

Targeting Microglia in Alzheimer’s Disease: Pathogenesis and Potential Therapeutic Strategies


Abstract
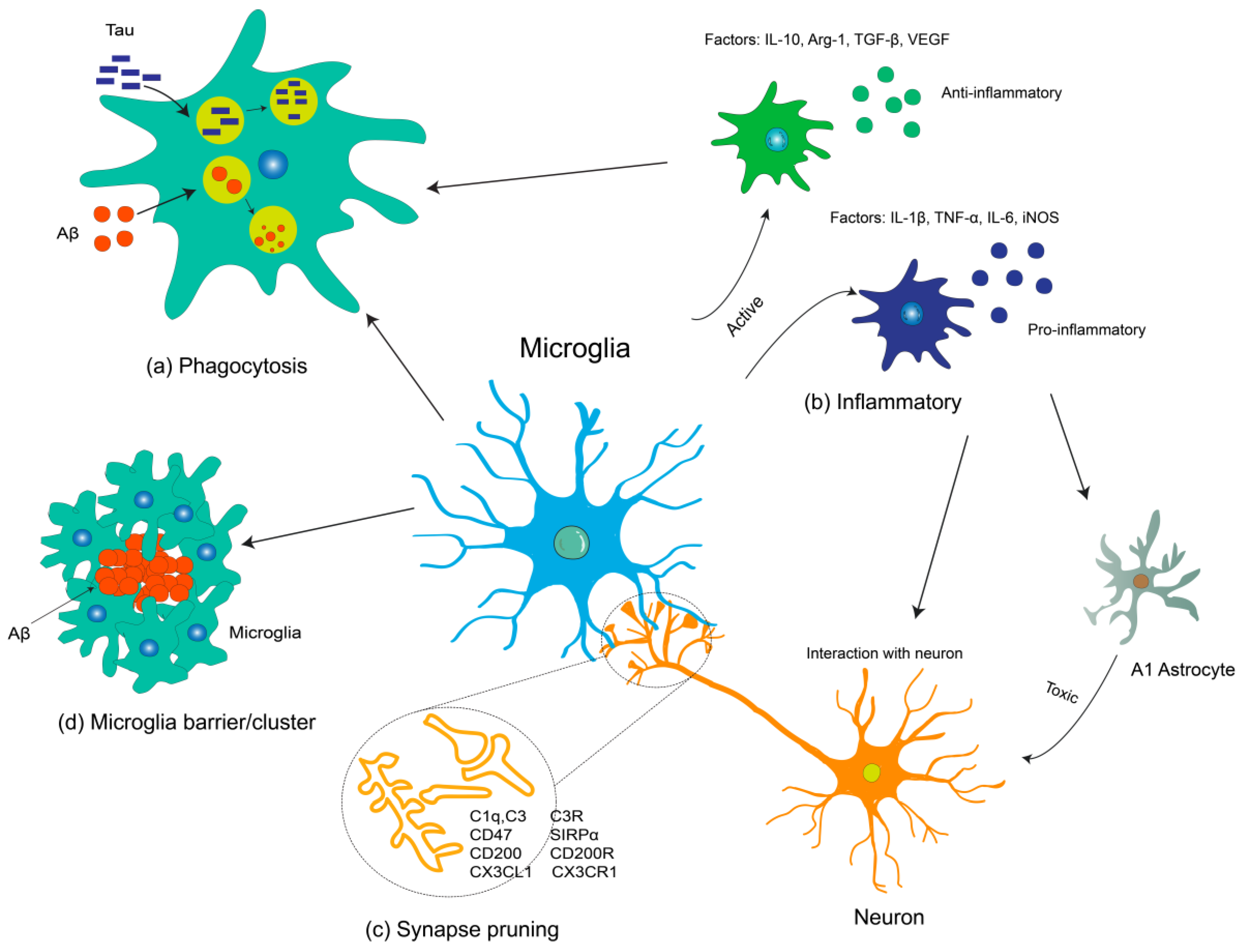
:1. Introduction
2. Microglia in AD Pathogenesis
3. Microglia-Related Treatment for AD
3.1. Enhancing Microglial Function

{kind=link}
{kind=link}
| Targets | Cellular Function in AD | Genetic Manipulations/Pharmacological Interventions | Target/Mechanism | Mechanism of Action | References |
|---|---|---|---|---|---|
| CD33 | Negative microglia phagocytosis | CD33 knockout; AAV-mediated miRCD33 Inhibitor: P22; Lintuzumab; AL003 | Blocked CD33 expression | Promoted Aβ phagocytosis and clearance; decreased neuroinflammatory | [76,78,95,96] |
| Complement C3 | Microglia-mediated synaptic refinement | C3 knockout | Ameliorated C3b/CR3 complement activation | Decreased inflammatory cytokines; promoted Aβ phagocytosis and inhibited synapse loss | [54,56] |
| GRN, a secreted pleiotropic growth factor | Microglia-mediated phagocytosis | Selectively reducing microglial expression of PGRN; Lentivirus-mediated PGRN overexpression | Exacerbated microglial activation | Impaired phagocytosis; increased plaque load and exacerbated cognitive deficits Lowered plaque load; prevented spatial memory deficits | [84] |
| MicroRNA-155 (miR155) and IFN-γ | Mediated a protective microglial state | Deletion of miR-155; blocked IFN-γ signaling | Induced a pre-MGnD activation state via IFN-γ signaling Attenuated MGnD induction and microglial phagocytosis | Restricted neurodegenerative pathology and preserved cognitive function | [86] |
| Piezo1 | Microglial mechanosensor of Aβ fibril | Piezo1 deletion; pharmacological activation of Piezo1 | Modulated the microglial mechanosensing pathways | Exacerbated Aβ pathology and cognitive decline; ameliorated brain Aβ burden and cognitive impairment | [88] |
| RIPK1 | Microglia-mediated phagocytosis | RIPK1 deletion or Inhibitor | Enhanced the phagocytosis; reduced the inflammatory response | Reduced amyloid burden; the levels of inflammatory cytokines; and memory deficits | [90] |
| SYK | Regulator of microglia activation and phagocytosis | SYK deletion An antibody against CLEC7A | Restricted microglia phagocytosis; altered AKT/GSK3β-signaling; Directly activates SYK | Exacerbated Aβ deposition; cognitive defects Rescued microglia activation | [64,92] |
| TAM receptor | TAM-driven microglial phagocytosis | TAM deficient | Reduced microglia detect and engulf Aβ plaques | Developed fewer dense-core plaques | [94] |
| TREM2 | Positive microglia phagocytosis and enclosed to Aβ Activating TG-2, receptor for sTREM2 on neuron | TREM2 overexpression; soluble TREM2; soluble TREM2; agonist: AL002; 4D9. Transplantation of Trem2+/+ CDMC | Reprogramed microglia responsivity; enhanced microglia phagocytosis; sTREM2-TG2 interaction mediates the cross-talk between microglia and neurons; enhanced microglia phagocytosis; restores microglial function with Syk signaling-dependent transcription | Ameliorates amyloid pathology and behavioral deficits; enhanced Aβ clearance and rescued spatial memory; ameliorated tau phosphorylation and cognitive deficits; promote Aβ uptake and clearance, decrease neuroinflammatory; ameliorates amyloid pathology | [68,69,71,72,73,74] |
3.2. Regulation of Neuroinflammation
| Targets | Cellular Function in AD | Genetic Manipulations/Pharmacological Interventions | Target/Mechanism | Mechanism of Action | References |
|---|---|---|---|---|---|
