
A Shared Receptor Suggests a Common Ancestry between an Insecticidal Bacillus thuringiensis Cry Protein and an Anti-Cancer Parasporin


Abstract
1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Constructs
2.1.1. Bacillus thuringiensis Strain 4D7
2.1.2. Toxin Expression Plasmids
2.2. Growth, Solubilization and Activation of Cry Toxins
2.3. Cell Culture
2.4. Fluorescence Microscopy Using GFP-Labelled Cry41Aa
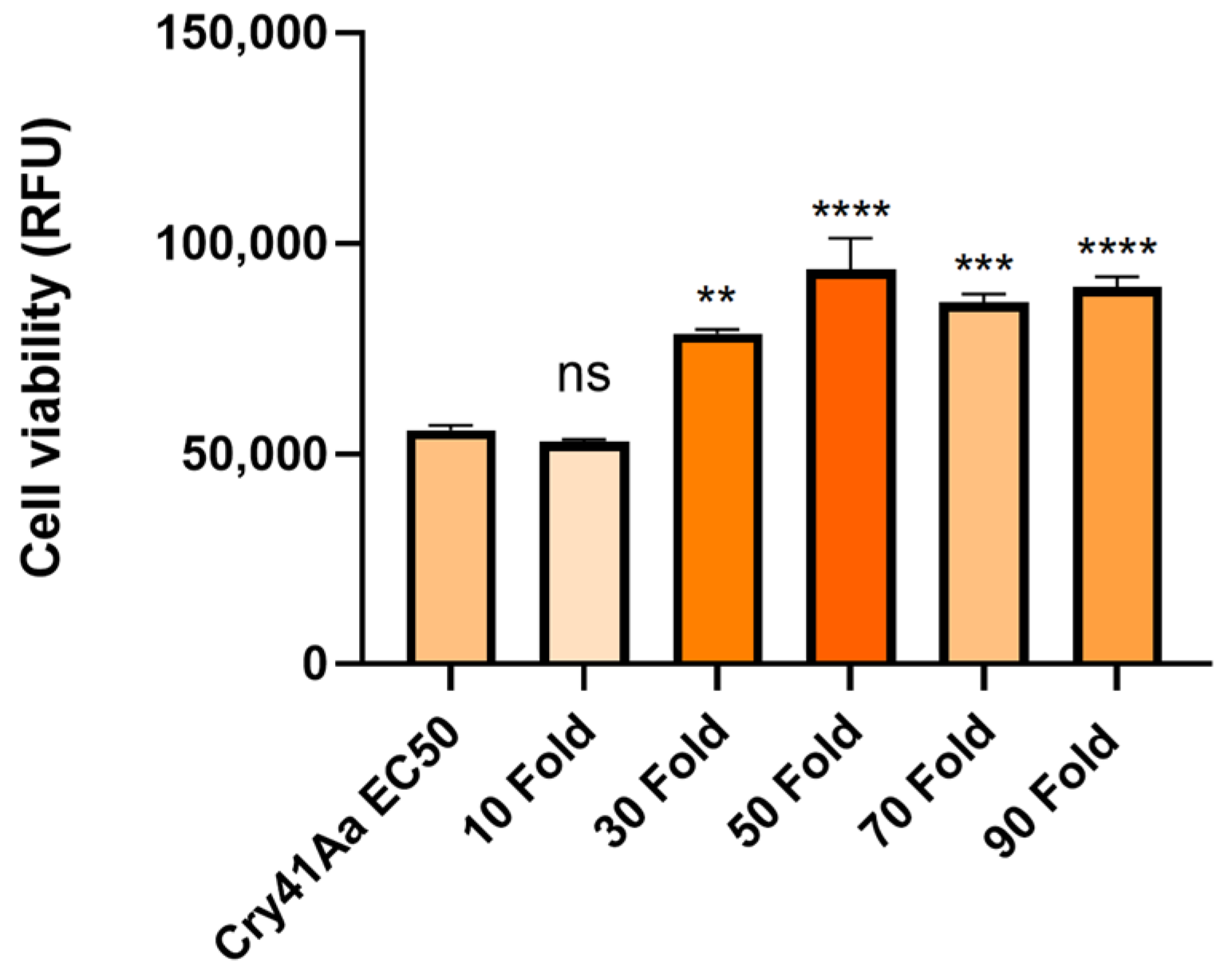
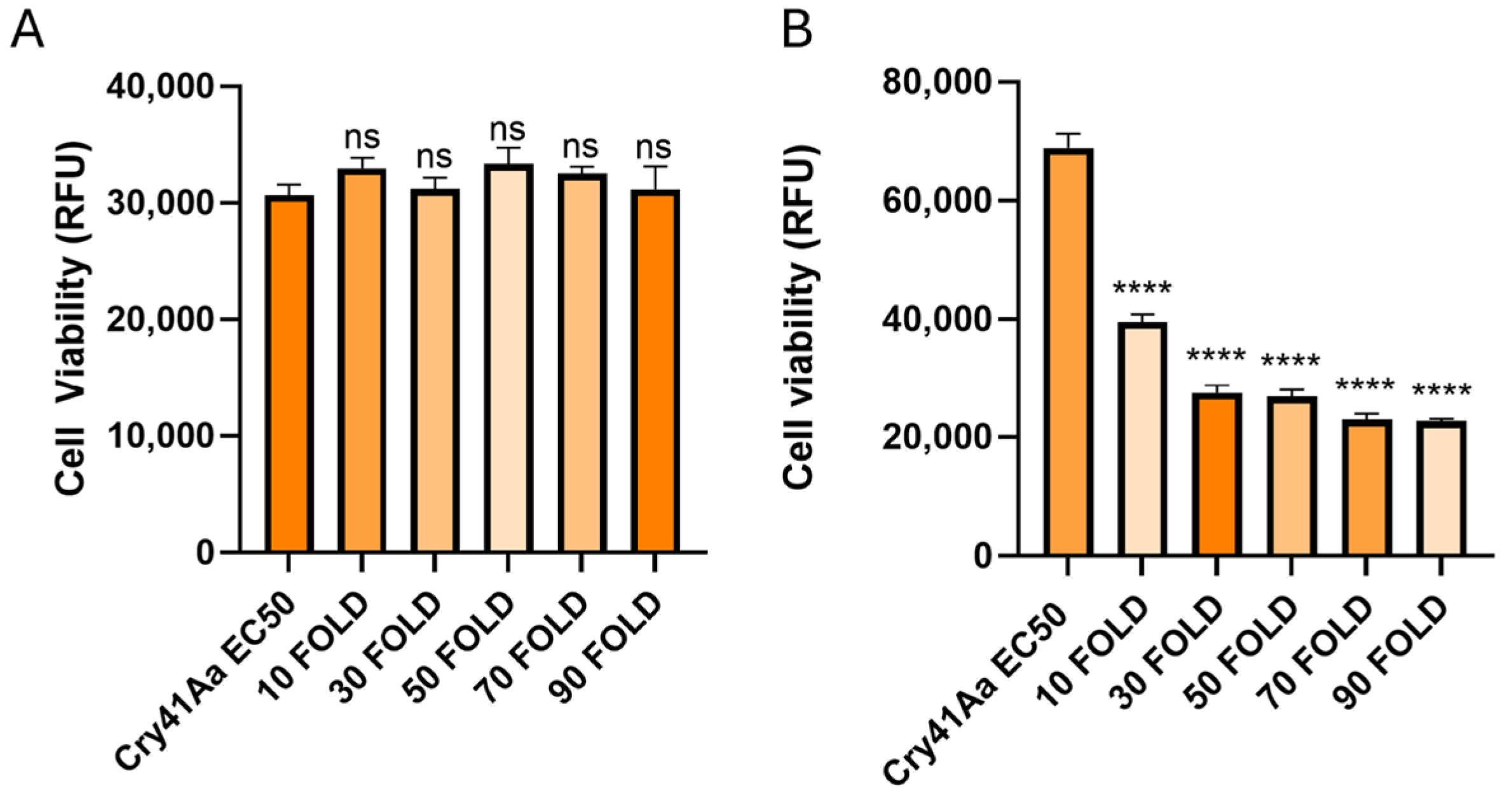
2.5. Cellular Competition Assays
2.6. Cellular Binding Assays
2.7. Western Blotting
2.8. Aedes aegypti Bioassays
2.9. Data Analysis
3. Results
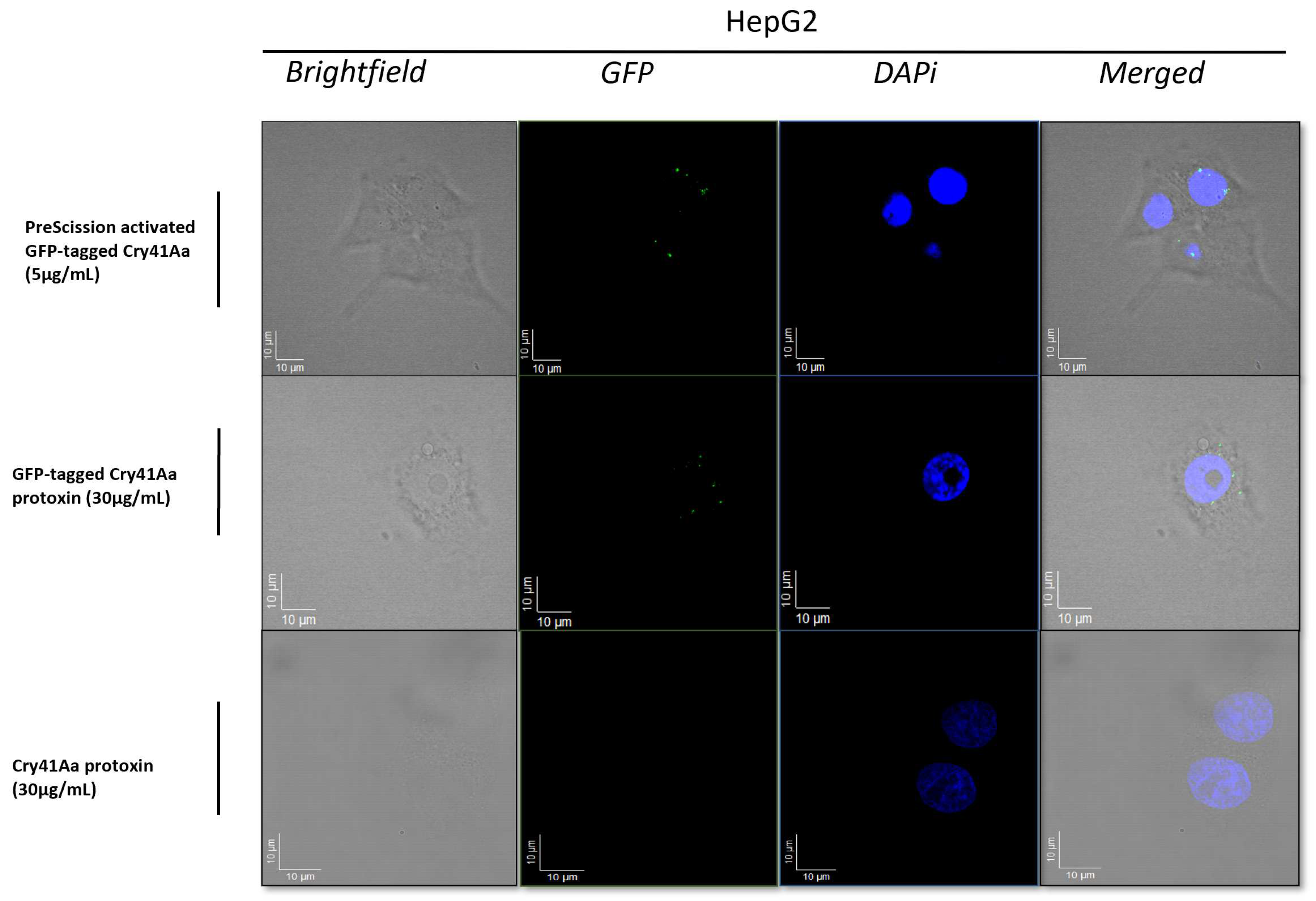
3.1. Fluorescence Microscopy Analysis of Binding of the GFP-Tagged Cry41Aa to the Surface of HepG2 Cells
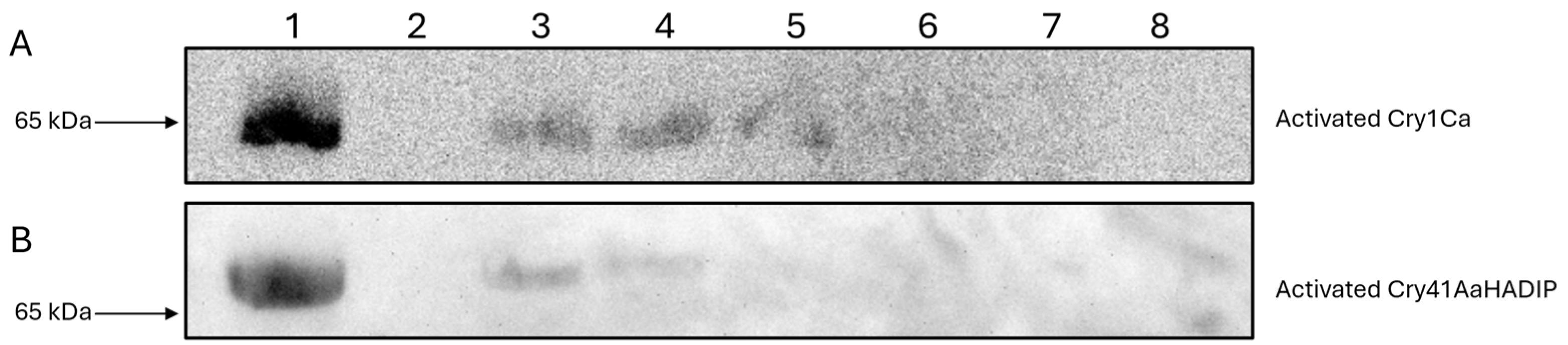
3.2. Immunoblotting Confirms Cry41Aa’s Binding Capability
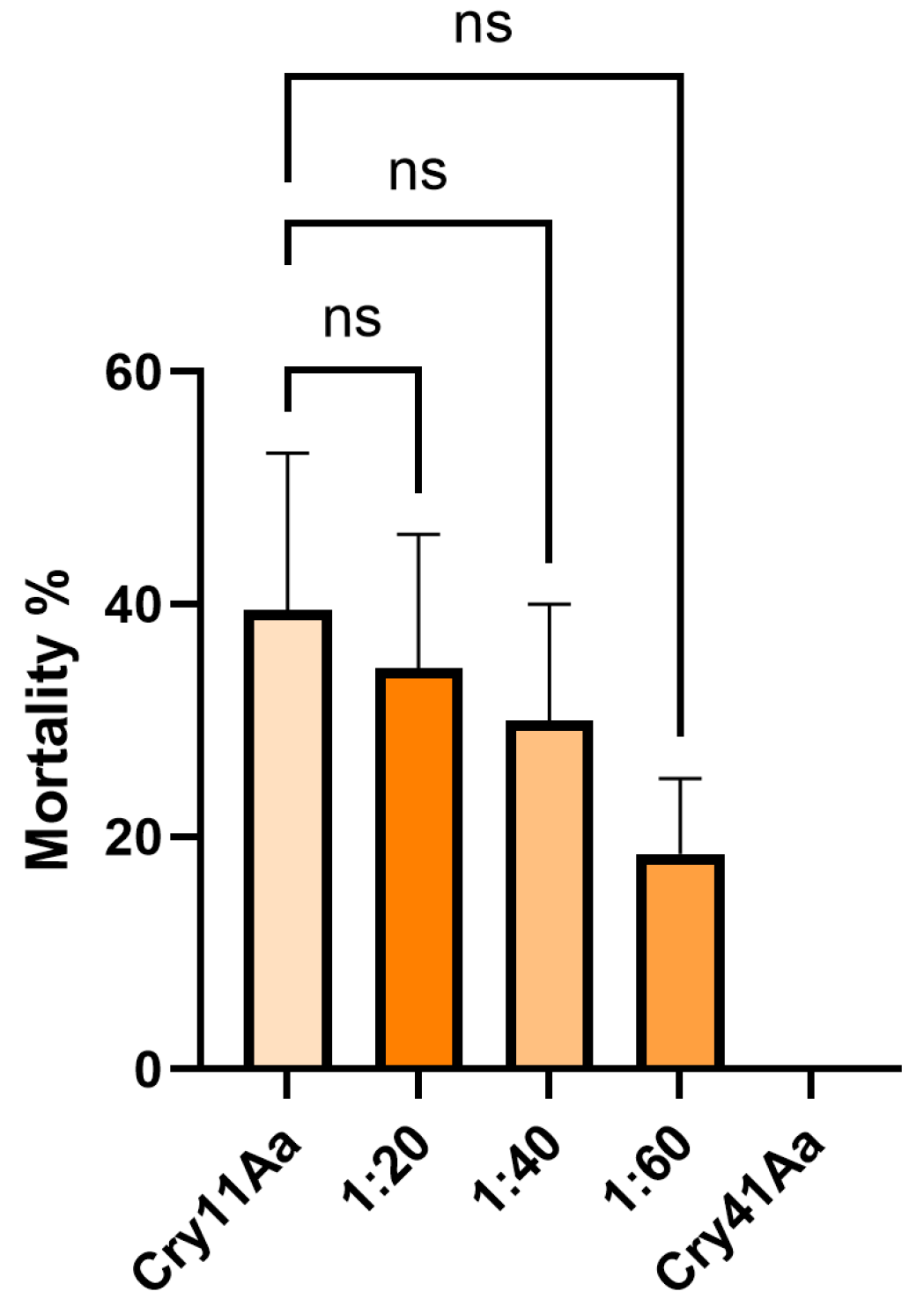
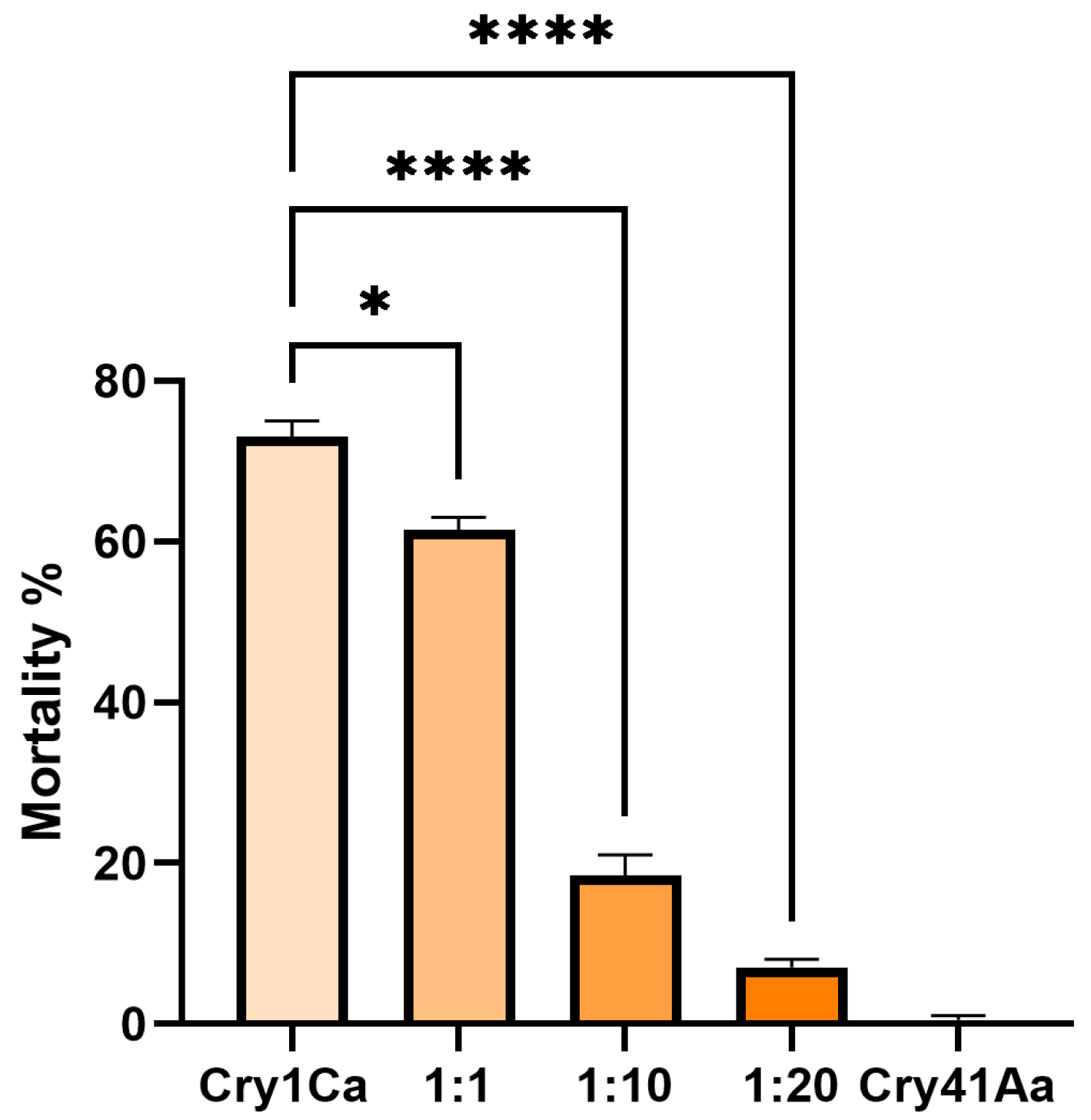
3.3. Cry41Aa and Insecticidal Cry Toxin Synergism
3.4. Cry41Aa Inhibits Cry1Ca Activity in Aedes aegypti Larvae
