
Protein Thermal Stability Changes Induced by the Global Methylation Inhibitor 3-Deazaneplanocin A (DZNep)

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Treatment
2.2. Western Blots
2.3. Proteome Integral Solubility Assay
2.4. RNAseq
2.5. Proteomics Sample Preparation and Nano-LC-MS/MS
2.6. Proteomic Data Analysis
2.7. Data Analysis
3. Results
3.1. Conducting PISA, Global Proteomics, and Transcriptomics on DZNep-Treated Cells
3.2. Global Proteomics and Transcriptomics Reveal Changes in Metabolic Processes and Nucleosome Organization
3.3. DZNep Impacts the Thermal Stability of Hundreds of Proteins
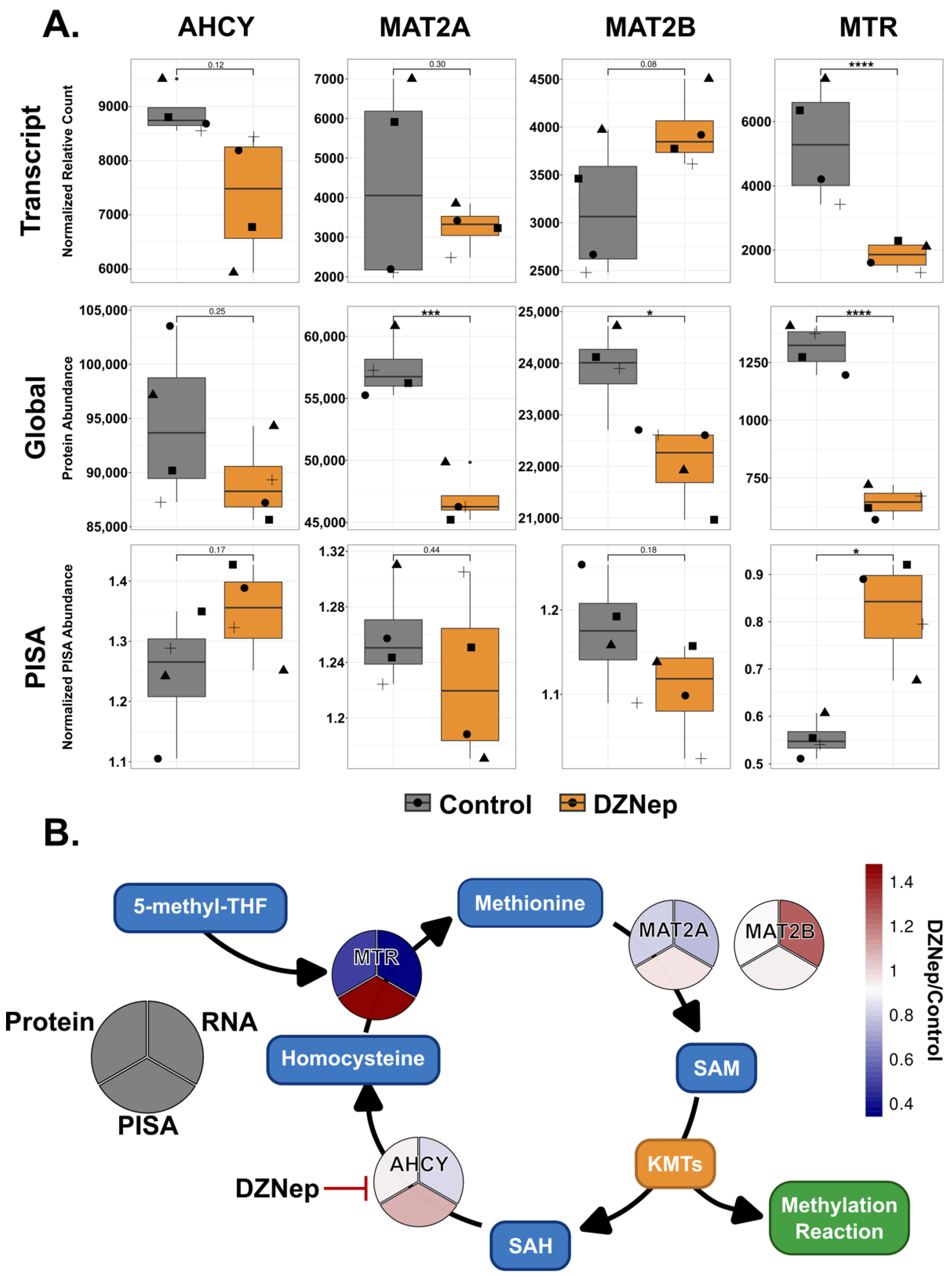
3.4. DZNep Globally Impacts Components of the Methionine Cycle
3.5. DZNep Impacts the Thermal Stability of the Aminoacyl-tRNA Synthetase Multienzyme Complex
3.6. DZNep Impacts the Thermal Stability of ATP Binders and Methyltransferases
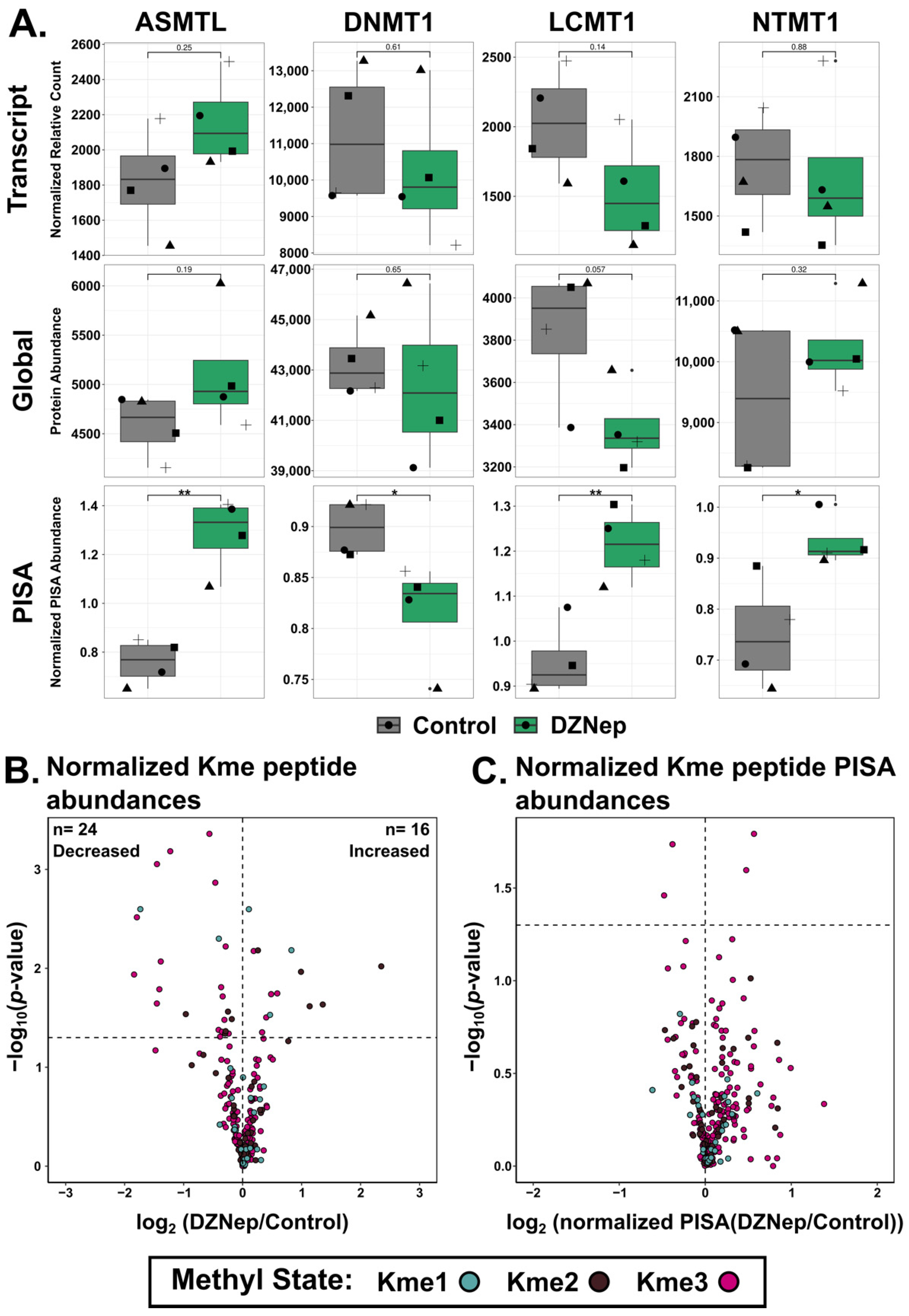
3.7. DZNep Treatment Alters the Abundance of Methylated Proteins and Peptides
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berryhill, C.A.; Hanquier, J.N.; Doud, E.H.; Cordeiro-Spinetti, E.; Dickson, B.M.; Rothbart, S.B.; Mosley, A.L.; Cornett, E.M. Global Lysine Methylome Profiling Using Systematically Characterized Affinity Reagents. Sci. Rep. 2023, 13, 377. [Google Scholar] [CrossRef] [PubMed]
- Cornett, E.M.; Ferry, L.; Defossez, P.-A.; Rothbart, S.B. Lysine Methylation Regulators Moonlighting outside the Epigenome. Mol. Cell 2019, 75, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.; Kaniskan, H.Ü.; Jin, J.; Gozani, O. Epigenetics and beyond: Targeting Writers of Protein Lysine Methylation to Treat Disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Nakamura, Y. Dysregulation of Protein Methyltransferases in Human Cancer: An Emerging Target Class for Anticancer Therapy. Cancer Sci. 2016, 107, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Peitta, G.; Simile, M.M.; Feo, F. Alterations of Methionine Metabolism as Potential Targets for the Prevention and Therapy of Hepatocellular Carcinoma. Medicina 2019, 55, 296. [Google Scholar] [CrossRef] [PubMed]