| TNF-α | Exacerbate inflammation | TNF-α AAV-mediated overexpression Antibodies: XENP345, Thalidomide | Enhanced the microglia response Decrease neuroinflammatory | Induced robust glial activation attenuated plaque deposition Reduced Aβ plaques, and inhibited inflammatory cytokines and APP processing | [108,109,110,132] |
| P2X7 receptor | Exacerbate inflammation | Inhibitor: Brilliant blue G | Reduced neuroinflammatory | Attenuated gliosis; diminished leakiness of blood–brain barrier | [133] |
| NLRP3 | Exacerbate inflammation | NLRP3 knockout; Casp 1 knockout; loss of NLRP3; inhibitor: JC124 | Reduced NLRP3 inflammasome activation; decreased the inflammasome | Ameliorated amyloid pathology and skewed microglial cells to an M2 phenotype; reduced Tau pathology; ameliorated the amyloid pathology and improved spatial memory | [117,118,134] |
| Extracellular ASC speck | Bound to Aβ and cross-seed Aβ | Injection of ASC specks; antibody-neutralizing extracellular ASC | Inflammasome-dependent formation of ASC specks | Blocked amyloid pathology | [120] |
| IL-1β | Exacerbated inflammation | IL-1β transgenic antibodies: Canakinumab | Enhanced the microglia response; decreased neuroinflammatory | Mediated chronic neuroinflammation and ameliorated amyloid pathology; decreased NF-κB activity and reduced tau pathology | [121,135] |
| miR-25802 | Microglia-mediated neuroinflammation | Overexpression of miR-25802; inhibition of miR-25802 | miR-25802/KLF4/NF-κB signaling axis | Aggravated AD-related pathology, including cognitive disability, Aβ deposition, and microglial pro-inflammatory state; ameliorated AD-related pathology, improved spatial memory, and microglial anti-inflammatory state | [136] |
| TLR2 | Exacerbated inflammation | TLR2 knockout | Reduced neuroinflammatory | Shifted M1 microglia to M2 inflammatory activation | [126] |
| TLR4 | Exacerbated inflammation | A loss-of-function TLR4 mutation; Inhibitor: TAK242 | Reduced microglial activation Decreased neuroinflammatory | Increased Aβ deposits and exacerbated cognitive deficits Promoted M2 microglial polarization and suppressed inflammatory cytokines | [123,137] |
| IL-10 | Mediated inflammation | IL-10 knockout | Decreased neuroinflammatory | Ameliorated amyloid pathology and promoted cognitive deficits | [127] |
| IL-4 | Mediated inflammation | IL-4 AAV-mediated overexpression | Decreased neuroinflammation; acute suppression of glial clearance mechanisms | Reduced microgliosis; attenuated amyloid pathology | [129] |
4. Microglia-Targeted Modulation by Natural Products for the Prevention and Treatment of AD
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| AD | Alzheimer’s disease |
| Aβ | amyloid-β |
| ABCA7 | ATP-binding cassette, sub-family A (ABC1), member 7 |
| ALS | amyotrophic lateral sclerosis. |
| APP | amyloid precursor protein |
| APOC2 | apolipoprotein C 2 |
| ARM | activated response microglia |
| Arg-1 | arginase 1 |
| ASC | apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain |
| BBB | blood-brain barrier |
| BDNF | brain-derived neurotrophic factor |
| BIN1 | bridging integrator 1 |
| BGW | Bayesian genome-wide |
| ccl2 | chemokine (C-C motif) ligand 2 |
| CD63 | cluster of differentiation 63 |
| CD2AP | CD2-associated protein |
| CDMCs | circulation-derived myeloid cells |
| CD47 | cluster of differentiation 47 |
| CD200 | cluster of differentiation 200 |
| Clec7a | C-type lectin domain family 7A |
| CNS | central neuronal system |
| CR1 | complement component (3b/4b) receptor 1 |
| CST7 | cystatin 7 |
| CSF1R | Colony-stimulating factor 1 receptor |