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- de Maagd, R.; Bravo, A.; Crickmore, N. How Bacillus thuringiensis has evolved specific toxins to colonize the insect world. Trends Genet. 2001, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Jurat-Fuentes, J.L.; Crickmore, N. Specificity determinants for Cry insecticidal proteins: Insights from their mode of action. J. Invertebr. Pathol. 2017, 142, 5–10. [Google Scholar] [CrossRef]
- Krishnan, V.; Domanska, B.; Elhigazi, A.; Afolabi, F.; West, M.J.; Crickmore, N. The human cancer cell active toxin Cry41Aa from Bacillus thuringiensis acts like its insecticidal counterparts. Biochem. J. 2017, 474, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Souissi, W.; Kaloki, A.; Etherington, S.; Domanska, B.; West, M.J.; Crickmore, N. Differential proteolytic activation of the Bacillus thuringiensis Cry41Aa parasporin modulates its anticancer effect. Biochem. J. 2019, 476, 3805–3816. [Google Scholar] [CrossRef] [PubMed]
- Sato, R. Utilization of Diverse Molecules as Receptors by Cry Toxin and the Promiscuous Nature of Receptor-Binding Sites Which Accounts for the Diversity. Biomolecules 2024, 14, 425. [Google Scholar] [CrossRef]
- Akiba, T.; Okumura, S. Parasporins 1 and 2: Their structure and activity. J. Invertebr. Pathol. 2017, 142, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Endo, H. Molecular and Kinetic Models for Pore Formation of Bacillus thuringiensis Cry Toxin. Toxins 2022, 14, 433. [Google Scholar] [CrossRef] [PubMed]
- Arantes, O.; Lereclus, D. Construction of cloning vectors for Bacillus thuringiensis. Gene 1991, 108, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, F.A.; Crickmore, N. N-terminal proteolysis determines the differential activity of Bacillus thuringiensis Cry2A toxins towards Aedes aegypti. J. Invertebr. Pathol. 2024, 204, 108100. [Google Scholar] [CrossRef]
- Jerga, A.; Evdokimov, A.G.; Moshiri, F.; Haas, J.A.; Chen, M.; Clinton, W.; Fu, X.; Halls, C.; Jimenez-Juarez, N.; Kretzler, C.N.; et al. Disabled insecticidal proteins: A novel tool to understand differences in insect receptor utilization. Insect Biochem. Mol. Biol. 2019, 105, 79–88. [Google Scholar] [CrossRef]
