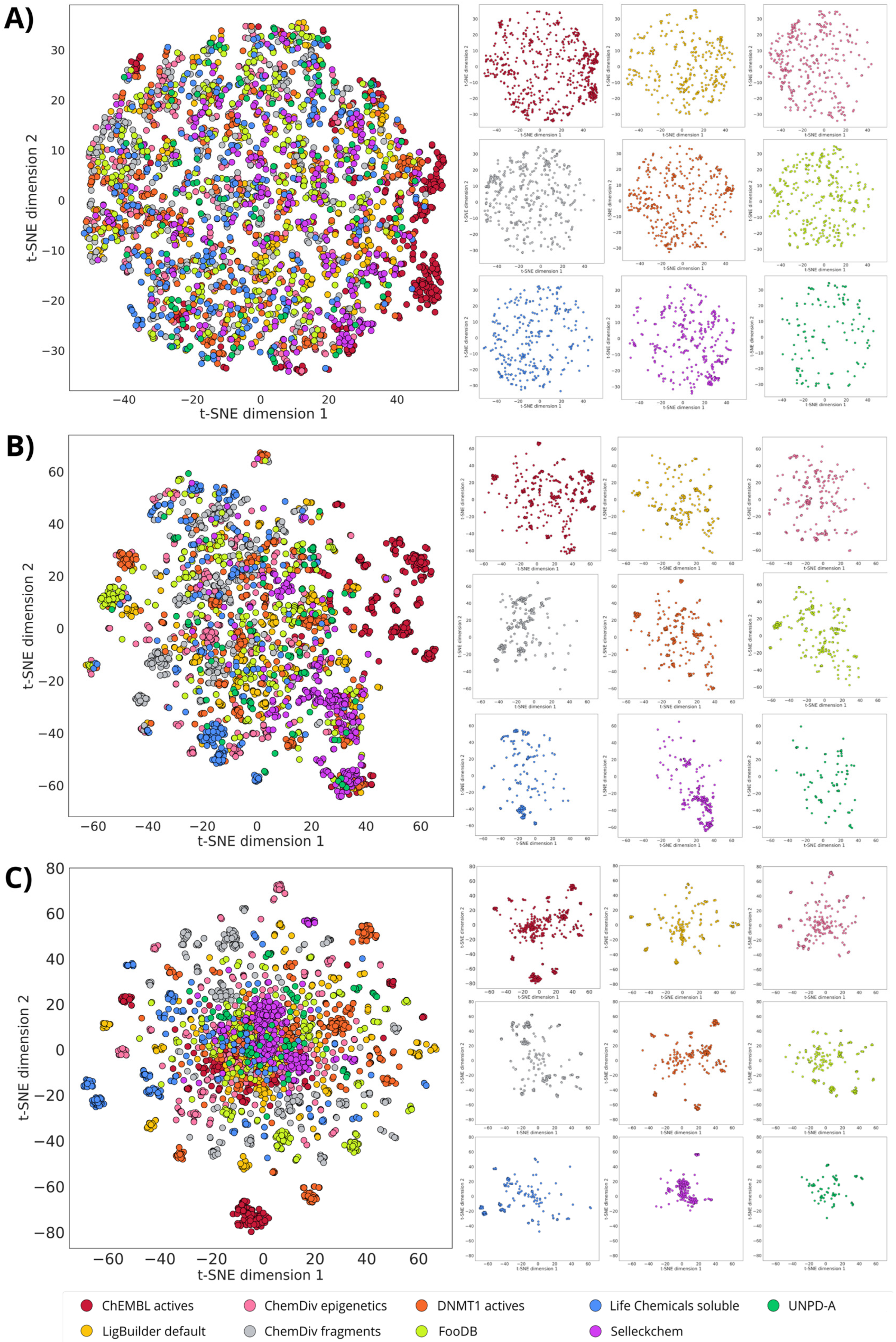
2.4. Visualization of the Chemical Space
2.5. Drug Likeness and Synthetic Accessibility
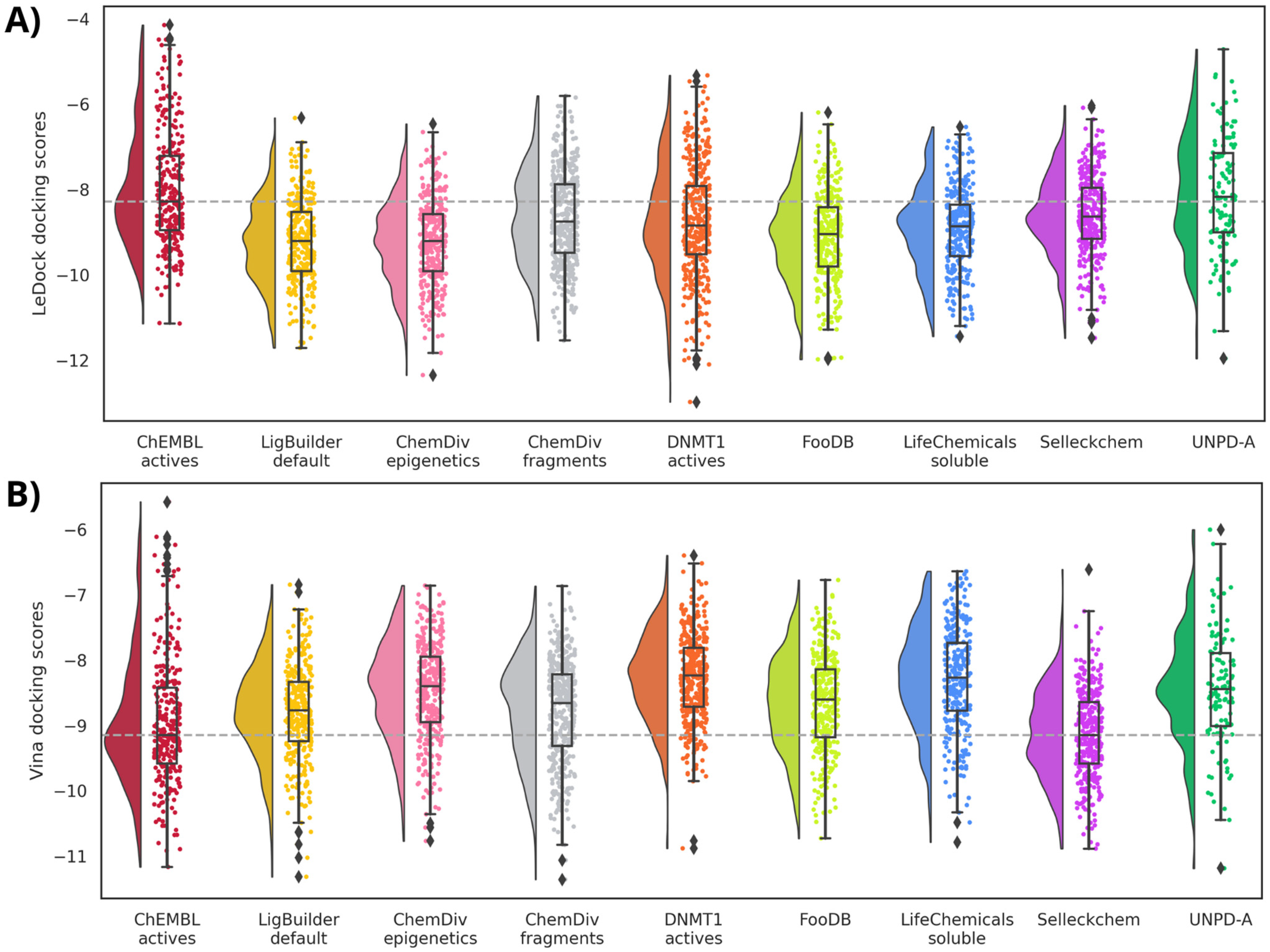
2.6. Molecular Docking
2.6.1. Docking with Vina
2.6.2. Docking with LeDock
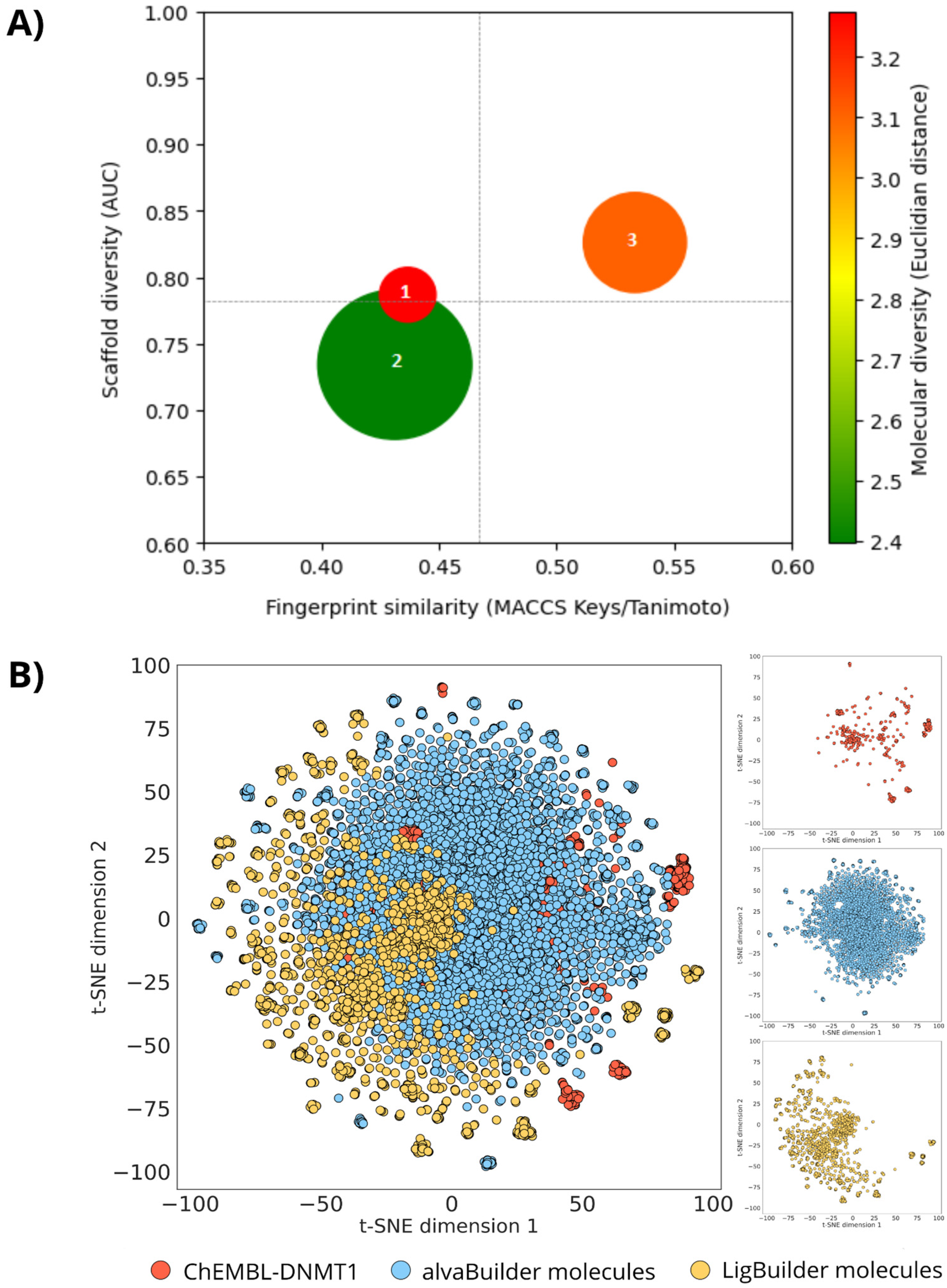
2.7. Global Diversity and Scaffold Analysis
Molecular Descriptors
2.8. Classification Models
2.8.1. Dataset
2.8.2. Molecular Representations
2.8.3. Machine Learning Methods
2.9. Molecular Dynamics
3. Results and Discussion
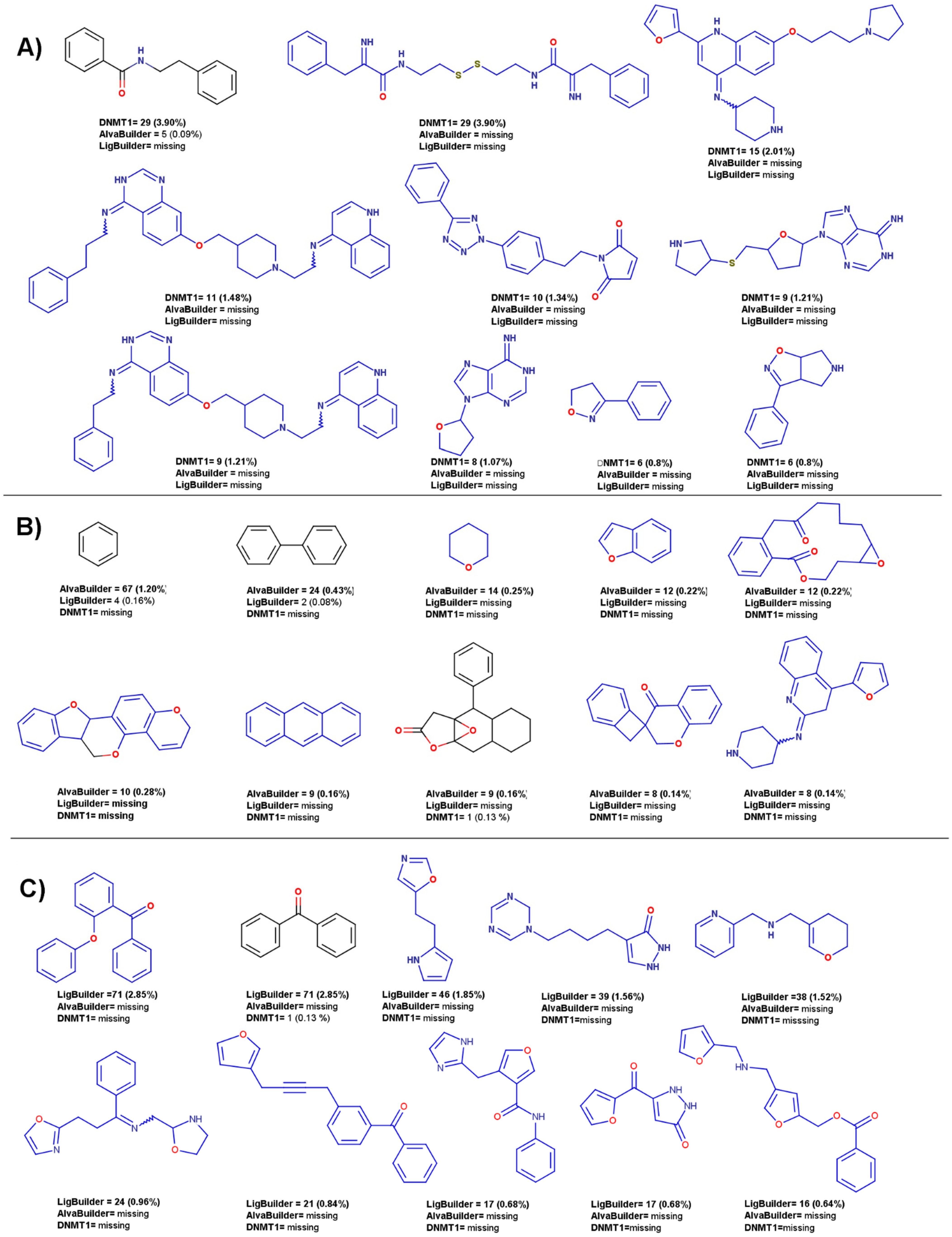
3.1. Ligand-Based De Novo Design with alvaBuilder