- Haws, S.A.; Yu, D.; Ye, C.; Wille, C.K.; Nguyen, L.C.; Krautkramer, K.A.; Tomasiewicz, J.L.; Yang, S.E.; Miller, B.R.; Liu, W.H.; et al. Methyl-Metabolite Depletion Elicits Adaptive Responses to Support Heterochromatin Stability and Epigenetic Persistence. Mol. Cell 2020, 78, 210–223.e8. [Google Scholar] [CrossRef] [PubMed]
- Glazer, R.I.; Knode, M.C.; Tseng, C.K.H.; Haines, D.R.; Marquez, V.E. 3-Deazaneplanocin A: A New Inhibitor of S-Adenosylhomocysteine Synthesis and Its Effects in Human Colon Carcinoma Cells. Biochem. Pharmacol. 1986, 35, 4523–4527. [Google Scholar] [CrossRef] [PubMed]
- Clercq, E.D.; Cools, M.; Balzarini, J.; Marquez, V.E.; Borcherding, D.R.; Borchardt, R.T.; Drach, J.C.; Kitaoka, S.; Konno, T. Broad-Spectrum Antiviral Activities of Neplanocin A, 3-Deazaneplanocin A, and Their 5′-nor Derivatives. Antimicrob. Agents Chemother. 1989, 33, 1291–1297. [Google Scholar] [CrossRef]
- Tseng, C.K.; Marquez, V.E.; Fuller, R.W.; Goldstein, B.M.; Haines, D.R.; McPherson, H.; Parsons, J.L.; Shannon, W.M.; Arnett, G.; Hollingshead, M. Synthesis of 3-Deazaneplanocin A, a Powerful Inhibitor of S-Adenosylhomocysteine Hydrolase with Potent and Selective in Vitro and in Vivo Antiviral Activities. J. Med. Chem. 1989, 32, 1442–1446. [Google Scholar] [CrossRef]
- Xie, Z.; Bi, C.; Cheong, L.L.; Liu, S.C.; Huang, G.; Zhou, J.; Yu, Q.; Chen, C.-S.; Chng, W.J. Determinants of Sensitivity to DZNep Induced Apoptosis in Multiple Myeloma Cells. PLoS ONE 2011, 6, e21583. [Google Scholar] [CrossRef]
- Cheng, L.L.; Itahana, Y.; Lei, Z.D.; Chia, N.-Y.; Wu, Y.; Yu, Y.; Zhang, S.L.; Thike, A.A.; Pandey, A.; Rozen, S.; et al. TP53 Genomic Status Regulates Sensitivity of Gastric Cancer Cells to the Histone Methylation Inhibitor 3-Deazaneplanocin A (DZNep). Clin. Cancer Res. 2012, 18, 4201–4212. [Google Scholar] [CrossRef]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.M.; Tan, P.B.O.; Liu, E.T.; Yu, Q. Pharmacologic Disruption of Polycomb-Repressive