- Crickmore, N.; Ellar, D.J. Involvement of a possible chaperonin in the efficient expression of a cloned CryIIA delta-endotoxin gene in Bacillus thuringiensis. Mol. Microbiol. 1992, 6, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Bravo, A.; Sánchez, J.; Kouskoura, T.; Crickmore, N. N-terminal Activation Is an Essential Early Step in the Mechanism of Action of the Bacillus thuringiensis Cry1Ac Insecticidal Toxin. J. Biol. Chem. 2002, 277, 23985–23987. [Google Scholar] [CrossRef] [PubMed]
- González-Villarreal, S.E.; García-Montelongo, M.; Ibarra, J.E. Insecticidal Activity of a Cry1Ca toxin of Bacillus thuringiensis Berliner (Firmicutes: Bacillaceae) and Its Synergism with the Cyt1Aa Toxin Against Aedes aegypti (Diptera: Culicidae). J. Med. Entomol. 2020, 57, 1852–1856. [Google Scholar] [CrossRef]
- Schwartz, J.L.; Juteau, M.; Grochulski, P.; Cygler, M.; Préfontaine, G.; Brousseau, R.; Masson, L. Restriction of intramolecular movements within the CrylAa toxin molecule of Bacillus thuringiensis through disulfide bond engineering. FEBS Lett. 1997, 410, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, S.; Quiliche, J.P.J.; Gómez, I.; Sánchez, J.; Soberón, M.; Bravo, A. Rearrangement of N-Terminal α-Helices of Bacillus thuringiensis Cry1Ab Toxin Essential for Oligomer Assembly and Toxicity. Toxins 2020, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Bravo, A.; Gill, S.S.; Soberón, M. Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control. Toxicon 2007, 49, 423–435. [Google Scholar] [CrossRef]
- Tabashnik, B.E.; Zhang, M.; Fabrick, J.A.; Wu, Y.; Gao, M.; Huang, F.; Wei, J.; Zhang, J.; Yelich, A.; Unnithan, G.C.; et al. Dual mode of action of Bt proteins: Protoxin efficacy against resistant insects. Sci. Rep. 2015, 5, 15107. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Chang, F.N. Synergism in mosquitocidal activity of 26 and 65 kDa proteins from Bacillus thuringiensis subsp. israelensis crystal. FEBS Lett. 1985, 190, 232–236. [Google Scholar] [CrossRef]