3.2. Structure-Based De Novo Design with LigBuilder
3.3. Global Diversity and Scaffold Analysis
3.4. Classification Models
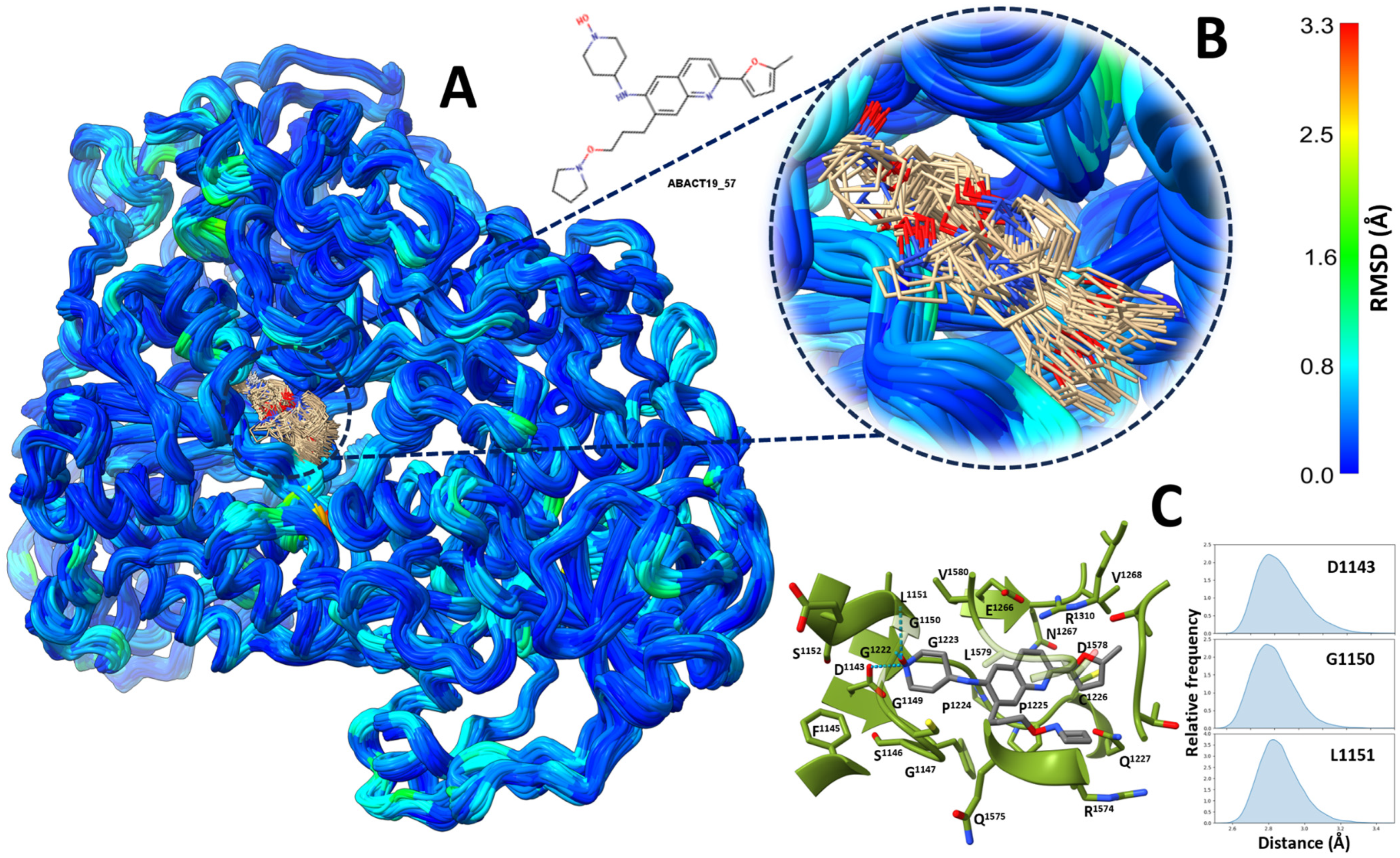
3.5. Molecular Dynamics
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jiménez-Luna, J.; Grisoni, F.; Weskamp, N.; Schneider, G. Artificial Intelligence in Drug Discovery: Recent Advances and Future Perspectives. Expert Opin. Drug Discov. 2021, 16, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.I.; Jzerman, A.P.; van Westen, G.J.P. Computational Approaches for De Novo Drug Design: Past, Present, and Future. Methods Mol. Biol. 2021, 2190, 139–165. [Google Scholar] [PubMed]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef] [PubMed]
- Hartenfeller, M.; Schneider, G. Enabling Future Drug Discovery by de Novo Design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 742–759. [Google Scholar] [CrossRef]
- Focused and Targeted Libraries. Available online: https://www.chemdiv.com/catalog/focused-and-targeted-libraries/ (accessed on 2 November 2023).
- Active Reference Sets. Available online: https://www.chemdiv.com/catalog/sets/ (accessed on 2 November 2023).