| CX3CR1 | CX3C chemokine receptor 1 |
| CX3CL1 | CX3C chemokine ligand 1 |
| DAM | disease-associated microglia |
| DM | dark microglia |
| Fabp5 | Fatty acid-binding protein 5 |
| GDNF | glial cell-derived neurotrophic factor |
| Gpnmb | Glycoprotein-NMB |
| GRN | Granulin |
| GWASs | genome-wide association studies |
| INF-γ | Interferon-γ |
| iNOS | inducible nitric oxide synthase |
| INPP5D | inositol polyphosphate-5-phosphatase |
| ITAM | immunoreceptor tyrosine-based activation motif |
| Itgax | integrin alpha X |
| LBP | Lycium barbarum polysaccharide |
| LDAM | lipid droplet-accumulating microglia |
| Lgals3 | Galectin-3 |
| MAPT | microtubule-associated protein tau |
| MGnD | microglial neurodegenerative-phenotype |
| MS | multiple sclerosis |
| NCL3 | neuronal ceroid lipofuscinosis 3 |
| NGF | nerve growth factor |
| NPC2 | Niemann-Pick Disease Type C2 |
| OR | odds ratio |
| PICALM | phosphatidylinositol binding clathrin assembly protein |
| PLCγ | phospholipase C-γ |
| PLX3397 | Pexidartinib |
| PD | Parkinson’s disease |
| P2RY12 | P2Y purinoceptor 12 |
| RA | rheumatoid arthritis |
| RAK1 | receptor for activated C kinase1 |
| RAP1B | RAS related protein 1b |
| RCTs | randomized controlled trials |
| SFFV | spleen focus forming virus |
| scRNA-seq | single-cell RNA sequencing |
| SGIP1 | SH3GL interacting endocytic adaptor 1 |
| SIRPα | signal regulatory protein alpha |
| SLC33A1 | solute carrier family 33 member 1 |
| SNPs | Single nucleotide polymorphisms |
| SNX17 | sorting nexin 17 |
| snRNA-seq | single nucleus RNA sequencing |
| SPI1 | proviral integration oncogene 1 |
| SOCS1 | suppressing cell signaling protein 1 |
| Syk | spleen tyrosine kinase |
| TLR2/4 | Toll-like receptor 2/4 |
| TMEM119 | transmembrane protein 119 |
| TNF-α | tumor necrosis factor-α |
| TREM2 | triggering receptor expressed on myeloid cells 2 |
| TWAS | transcriptome-wide association study |
| VEGF | vascular endothelial growth factor |
| WAM | white matter-associate microglia |
References
- Fujikawa, R.; Tsuda, M. The Functions and Phenotypes of Microglia in Alzheimer’s Disease. Cells 2023, 12, 1207. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; De Strooper, B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2024. Alzheimers Dement 2024, 10, 12465. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Fruhwürth, S.; Zetterberg, H.; Paludan, S.R. Microglia and amyloid plaque formation in Alzheimer’s disease—Evidence, possible mechanisms, and future challenges. J. Neuroimmunol. 2024, 390, 578342. [Google Scholar] [CrossRef] [PubMed]
- Brase, L.; You, S.F.; D’Oliveira Albanus, R.; Del-Aguila, J.L.; Dai, Y.; Novotny, B.C.; Soriano-Tarraga, C.; Dykstra, T.; Fernandez, M.V.; Budde, J.P.; et al. Single-nucleus RNA-sequencing of autosomal dominant Alzheimer disease and risk variant carriers. Nat. Commun. 2023, 14, 2314. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Victor, M.B.; Park, Y.P.; Xiong, X.; Scannail, A.N.; Leary, N.; Prosper, S.; Viswanathan, S.; Luna, X.; Boix, C.A.; et al. Human microglial state dynamics in Alzheimer’s disease progression. Cell 2023, 186, 4386–4403.e29. [Google Scholar] [CrossRef]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 2023, 15, 1201982. [Google Scholar] [CrossRef]
- Guo, S.; Yang, J. Bayesian genome-wide TWAS with reference transcriptomic data of brain and blood tissues identified 141 risk genes for Alzheimer’s disease dementia. Alzheimers Res. Ther. 2024, 16, 120. [Google Scholar] [CrossRef] [PubMed]