Complex 2-Mediated Gene Repression Selectively Induces Apoptosis in Cancer Cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined Epigenetic Therapy with the Histone Methyltransferase EZH2 Inhibitor 3-Deazaneplanocin A and the Histone Deacetylase Inhibitor Panobinostat against Human AML Cells. Blood 2009, 114, 2733–2743. [Google Scholar] [CrossRef]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep Is a Global Histone Methylation Inhibitor That Reactivates Developmental Genes Not Silenced by DNA Methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef]
- Tam, E.K.W.; Li, Z.; Goh, Y.L.; Cheng, X.; Wong, S.Y.; Santhanakrishnan, S.; Chai, C.L.L.; Yao, S.Q. Cell-Based Proteome Profiling Using an Affinity-Based Probe (AfBP) Derived from 3-Deazaneplanocin A (DzNep). Chem. Asian J. 2013, 8, 1818–1828. [Google Scholar] [CrossRef]
- King, D.T.; Serrano-Negrón, J.E.; Zhu, Y.; Moore, C.L.; Shoulders, M.D.; Foster, L.J.; Vocadlo, D.J. Thermal Proteome Profiling Reveals the O-GlcNAc-Dependent Meltome. J. Am. Chem. Soc. 2021, 144, 3833–3842. [Google Scholar] [CrossRef]
- Huang, J.X.; Lee, G.; Cavanaugh, K.E.; Chang, J.W.; Gardel, M.L.; Moellering, R.E. High Throughput Discovery of Functional Protein Modifications by Hotspot Thermal Profiling. Nat. Methods 2019, 16, 894–901. [Google Scholar] [CrossRef]
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking Cancer Drugs in Living Cells by Thermal Profiling of the Proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef]
- Gaetani, M.; Sabatier, P.; Saei, A.A.; Beusch, C.M.; Yang, Z.; Lundström, S.L.; Zubarev, R.A. Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J. Proteome Res. 2019, 18, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Grecco, G.G.; Huang, J.Y.; Muñoz, B.; Doud, E.H.; Hines, C.D.; Gao, Y.; Rodriguez, B.; Mosley, A.L.; Lu, H.-C.; Atwood, B.K. Sex-Dependent Synaptic Remodeling of the Somatosensory Cortex in Mice with Prenatal Methadone Exposure. Adv. Drug Alcohol. Res. 2022, 2, 10400. [Google Scholar] [CrossRef] [PubMed]