- Zhang, Z.; Wang, G.; Li, Y.; Lei, D.; Xiang, J.; Ouyang, L.; Wang, Y.; Yang, J. Recent Progress in DNA Methyltransferase Inhibitors as Anticancer Agents. Front. Pharmacol. 2022, 13, 1072651. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xie, T.; Wang, Z.; Wang, X.; Zeng, S.; Kang, Y.; Hou, T. DNA Methyltransferases: Emerging Targets for the Discovery of Inhibitors as Potent Anticancer Drugs. Drug Discov. Today 2019, 24, 2323–2331. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, Y.; Li, L.; Tang, J.; Zhang, R. DNA Methylation Profiles in Cancer: Functions, Therapy, and beyond. Cancer Biol. Med. 2023, 21, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.; Hahn, K.; Kolesar, J.M. Azacitidine: A Novel Agent for Myelodysplastic Syndromes. Am. J. Health. Syst. Pharm. 2005, 62, 1567–1573. [Google Scholar] [CrossRef]
- Derissen, E.J.B.; Beijnen, J.H.; Schellens, J.H.M. Concise Drug Review: Azacitidine and Decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef]
- Zwergel, C.; Valente, S.; Mai, A. DNA Methyltransferases Inhibitors from Natural Sources. Curr. Top. Med. Chem. 2016, 16, 680–696. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Gómez-García, A.; Chávez-Ponce de León, D.E.; Sánchez-Cruz, N.; Ruiz-Rios, J.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Inhibitors of DNA Methyltransferases From Natural Sources: A Computational Perspective. Front. Pharmacol. 2018, 9, 1144. [Google Scholar] [CrossRef] [PubMed]
- Flores-Padilla, E.A.; Juárez-Mercado, K.E.; Naveja, J.J.; Kim, T.D.; Alain Miranda-Quintana, R.; Medina-Franco, J.L. Chemoinformatic Characterization of Synthetic Screening Libraries Focused on Epigenetic Targets. Mol. Inform. 2022, 41, e2100285. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Mercado, K.E.; Prieto-Martínez, F.D.; Sánchez-Cruz, N.; Peña-Castillo, A.; Prada-Gracia, D.; Medina-Franco, J.L. Expanding the Structural Diversity of DNA Methyltransferase Inhibitors. Pharmaceuticals 2020, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; López-López, E.; Martínez-Fernández, L.P. 7-Aminoalkoxy-Quinazolines from Epigenetic Focused Libraries Are Potent and Selective Inhibitors of DNA Methyltransferase 1. Molecules 2022, 27, 2892. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-S.; Zheng, Q.-C. Theoretical Research in Structure Characteristics of Different Inhibitors and Differences of Binding Modes with CBP Bromodomain. Bioorg. Med. Chem. 2018, 26, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Prado-Romero, D.L.; Medina-Franco, J.L. Advances in the Exploration of the Epigenetic Relevant Chemical Space. ACS Omega 2021, 6, 22478–22486. [Google Scholar] [CrossRef] [PubMed]
- Prado-Romero, D.L.; Gómez-García, A.; Cedillo-González, R.; Villegas-Quintero, H.; Avellaneda-Tamayo, J.F.; López-López, E.; Saldívar-González, F.I.; Chávez-Hernández, A.L.; Medina-Franco, J.L. Consensus Docking Aid to Model the Activity of an Inhibitor of DNA Methyltransferase 1 Inspired by de Novo Design. Front. Drug Des. Discov. 2023, 3, 1261094. [Google Scholar] [CrossRef]
- Lanka, G.; Banerjee, S.; Adhikari, N.; Ghosh, B. Fragment-Based Discovery of New Potential DNMT1 Inhibitors Integrating Multiple Pharmacophore Modeling, 3D-QSAR, Virtual Screening, Molecular Docking, ADME, and Molecular Dynamics Simulation Approaches. Mol. Divers. 2024. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A.; Bertola, M. AlvaBuilder: A Software for De Novo Molecular Design. J. Chem. Inf. Model. 2023, 64, 2136–2142. [Google Scholar] [CrossRef]
- Yuan, Y.; Pei, J.; Lai, L. LigBuilder V3: A Multi-Target de Novo Drug Design Approach. Front. Chem. 2020, 8, 142. [Google Scholar] [CrossRef]
- Lewell, X.Q.; Judd, D.B.; Watson, S.P.; Hann, M.M. RECAP—Retrosynthetic Combinatorial Analysis Procedure: A Powerful New Technique for Identifying Privileged Molecular Fragments with Useful Applications in Combinatorial Chemistry. J. Chem. Inf. Comput. Sci. 1998, 38, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lephar Research Software. Available online: http://www.lephar.com/software.htm (accessed on 12 July 2023).
- Sánchez-Cruz, N.; Medina-Franco, J.L. Epigenetic Target Fishing with Accurate Machine Learning Models. J. Med. Chem. 2021, 64, 8208–8220. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cruz, N.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Functional Group and Diversity Analysis of BIOFACQUIM: A Mexican Natural Product Database. F1000Research 2019, 8, 1–14. [Google Scholar] [CrossRef]
- DIFACQUIM. IFG_General: Repository for the Work Functional Group and Diversity Analysis of BIOFACQUIM: A Mexican Natural Product Database; Github: San Francisco, CA, USA, 2020. [Google Scholar]
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org (accessed on 5 December 2023).
- MolVS: Molecule Validation and Standardization—MolVS 0.1.1 Documentation. Available online: https://molvs.readthedocs.io/en/latest/ (accessed on 5 December 2023).
- Sun, J.; Jeliazkova, N.; Chupakin, V.; Golib-Dzib, J.-F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; et al. ExCAPE-DB: An Integrated Large Scale Dataset Facilitating Big Data Analysis in Chemogenomics. J. Cheminform. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- López-López, E.; Fernández-de Gortari, E.; Medina-Franco, J.L. Yes SIR! On the Structure-Inactivity Relationships in Drug Discovery. Drug Discov. Today 2022, 27, 2353–2362. [Google Scholar] [CrossRef]
- Mauri, A. alvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints. In Ecotoxicological QSARs; Roy, K., Ed.; Springer: New York, NY, USA, 2020; pp. 801–820. ISBN 9781071601501. [Google Scholar]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “Rule of Three” for Fragment-Based Lead Discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Alvascience, alvaMolecule (Software to View and Prepare Chemical Datasets) Version 1.0.4, 2020. Available online: https://www.alvascience.com/ (accessed on 15 November 2023).
- Leach, A.R.; Gillet, V.J. (Eds.) Selecting Diverse Sets of Compounds. In An Introduction to Chemoinformatics; Springer: Dordrecht, The Netherlands, 2007; pp. 119–139. ISBN 9781402062919. [Google Scholar]
- Jaccard, P. THE DISTRIBUTION OF THE FLORA IN THE ALPINE ZONE. New Phytol. 1912, 11, 37–50. [Google Scholar]
- Tanimoto, T.T. An Elementary Mathematical Theory of Classification and Prediction; International Business Machines Corporation: Armonk, NY, USA, 1958. [Google Scholar]
- DNMT-Targeted Library. Available online: https://www.chemdiv.com/catalog/focused-and-targeted-libraries/dnmt-targeted-library/ (accessed on 8 November 2023).