- Sudwarts, A.; Ramesha, S.; Gao, T.; Ponnusamy, M.; Wang, S.; Hansen, M.; Kozlova, A.; Bitarafan, S.; Kumar, P.; Beaulieu-Abdelahad, D.; et al. BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia. Mol. Neurodegener. 2022, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, S.; Baglietto-Vargas, D.; Caballero, C.; Moreno-Gonzalez, I.; Torres, M.; Sanchez-Varo, R.; Ruano, D.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: Age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 2008, 28, 11650–11661. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Deng, M.; Hu, G.; Li, N.; Yuan, H.; Zhou, Y. New Insights into Microglial Mechanisms of Memory Impairment in Alzheimer’s Disease. Biomolecules 2022, 12, 1722. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, X. Different phenotypes of microglia in animal models of Alzheimer disease. Immun. Ageing 2022, 19, 44. [Google Scholar] [CrossRef] [PubMed]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.; Joseph, B. Microglial subtypes: Diversity within the microglial community. Embo J. 2019, 38, e101997. [Google Scholar] [CrossRef] [PubMed]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef]
- Ajami, B.; Samusik, N.; Wieghofer, P.; Ho, P.P.; Crotti, A.; Bjornson, Z.; Prinz, M.; Fantl, W.J.; Nolan, G.P.; Steinman, L. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 2018, 21, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trümbach, D.; Wurst, W.; et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017, 18, 1186–1198. [Google Scholar] [CrossRef]
- Afridi, R.; Lee, W.H.; Suk, K. Microglia Gone Awry: Linking Immunometabolism to Neurodegeneration. Front. Cell Neurosci. 2020, 14, 246. [Google Scholar] [CrossRef]
- Javanmehr, N.; Saleki, K.; Alijanizadeh, P.; Rezaei, N. Microglia dynamics in aging-related neurobehavioral and neuroinflammatory diseases. J. Neuroinflamm. 2022, 19, 273. [Google Scholar] [CrossRef] [PubMed]
- Safaiyan, S.; Besson-Girard, S.; Kaya, T.; Cantuti-Castelvetri, L.; Liu, L.; Ji, H.; Schifferer, M.; Gouna, G.; Usifo, F.; Kannaiyan, N.; et al. White matter aging drives microglial diversity. Neuron 2021, 109, 1100–1117.e10. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Lee, S.J.; Mook-Jung, I. White matter-associated microglia: New players in brain aging and neurodegenerative diseases. Ageing Res. Rev. 2022, 75, 101574. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Alonso, A.; Schetters, S.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 2016, 139, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Sosna, J.; Philipp, S.; Albay, R., 3rd; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef]
- Liu, Z.; Condello, C.; Schain, A.; Harb, R.; Grutzendler, J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J. Neurosci. 2010, 30, 17091–17101. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Sun, B.; Zhou, Y.; Kauppinen, T.M.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Jiménez, J.M.; Mancilla, M.; Maccioni, R.B. Tau oligomers and fibrils induce activation of microglial cells. J. Alzheimers Dis. 2013, 37, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Ayyubova, G. Dysfunctional microglia and tau pathology in Alzheimer’s disease. Rev. Neurosci. 2023, 34, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.R.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Vanni, V.; Guadalupi, L.; Stampanoni Bassi, M.; Buttari, F.; Mandolesi, G.; et al. Tumor Necrosis Factor and Interleukin-1β Modulate Synaptic Plasticity during Neuroinflammation. Neural Plast. 2018, 2018, 8430123. [Google Scholar] [CrossRef]