- Monnerat, R.; Pereira, E.; Teles, B.; Martins, E.; Praça, L.; Queiroz, P.; Soberon, M.; Bravo, A.; Ramos, F.; Soares, C.M. Synergistic activity of Bacillus thuringiensis toxins against Simulium spp. larvae. J. Invertebr. Pathol. 2014, 121, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Crickmore, N.; Bone, E.J.; Williams, J.A.; Ellar, D.J. Contribution of the individual components of the δ-endotoxin crystal to the mosquitocidal activity of Bacillus thuringiensis subsp. israelensis. FEMS Microbiol. Lett. 1995, 131, 249–254. [Google Scholar] [CrossRef]
- Pinos, D.; Joya, N.; Herrero, S.; Ferré, J.; Hernández-Martínez, P. Hetero-oligomerization of Bacillus thuringiensis Cry1A proteins enhance binding to the ABCC2 transporter of Spodoptera exigua. Biochem. J. 2021, 478, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Pérez, C.; Fernandez, L.E.; Sun, J.; Folch, J.L.; Gill, S.S.; Soberon, M.; Bravo, A. Bacillus thuringiensis subsp. israelensis Cyt1Aa synergizes Cry11Aa toxin by functioning as a membrane-bound receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 18303–18308. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Martínez, P.; Bretsnyder, E.C.; Baum, J.A.; Haas, J.A.; Head, G.P.; Jerga, A.; Ferré, J. Comparison of in vitro and in vivo binding site competition of Bacillus thuringiensis Cry1 proteins in two important maize pests. Pest Manag. Sci. 2022, 78, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.E.; Aimanova, K.G.; Gill, S.S.; Bravo, A.; Soberón, M. A GPI-anchored alkaline phosphatase is a functional midgut receptor of Cry11Aa toxin in Aedes aegypti larvae. Biochem. J. 2006, 394, 77–84. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Insecticidal Cry Toxin | Insecticidal Cry Toxin Excess by Weight | Cry41Aa Concentration | Effect of Combination |
|---|---|---|---|
| Cry1Ca | 10–180 fold | 2 µg/mL | Synergism |
| Cry11Aa | 10–90 fold | 2 µg/mL | Synergism |
| Cry4Aa | 10–180 fold | 2 µg/mL | Synergism |
| Cry1Ac | 10–180 fold | 2 µg/mL | No effect |
| Cry1Da | 10–180 fold | 2 µg/mL | No effect |
| Cry2Aa | 10–90 fold | 2 µg/mL | No effect |