- Epigenetics Focused Set. Available online: https://www.chemdiv.com/catalog/focused-and-targeted-libraries/epigenetics-focused-set/ (accessed on 8 November 2023).
- Soluble Diversity Library. Available online: https://www.chemdiv.com/catalog/diversity-libraries/soluble-diversity-library/ (accessed on 24 February 2023).
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [PubMed]
- FooDB Version 1.0. Available online: https://foodb.ca/ (accessed on 1 December 2023).
- Diversity Screening Libraries. Available online: https://lifechemicals.com/screening-libraries/pre-plated-diversity-sets (accessed on 13 March 2023).
- Epigenetic Screening Libraries. Available online: https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/epigenetic-screening-libraries (accessed on 8 November 2023).
- Chávez-Hernández, A.L.; Medina-Franco, J.L. Natural Products Subsets: Generation and Characterization. Artif. Intell. Life Sci. 2023, 3, 100066. [Google Scholar] [CrossRef]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.-Z.; Xu, X. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pei, J.; Lai, L. LigBuilder 2: A Practical de Novo Drug Design Approach. J. Chem. Inf. Model. 2011, 51, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Fechner, U. Computer-Based de Novo Design of Drug-like Molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pei, J.; Lai, L. Binding Site Detection and Druggability Prediction of Protein Targets for Structure-Based Drug Design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications; Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. ISBN 9783540782391. [Google Scholar]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program for Chemistry Aware Data Visualization and Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Systèmes, D. BIOVIA Discovery Studio Visualizer. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 24 January 2024).
- The PyMOL Molecular Graphics System, Version 2.5 Schrödinger, LLC. Available online: https://pymol.org/ (accessed on 24 January 2024).
- Fragments Library. Available online: https://www.chemdiv.com/catalog/focused-and-targeted-libraries/fragments-library/ (accessed on 8 November 2023).
- Privileged Fragments Annotated Library. Available online: https://www.chemdiv.com/catalog/focused-and-targeted-libraries/privileged-fragments-annotated-library/ (accessed on 9 February 2024).
- Diversity Screening Subsets of Soluble Fragments. Available online: https://lifechemicals.com/fragment-libraries/soluble-fragment-diversity-subsets (accessed on 21 February 2024).
- Compound Libraries for High Throughput/Content Screening. Available online: https://www.selleckchem.com/screening/fragment-library.html (accessed on 25 August 2023).
- Molecular Operating Environment (MOE), 2022.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2024.
- Van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Gerber, P.R.; Müller, K. MAB, a Generally Applicable Molecular Force Field for Structure Modelling in Medicinal Chemistry. J. Comput. Aided Mol. Des. 1995, 9, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, S.; Gelatt, C.D., Jr.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef]