- McCracken, N.A.; Liu, H.; Runnebohm, A.M.; Wijeratne, H.R.S.; Wijeratne, A.B.; Staschke, K.A.; Mosley, A.L. Obtaining Functional Proteomics Insights from Thermal Proteome Profiling through Optimized Melt Shift Calculation and Statistical Analysis with InflectSSP. Mol. Cell. Proteom. 2023, 22, 100630. [Google Scholar] [CrossRef]
- Li, J.; Vranken, J.G.V.; Vaites, L.P.; Schweppe, D.K.; Huttlin, E.L.; Etienne, C.; Nandhikonda, P.; Viner, R.; Robitaille, A.M.; Thompson, A.H.; et al. TMTpro Reagents: A Set of Isobaric Labeling Mass Tags Enables Simultaneous Proteome-Wide Measurements across 16 Samples. Nat. Methods 2020, 17, 399–404. [Google Scholar] [CrossRef]
- Orsburn, B.C. Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes 2021, 9, 15. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 Years of PhosphoSitePlus®: Integrating Post-Translationally Modified Sites, Disease Variants and Isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hyeon, D.Y.; Kim, J.H.; Ahn, T.J.; Cho, Y.; Hwang, D.; Kim, S. Evolution of the Multi-TRNA Synthetase Complex and Its Role in Cancer. J. Biol. Chem. 2019, 294, 5340–5351. [Google Scholar] [CrossRef]
- Cui, H.; Kapur, M.; Diedrich, J.K.; Yates, J.R.; Ackerman, S.L.; Schimmel, P. Regulation of Ex-Translational Activities Is the Primary Function of the Multi-TRNA Synthetase Complex. Nucleic Acids Res. 2020, 49, 3603–3616. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, C.D.; Ibba, M. Aminoacyl-tRNA Synthetase Complexes: Molecular Multitasking Revealed. FEMS Microbiol. Rev. 2008, 32, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Park, B.-J.; Kang, Y.-S.; Kim, H.J.; Park, J.-H.; Kang, J.W.; Lee, S.W.; Han, J.M.; Lee, H.-W.; Kim, S. Downregulation of FUSE-Binding Protein and c-Myc by TRNA Synthetase Cofactor P38 Is Required for Lung Cell Differentiation. Nat. Genet. 2003, 34, 330–336. [Google Scholar] [CrossRef]
- Estève, P.-O.; Chang, Y.; Samaranayake, M.; Upadhyay, A.K.; Horton, J.R.; Feehery, G.R.; Cheng, X.; Pradhan, S. A Methylation and Phosphorylation Switch between an Adjacent Lysine and Serine Determines Human DNMT1 Stability. Nat. Struct. Mol. Biol. 2011, 18, 42–48. [Google Scholar] [CrossRef]