- Xiao, J.; Yao, R.; Xu, B.; Wen, H.; Zhong, J.; Li, D.; Zhou, Z.; Xu, J.; Wang, H. Inhibition of PDE4 Attenuates TNF-α-Triggered Cell Death Through Suppressing NF-κB and JNK Activation in HT-22 Neuronal Cells. Cell Mol. Neurobiol. 2020, 40, 421–435. [Google Scholar] [CrossRef]
- Avital, A.; Goshen, I.; Kamsler, A.; Segal, M.; Iverfeldt, K.; Richter-Levin, G.; Yirmiya, R. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 2003, 13, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yao, H.; Liu, W.; Ya, B.; Cheng, H.; Xing, Z.; Wu, Y. Microglia Polarization in Alzheimer’s Disease: Mechanisms and a Potential Therapeutic Target. Front. Aging Neurosci. 2021, 13, 772717. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; McPherson, C.A.; Harry, G.J. Heterogeneity of microglia and TNF signaling as determinants for neuronal death or survival. Neurotoxicology 2009, 30, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Stevens, B. Microglia: Phagocytosing to Clear, Sculpt, and Eliminate. Dev. Cell 2016, 38, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Jung, Y.J.; Scerba, M.T.; Hwang, I.; Kim, Y.K.; Kim, S.; Modrow, S.; Tweedie, D.; Hsueh, S.C.; Liu, D.; et al. Role of chronic neuroinflammation in neuroplasticity and cognitive function: A hypothesis. Alzheimers Dement. 2022, 18, 2327–2340. [Google Scholar] [CrossRef]
- Yang, L.; Duan, R.; Liu, T.C.-Y.; Parker, E.; Wu, C.; Deng, Q. Microglia and Astrocytes in Alzheimer’s Disease: Significance and Summary of Recent Advances. Aging Dis. 2023, 15, 4–32. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Loeffler, D.A. Antibody-Mediated Clearance of Brain Amyloid-β: Mechanisms of Action, Effects of Natural and Monoclonal Anti-Aβ Antibodies, and Downstream Effects. J. Alzheimers Dis. Rep. 2023, 7, 873–899. [Google Scholar] [CrossRef] [PubMed]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wen, J.; Yang, H.; Jones, I.R.; Zhu, X.; Liu, W.; Li, B.; Clelland, C.D.; Luo, W.; Wong, M.Y.; et al. Functional characterization of Alzheimer’s disease genetic variants in microglia. Nat. Genet. 2023, 55, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sudan, R.; Peng, V.; Zhou, Y.; Du, S.; Yuede, C.M.; Lei, T.; Hou, J.; Cai, Z.; Cella, M.; et al. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 2022, 185, 4153–4169.e19. [Google Scholar] [CrossRef] [PubMed]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Gao, Y.; Zhu, Y.; Sheng, X.; et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 1365. [Google Scholar] [CrossRef]
- Schlepckow, K.; Morenas-Rodríguez, E.; Hong, S.; Haass, C. Stimulation of TREM2 with agonistic antibodies-an emerging therapeutic option for Alzheimer’s disease. Lancet Neurol. 2023, 22, 1048–1060. [Google Scholar] [CrossRef]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef] [PubMed]
- Schlepckow, K.; Monroe, K.M.; Kleinberger, G.; Cantuti-Castelvetri, L.; Parhizkar, S.; Xia, D.; Willem, M.; Werner, G.; Pettkus, N.; Brunner, B.; et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med. 2020, 12, e11227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, L.; Yang, J.; Meng, L.; Chen, J.; Zhou, L.; Wang, J.; Xiong, M.; Zhang, Z. Soluble TREM2 ameliorates tau phosphorylation and cognitive deficits through activating transgelin-2 in Alzheimer’s disease. Nat. Commun. 2023, 14, 6670. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.; Neumayer, G.; Shibuya, Y.; Mader, M.M.; Wernig, M. A cell therapy approach to restore microglial Trem2 function in a mouse model of Alzheimer’s disease. Cell Stem Cell 2023, 30, 1043–1053.e6. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement. 2021, 7, e12179. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Brouwers, N.; Van Cauwenberghe, C.; Engelborghs, S.; Lambert, J.C.; Bettens, K.; Le Bastard, N.; Pasquier, F.; Montoya, A.G.; Peeters, K.; Mattheijssens, M.; et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol. Psychiatry 2012, 17, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.C.; Yu, J.T.; Jiang, T.; Wang, P.; Cao, L.; Tan, L. CR1 in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, H.; Tatton, N.; McCaughey, S.; Kurnellas, M.; Rosenthal, A. Progranulin as a therapeutic target in neurodegenerative diseases. Trends Pharmacol. Sci. 2022, 43, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.S.; Min, S.W.; Krabbe, G.; Wang, C.; Zhou, Y.; Asgarov, R.; Li, Y.; Martens, L.H.; Elia, L.P.; Ward, M.E.; et al. Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 2014, 20, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Rastegar-Moghaddam, S.H.; Ebrahimzadeh-Bideskan, A.; Shahba, S.; Malvandi, A.M.; Mohammadipour, A. Roles of the miR-155 in Neuroinflammation and Neurological Disorders: A Potent Biological and Therapeutic Target. Cell Mol. Neurobiol. 2023, 43, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Herron, S.; Silveira, S.; Kleemann, K.; Gauthier, C.; Mallah, D.; Cheng, Y.; Margeta, M.A.; Pitts, K.M.; Barry, J.L.; et al. Identification of a protective microglial state mediated by miR-155 and interferon-γ signaling in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Zong, B.; Yu, F.; Zhang, X.; Pang, Y.; Zhao, W.; Sun, P.; Li, L. Mechanosensitive Piezo1 channel in physiology and pathophysiology of the central nervous system. Ageing Res. Rev. 2023, 90, 102026. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, Q.; Zhu, H.; Hou, L.; Liu, W.; Yang, Q.; Shen, H.; Chai, G.; Zhang, B.; Chen, S.; et al. Microglial Piezo1 senses Aβ fibril stiffness to restrict Alzheimer’s disease. Neuron 2023, 111, 15–29.e18. [Google Scholar] [CrossRef]
- Davis, H.; Attwell, D. Plaque attack: Microglia have hard feelings toward amyloid-β. Neuron 2023, 111, 1–2. [Google Scholar] [CrossRef]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef]
- Pauwels, E.K.J.; Boer, G.J. Friends and Foes in Alzheimer’s Disease. Med. Princ. Pract. 2023, 32, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ennerfelt, H.; Frost, E.L.; Shapiro, D.A.; Holliday, C.; Zengeler, K.E.; Voithofer, G.; Bolte, A.C.; Lammert, C.R.; Kulas, J.A.; Ulland, T.K.; et al. SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell 2022, 185, 4135–4152.e22. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Li, Y.; Zhang, Z.; Yuan, Y. An insight into the TAM system in Alzheimer’s disease. Int. Immunopharmacol. 2023, 116, 109791. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.N.; Gooi, J.H.; Doughty, L.; Nero, T.L.; Markulic, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Abeta Phagocytosis. iScience 2019, 19, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Chiles, J., 3rd; Xi, H.S.; Medway, C.; Simpson, J.; Potluri, S.; Howard, D.; Liang, Y.; Paumi, C.M.; Mukherjee, S.; et al. Genetics of CD33 in Alzheimer’s disease and acute myeloid leukemia. Hum. Mol. Genet. 2015, 24, 3557–3570. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Lim, L.W. Glial cells in Alzheimer’s disease: From neuropathological changes to therapeutic implications. Ageing Res. Rev. 2022, 78, 101622. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Zhang, J.; Liu, H.; Babu-Khan, S.; Vassar, R.; Biere, A.L.; Citron, M.; Landreth, G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J. Neurosci. 2003, 23, 7504–7509. [Google Scholar] [CrossRef]