- González-Medina, M.; Prieto-Martínez, F.D.; Owen, J.R.; Medina-Franco, J.L. Consensus Diversity Plots: A Global Diversity Analysis of Chemical Libraries. J. Cheminform. 2016, 8, 63. [Google Scholar] [CrossRef]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Medina-Franco, J.; Martà nez-Mayorga, K.; Bender, A.; Scior, T. Scaffold Diversity Analysis of Compound Data Sets Using an Entropy-Based Measure. QSAR Comb. Sci. 2009, 28, 1551–1560. [Google Scholar] [CrossRef]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: A Drug Discovery Platform Spanning Multiple Bioactivity Data Types and Time Periods. Nucleic Acids Res. 2024, 52, D1180–D1192. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Altman, N.S. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am. Stat. 1992, 46, 175. [Google Scholar] [CrossRef]
- Ho, T.K. The Random Subspace Method for Constructing Decision Forests. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 832–844. [Google Scholar]
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Hopfield, J.J. Neural Networks and Physical Systems with Emergent Collective Computational Abilities. Proc. Natl. Acad. Sci. USA 1982, 79, 2554–2558. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-Learn: Machine Learning in Python. arXiv 2012, arXiv:1201.0490. [Google Scholar]
- Tetko, I.V.; Sushko, I.; Pandey, A.K.; Zhu, H.; Tropsha, A.; Papa, E.; Oberg, T.; Todeschini, R.; Fourches, D.; Varnek, A. Critical Assessment of QSAR Models of Environmental Toxicity against Tetrahymena Pyriformis: Focusing on Applicability Domain and Overfitting by Variable Selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Cherkasov, A.; Li, J.; Gramatica, P.; Hansen, K.; Schroeter, T.; Müller, K.-R.; et al. Applicability Domains for Classification Problems: Benchmarking of Distance to Models for Ames Mutagenicity Set. J. Chem. Inf. Model. 2010, 50, 2094–2111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-M.; Liu, S.; Lin, K.; Luo, Y.; Perry, J.J.; Wang, Y.; Song, J. Crystal Structure of Human DNA Methyltransferase 1. J. Mol. Biol. 2015, 427, 2520–2531. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M., Jr. Structural Survey of Zinc Containing Proteins and the Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Betz, R.M.; Cerutti, D.S.; Cheatham, I.T.E.; Cisneros, G.A.; et al. Amber 22; University of California, San Francisco: San Francisco, CA, USA, 2022. [Google Scholar]
- Cofas-Vargas, L.F.; Mendoza-Espinosa, P.; Avila-Barrientos, L.P.; Prada-Gracia, D.; Riveros-Rosas, H.; García-Hernández, E. Exploring the Druggability of the Binding Site of Aurovertin, an Exogenous Allosteric Inhibitor of FOF1-ATP Synthase. Front. Pharmacol. 2022, 13, 1012008. [Google Scholar] [CrossRef] [PubMed]
- Medrano-Cerano, J.L.; Cofas-Vargas, L.F.; Leyva, E.; Rauda-Ceja, J.A.; Calderón-Vargas, M.; Cano-Sánchez, P.; Titaux-Delgado, G.; Melchor-Meneses, C.M.; Hernández-Arana, A.; Del Río-Portilla, F.; et al. Decoding the Mechanism Governing the Structural Stability of Wheat Germ Agglutinin and Its Isolated Domains: A Combined Calorimetric, NMR, and MD Simulation Study. Protein Sci. 2024, 33, e5020. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Man, V.H.; Yang, W.; Lee, T.-S.; Wang, J. A Fast and High-Quality Charge Model for the next Generation General AMBER Force Field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Capecchi, A.; Probst, D.; Reymond, J.-L. One Molecular Fingerprint to Rule Them All: Drugs, Biomolecules, and the Metabolome. J. Cheminform. 2020, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A New Class of Quinoline-Based DNA Hypomethylating Agents Reactivates Tumor Suppressor Genes by Blocking DNA Methyltransferase 1 Activity and Inducing Its Degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar] [CrossRef] [PubMed]