- Cao, X.-J.; Arnaudo, A.M.; Garcia, B.A. Large-Scale Global Identification of Protein Lysine Methylation in vivo. Epigenetics 2013, 8, 477–485. [Google Scholar] [CrossRef]
- Moore, K.E.; Carlson, S.M.; Camp, N.D.; Cheung, P.; James, R.G.; Chua, K.F.; Wolf-Yadlin, A.; Gozani, O. A General Molecular Affinity Strategy for Global Detection and Proteomic Analysis of Lysine Methylation. Mol. Cell 2013, 50, 444–456. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, Z.; Sun, M.; Wan, X.; Liu, P.; He, T.; Tan, M.; Zhao, Y. A Chemical Proteomics Approach for Global Analysis of Lysine Monomethylome Profiling. Mol. Cell. Proteom. 2015, 14, 329–339. [Google Scholar] [CrossRef]
- Bergant, V.; Yamada, S.; Grass, V.; Tsukamoto, Y.; Lavacca, T.; Krey, K.; Mühlhofer, M.; Wittmann, S.; Ensser, A.; Herrmann, A.; et al. Attenuation of SARS-CoV-2 Replication and Associated Inflammation by Concomitant Targeting of Viral and Host Cap 2′-O-ribose Methyltransferases. EMBO J. 2022, 41, e111608. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berryhill, C.A.; Doud, E.H.; Hanquier, J.N.; Smith-Kinnaman, W.R.; McCourry, D.L.; Mosley, A.L.; Cornett, E.M. Protein Thermal Stability Changes Induced by the Global Methylation Inhibitor 3-Deazaneplanocin A (DZNep). Biomolecules 2024, 14, 817. https://doi.org/10.3390/biom14070817
Berryhill CA, Doud EH, Hanquier JN, Smith-Kinnaman WR, McCourry DL, Mosley AL, Cornett EM. Protein Thermal Stability Changes Induced by the Global Methylation Inhibitor 3-Deazaneplanocin A (DZNep). Biomolecules. 2024; 14(7):817. https://doi.org/10.3390/biom14070817
Chicago/Turabian StyleBerryhill, Christine A., Emma H. Doud, Jocelyne N. Hanquier, Whitney R. Smith-Kinnaman, Devon L. McCourry, Amber L. Mosley, and Evan M. Cornett. 2024. "Protein Thermal Stability Changes Induced by the Global Methylation Inhibitor 3-Deazaneplanocin A (DZNep)" Biomolecules 14, no. 7: 817. https://doi.org/10.3390/biom14070817