| Cry2Ab | 10–90 fold | 2 µg/mL | No effect |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryce-Sharron, N.; Nasiri, M.; Powell, T.; West, M.J.; Crickmore, N. A Shared Receptor Suggests a Common Ancestry between an Insecticidal Bacillus thuringiensis Cry Protein and an Anti-Cancer Parasporin. Biomolecules 2024, 14, 795. https://doi.org/10.3390/biom14070795
Bryce-Sharron N, Nasiri M, Powell T, West MJ, Crickmore N. A Shared Receptor Suggests a Common Ancestry between an Insecticidal Bacillus thuringiensis Cry Protein and an Anti-Cancer Parasporin. Biomolecules. 2024; 14(7):795. https://doi.org/10.3390/biom14070795
Chicago/Turabian StyleBryce-Sharron, Nicole, Mojtaba Nasiri, Tom Powell, Michelle J. West, and Neil Crickmore. 2024. "A Shared Receptor Suggests a Common Ancestry between an Insecticidal Bacillus thuringiensis Cry Protein and an Anti-Cancer Parasporin" Biomolecules 14, no. 7: 795. https://doi.org/10.3390/biom14070795
APA StyleBryce-Sharron, N., Nasiri, M., Powell, T., West, M. J., & Crickmore, N. (2024). A Shared Receptor Suggests a Common Ancestry between an Insecticidal Bacillus thuringiensis Cry Protein and an Anti-Cancer Parasporin. Biomolecules, 14(7), 795. https://doi.org/10.3390/biom14070795