- Gros, C.; Fleury, L.; Nahoum, V.; Faux, C.; Valente, S.; Labella, D.; Cantagrel, F.; Rilova, E.; Bouhlel, M.A.; David-Cordonnier, M.-H.; et al. New Insights on the Mechanism of Quinoline-Based DNA Methyltransferase Inhibitors. J. Biol. Chem. 2015, 290, 6293–6302. [Google Scholar] [CrossRef] [PubMed]
- Gamage, S.A.; Brooke, D.G.; Redkar, S.; Datta, J.; Jacob, S.T.; Denny, W.A. Structure-Activity Relationships for 4-Anilinoquinoline Derivatives as Inhibitors of the DNA Methyltransferase Enzyme DNMT1. Bioorg. Med. Chem. 2013, 21, 3147–3153. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; Sánchez-Arias, J.A.; San José-Enériz, E.; Agirre, X.; de Miguel, I.; Garate, L.; Miranda, E.; Sáez, E.; Roa, S.; Martínez-Climent, J.A.; et al. Detailed Exploration around 4-Aminoquinolines Chemical Space to Navigate the Lysine Methyltransferase G9a and DNA Methyltransferase Biological Spaces. J. Med. Chem. 2018, 61, 6546–6573. [Google Scholar] [CrossRef] [PubMed]
- López-López, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity Landscape and Molecular Modeling to Explore the SAR of Dual Epigenetic Inhibitors: A Focus on G9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; San José-Enériz, E.; Agirre, X.; Sánchez-Arias, J.A.; Vilas-Zornoza, A.; Ugarte, A.; de Miguel, I.; Miranda, E.; Garate, L.; Fraga, M.; et al. Discovery of Reversible DNA Methyltransferase and Lysine Methyltransferase G9a Inhibitors with Antitumoral in Vivo Efficacy. J. Med. Chem. 2018, 61, 6518–6545. [Google Scholar] [CrossRef]
- Liu, K.; Kokubo, H. Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-Docking Study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef]
- Avila-Barrientos, L.P.; Cofas-Vargas, L.F.; Agüero-Chapin, G.; Hernández-García, E.; Ruiz-Carmona, S.; Valdez-Cruz, N.A.; Trujillo-Roldán, M.; Weber, J.; Ruiz-Blanco, Y.B.; Barril, X.; et al. Computational Design of Inhibitors Targeting the Catalytic β Subunit of Escherichia Coli FOF1-ATP Synthase. Antibiotics 2022, 11, 557. [Google Scholar] [CrossRef]
- Martínez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Miljković, F.; Medina-Franco, J.L. Artificial Intelligence-Open Science Symbiosis in Chemoinformatics. Artif. Intell. Life Sci. 2024, 5, 100096. [Google Scholar] [CrossRef]

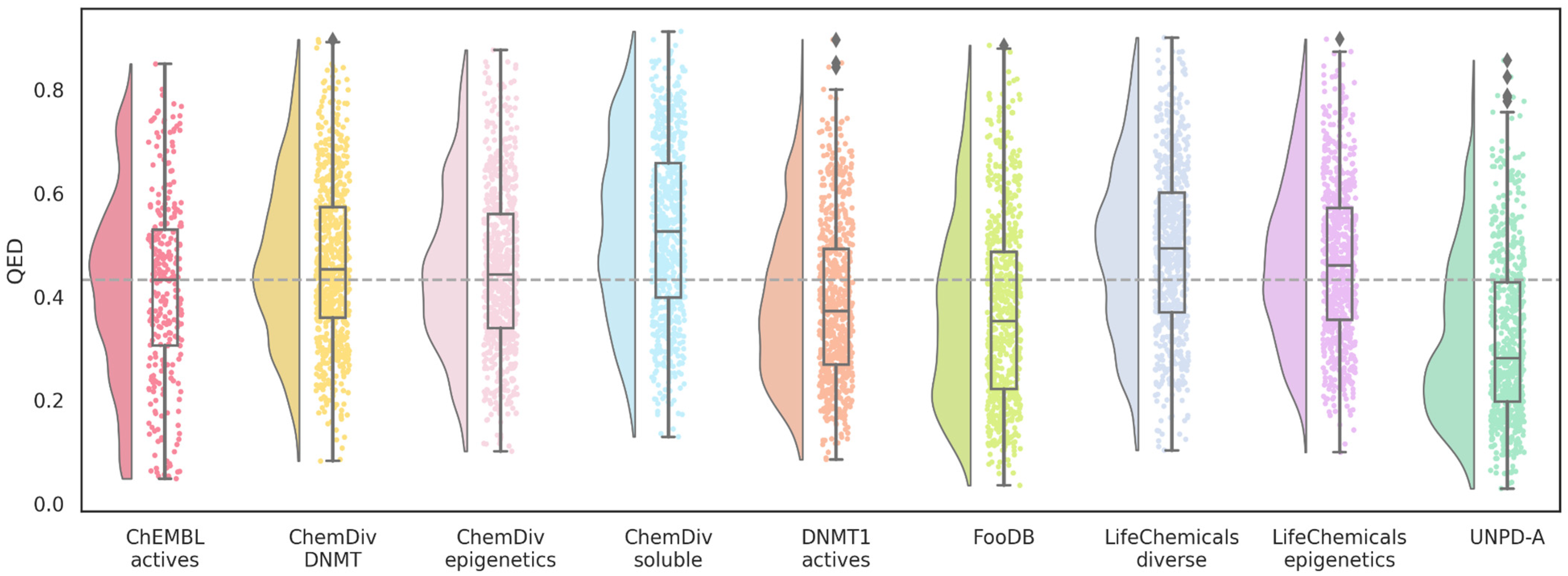
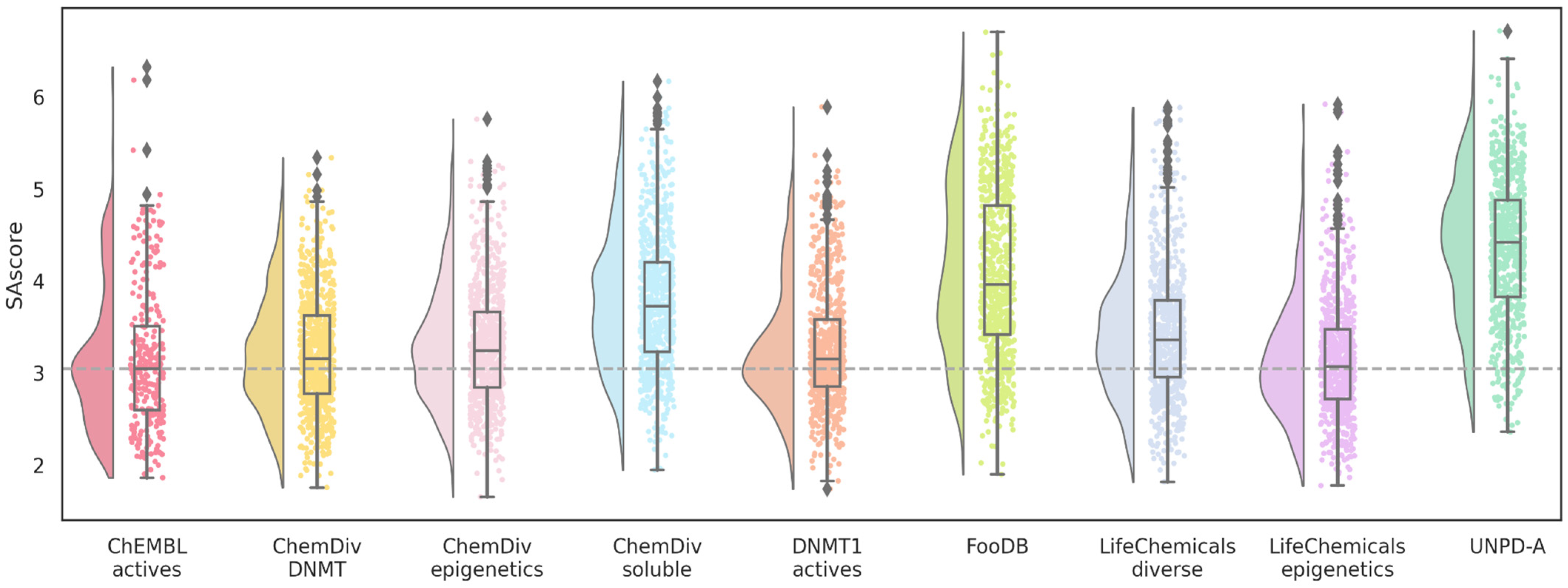
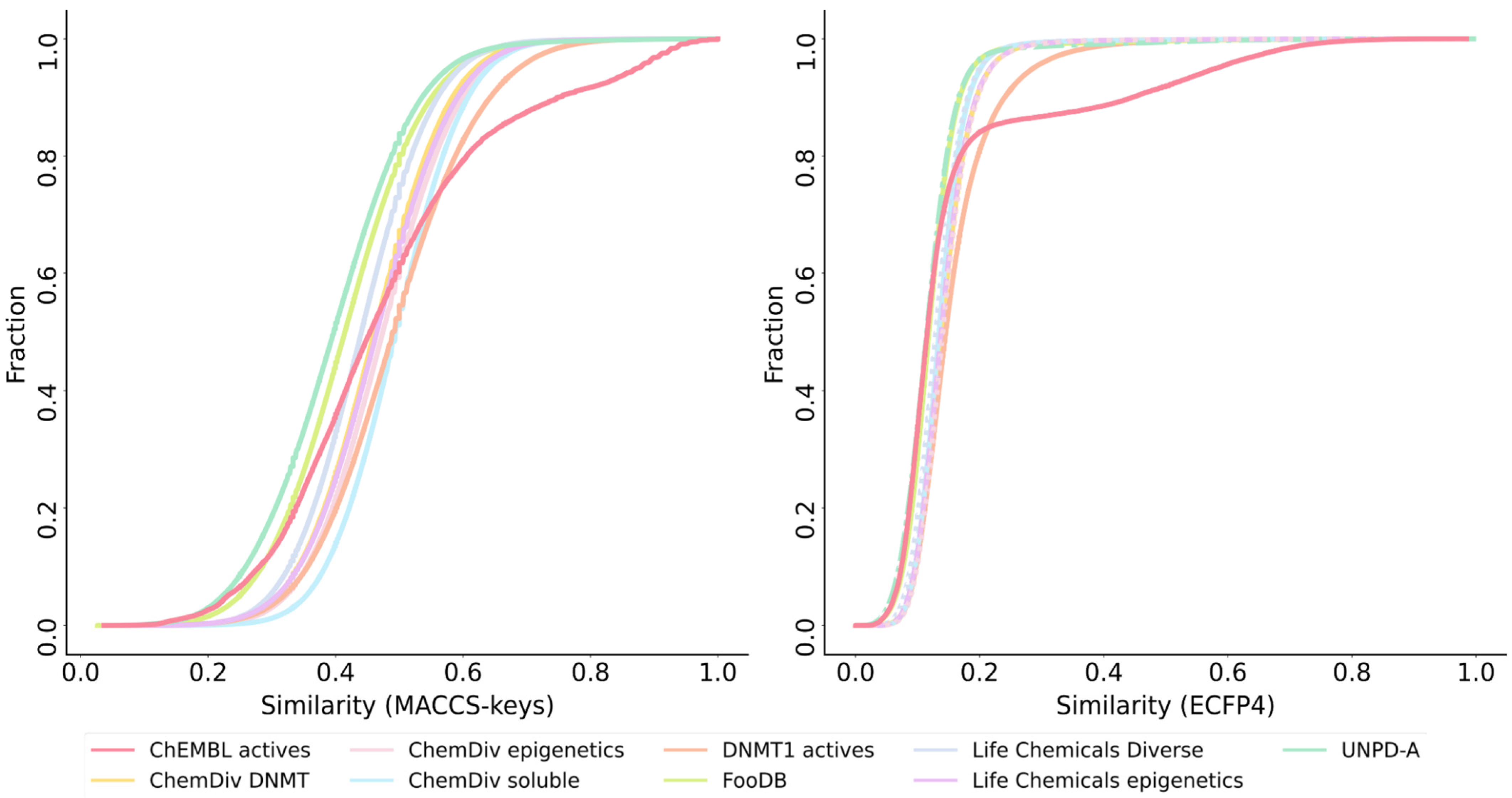
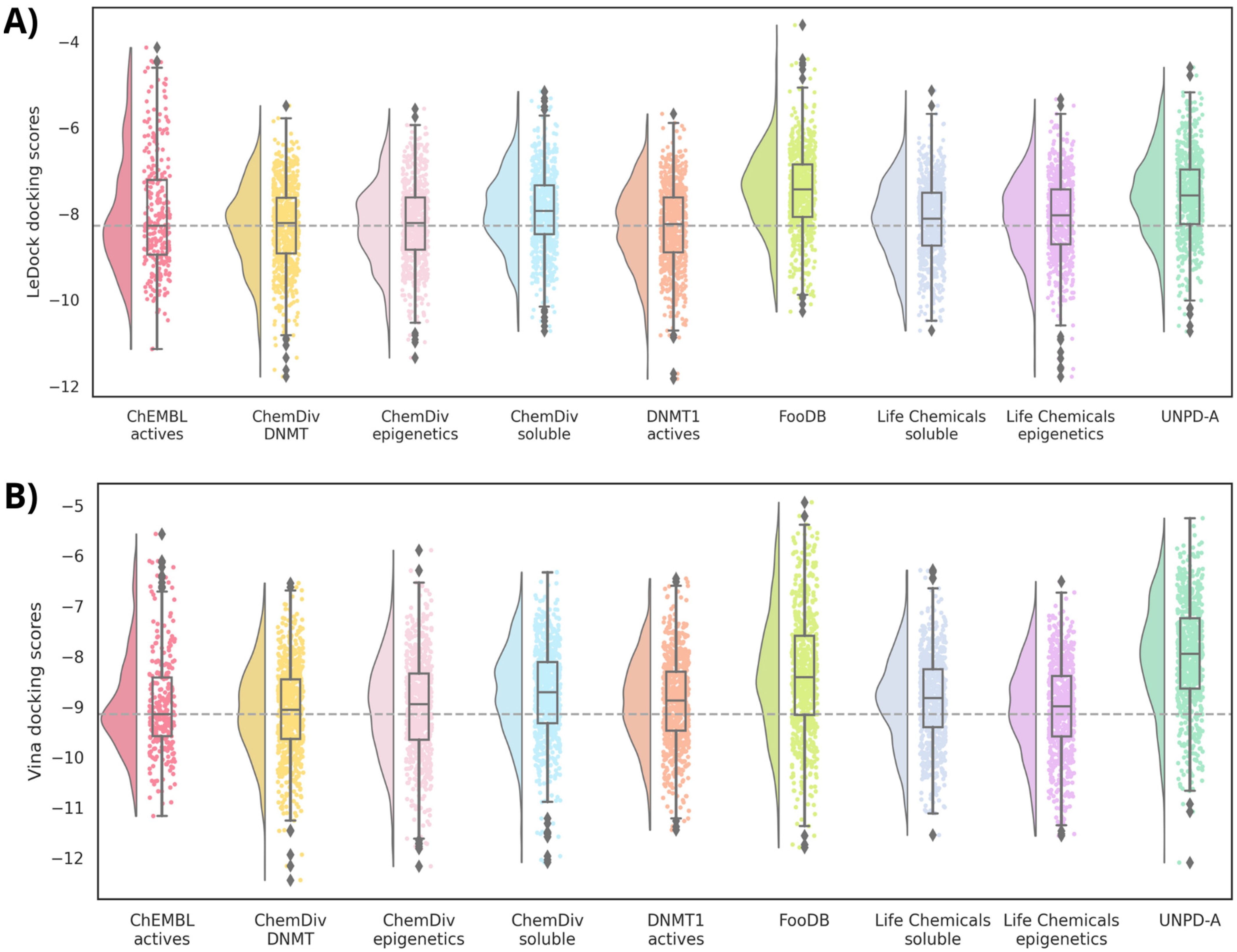




| Database | Description | Number of Compounds |
|---|---|---|
| ChemDiv DNMT-targeted library [42] | Small molecules targeting DNMTs. | 33,936 |
| ChemDiv Epigenetics Focused Set [43] | Drug-like compounds targeting families of epigenetic proteins. | 25,883 |
| ChemDiv Soluble Diversity Library [44] | Soluble drug-like compounds focused against various biological targets. | 15,500 |