- Eriksen, J.L.; Sagi, S.A.; Smith, T.E.; Weggen, S.; Das, P.; McLendon, D.C.; Ozols, V.V.; Jessing, K.W.; Zavitz, K.H.; Koo, E.H.; et al. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J. Clin. Investig. 2003, 112, 440–449. [Google Scholar] [CrossRef]
- Kummer, M.P.; Schwarzenberger, R.; Sayah-Jeanne, S.; Dubernet, M.; Walczak, R.; Hum, D.W.; Schwartz, S.; Axt, D.; Heneka, M.T. Pan-PPAR modulation effectively protects APP/PS1 mice from amyloid deposition and cognitive deficits. Mol. Neurobiol. 2015, 51, 661–671. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef]
- Garrido-Mesa, N.; Zarzuelo, A.; Gálvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Cooper, J.D.; Hanger, D.P.; Noble, W. Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front. Psychiatry 2010, 1, 136. [Google Scholar] [CrossRef]
- Parachikova, A.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M.; Green, K.N. Reductions in amyloid-beta-derived neuroinflammation, with minocycline, restore cognition but do not significantly affect tau hyperphosphorylation. J. Alzheimers Dis. 2010, 21, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef]
- Plantone, D.; Pardini, M.; Righi, D.; Manco, C.; Colombo, B.M.; De Stefano, N. The Role of TNF-α in Alzheimer’s Disease: A Narrative Review. Cells 2023, 13, 54. [Google Scholar] [CrossRef]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic Potential of TNF-α Inhibition for Alzheimer’s Disease Prevention. J. Alzheimers Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef]
- McAlpine, F.E.; Lee, J.K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Cheng, X.; Staufenbiel, M.; Li, R.; Shen, Y. Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of β-secretase in a mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e55091. [Google Scholar] [CrossRef]
- Tweedie, D.; Ferguson, R.A.; Fishman, K.; Frankola, K.A.; Van Praag, H.; Holloway, H.W.; Luo, W.; Li, Y.; Caracciolo, L.; Russo, I.; et al. Tumor necrosis factor-α synthesis inhibitor 3,6’-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 106. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Frailes, C.; Di Lauro, C.; Bianchi, C.; de Diego-García, L.; Sebastián-Serrano, Á.; Boscá, L.; Díaz-Hernández, M. Amyloid Peptide Induced Neuroinflammation Increases the P2X7 Receptor Expression in Microglial Cells, Impacting on Its Functionality. Front. Cell Neurosci. 2019, 13, 143. [Google Scholar] [CrossRef]
- Lee, H.G.; Won, S.M.; Gwag, B.J.; Lee, Y.B. Microglial P2X7 receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp. Mol. Med. 2011, 43, 7–14. [Google Scholar] [CrossRef]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef]
- Daniels, M.J.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef] [PubMed]
- Momtazmanesh, S.; Perry, G.; Rezaei, N. Toll-like receptors in Alzheimer’s disease. J. Neuroimmunol. 2020, 348, 577362. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.L.; Hennessy, E.; Rubio-Araiz, A.; Keogh, B.; McCormack, W.; McGuirk, P.; Reilly, M.; Lynch, M.A. Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer’s disease. Brain Behav. Immun. 2016, 58, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef]