| ChEMBL actives [45] | Inhibitors with IC50 ≤ 10 μM from ChEMBL 31. | 285 |
| FooDB [46] | Food chemicals. | 68,658 |
| Life Chemicals Diversity Set [47] | A subset of 5000 drug-like compounds with a wide range of chemical structures. | 5120 |
| Life Chemicals Epigenetic Focused Library [48] | Drug-like compounds selected by 2D fingerprint similarity search with known epigenetic modulators. | 3578 |
| UNPD-A [49,50] | A diverse subset from the Universal Natural Product Database. | 14,994 |
| Dataset | Code CDP | Size | ECFP4 a | MACCS Keys a | N/M | AUC | F50 | SSE10 | Molecular Properties |
|---|---|---|---|---|---|---|---|---|---|
| DNMT1 | 1 | 743 | 0.140 | 0.437 | 0.634 | 0.787 | 0.045 | 0.927 | 3.275 |
| alvaBuilder | 2 | 5575 | 0.148 | 0.431 | 0.795 | 0.734 | 0.138 | 0.739 | 2.396 |
| LigBuilder | 3 | 2491 | 0.171 | 0.533 | 0.501 | 0.826 | 0.020 | 0.939 | 3.108 |
| Quartile | Number of Active Compounds | SVM + RDK | FFNN + Morgan | |||
|---|---|---|---|---|---|---|
| Active | Inactive | Precision | Recall | Precision | Recall | |
| Q1 | 44 | 1 | 0.978 | 0.978 | 0.978 | 1.000 |
| Q2 | 40 | 5 | 0.974 | 0.950 | 0.950 | 0.950 |
| Q3 | 20 | 25 | 0.667 | 0.300 | 0.667 | 0.600 |
| Q4 | 9 | 36 | 0.667 | 0.222 | 0.600 | 0.333 |
| Database | Quartile | Proportion | ||
|---|---|---|---|---|
| Q2 | Q3 | Q4 | ||
| alvaBuilder—ChemDiv DNMT | 1 | 6 | 8 | 0.024 |
| Database | Quartile | Proportion | ||
|---|---|---|---|---|
| Q2 | Q3 | Q4 | ||
| alvaBuilder | ||||
| ChemDiv DNMT | 0 | 0 | 130 | 0.186 |
| ChemDiv epigenetics | 0 | 0 | 110 | 0.157 |
| ChemDiv soluble | 0 | 0 | 97 | 0.139 |
| DNMT1 actives | 1 | 12 | 141 | 0.220 |
| FooDB | 0 | 0 | 97 | 0.139 |
| Life Chemicals diverse | 0 | 0 | 132 | 0.189 |
| Life Chemicals epigenetics | 0 | 0 | 111 | 0.159 |
| UNPD-A | 0 | 0 | 137 | 0.202 |
| LigBuilder | ||||
| ChemDiv epigenetics | 0 | 0 | 16 | 0.049 |
| ChemDiv fragments | 0 | 0 | 3 | 0.007 |
| ChemDiv soluble | 0 | 0 | 7 | 0.024 |
| LigBuilder default | 0 | 0 | 13 | 0.042 |
| DNMT1 actives | 0 | 0 | 37 | 0.094 |
| FooDB | 0 | 0 | 7 | 0.022 |
| Selleckchem | 0 | 0 | 9 | 0.027 |
| UNPD-A | 0 | 0 | 6 | 0.051 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prado-Romero, D.L.; Saldívar-González, F.I.; López-Mata, I.; Laurel-García, P.A.; Durán-Vargas, A.; García-Hernández, E.; Sánchez-Cruz, N.; Medina-Franco, J.L. De Novo Design of Inhibitors of DNA Methyltransferase 1: A Critical Comparison of Ligand- and Structure-Based Approaches. Biomolecules 2024, 14, 775. https://doi.org/10.3390/biom14070775
Prado-Romero DL, Saldívar-González FI, López-Mata I, Laurel-García PA, Durán-Vargas A, García-Hernández E, Sánchez-Cruz N, Medina-Franco JL. De Novo Design of Inhibitors of DNA Methyltransferase 1: A Critical Comparison of Ligand- and Structure-Based Approaches. Biomolecules. 2024; 14(7):775. https://doi.org/10.3390/biom14070775
Chicago/Turabian StylePrado-Romero, Diana L., Fernanda I. Saldívar-González, Iván López-Mata, Pedro A. Laurel-García, Adrián Durán-Vargas, Enrique García-Hernández, Norberto Sánchez-Cruz, and José L. Medina-Franco. 2024. "De Novo Design of Inhibitors of DNA Methyltransferase 1: A Critical Comparison of Ligand- and Structure-Based Approaches" Biomolecules 14, no. 7: 775. https://doi.org/10.3390/biom14070775