- Kiyota, T.; Okuyama, S.; Swan, R.J.; Jacobsen, M.T.; Gendelman, H.E.; Ikezu, T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J. 2010, 24, 3093–3102. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Tianbai, L.; Herring, A.; Ceballos-Diaz, C.; Das, P.; Golde, T.E. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol. Neurodegener. 2012, 7, 36. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Jansen-West, K.; Beccard, A.; Ceballos-Diaz, C.; Levites, Y.; Verbeeck, C.; Zubair, A.C.; Dickson, D.; Golde, T.E.; Das, P. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010, 24, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Herring, A.; Ceballos-Diaz, C.; Das, P.; Golde, T.E. Hippocampal expression of murine TNFα results in attenuation of amyloid deposition in vivo. Mol. Neurodegener. 2011, 6, 16. [Google Scholar] [CrossRef]
- Ryu, J.K.; McLarnon, J.G. Block of purinergic P2X7 receptor is neuroprotective in an animal model of Alzheimer’s disease. Neuroreport 2008, 19, 1715–1719. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.N.; Johnson, R.E.; O’Banion, M.K. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Investig. 2007, 117, 1595–1604. [Google Scholar] [CrossRef]
- Zhao, K.; Liu, J.; Sun, T.; Zeng, L.; Cai, Z.; Li, Z.; Liu, R. The miR-25802/KLF4/NF-κB signaling axis regulates microglia-mediated neuroinflammation in Alzheimer’s disease. Brain Behav. Immun. 2024, 118, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Jin, J.; Lim, J.E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.D.; Tahara, K.; Lalonde, R.; Fukuchi, K. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 92. [Google Scholar] [CrossRef]
- Boxer, A.L.; Sperling, R. Accelerating Alzheimer’s therapeutic development: The past and future of clinical trials. Cell 2023, 186, 4757–4772. [Google Scholar] [CrossRef]
- Shal, B.; Ding, W.; Ali, H.; Kim, Y.S.; Khan, S. Anti-neuroinflammatory Potential of Natural Products in Attenuation of Alzheimer’s Disease. Front. Pharmacol. 2018, 9, 548. [Google Scholar] [CrossRef]
- Essa, M.M.; Vijayan, R.K.; Castellano-Gonzalez, G.; Memon, M.A.; Braidy, N.; Guillemin, G.J. Neuroprotective effect of natural products against Alzheimer’s disease. Neurochem. Res. 2012, 37, 1829–1842. [Google Scholar] [CrossRef]
- Kim, M.J.; Seong, A.R.; Yoo, J.Y.; Jin, C.H.; Lee, Y.H.; Kim, Y.J.; Lee, J.; Jun, W.J.; Yoon, H.G. Gallic acid, a histone acetyltransferase inhibitor, suppresses beta-amyloid neurotoxicity by inhibiting microglial-mediated neuroinflammation. Mol. Nutr. Food Res. 2011, 55, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Wang, L.; Liu, X.; Cong, X.; Yan, S.S.; Wang, Y.; Zhang, W. Geniposide attenuates oligomeric Abeta(1-42)-induced inflammatory response by targeting RAGE-dependent signaling in BV2 cells. Curr. Alzheimer Res. 2014, 11, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Liang, Q.; Ge, G. Gypenoside Attenuates β Amyloid-Induced Inflammation in N9 Microglial Cells via SOCS1 Signaling. Neural Plast. 2016, 2016, 6362707. [Google Scholar] [CrossRef] [PubMed]
- Manthey, A.L.; Chiu, K.; So, K.F. Effects of Lycium barbarum on the Visual System. Int. Rev. Neurobiol. 2017, 135, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Baum, L.; Yu, S.; Lin, Y.; Xiong, G.; Chang, R.C.; So, K.F.; Chiu, K. Preservation of Retinal Function Through Synaptic Stabilization in Alzheimer’s Disease Model Mouse Retina by Lycium Barbarum Extracts. Front. Aging Neurosci. 2021, 13, 788798. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Yu, M.S.; Lai, C.S.; So, K.F.; Yuen, W.H.; Chang, R.C. Characterizing the neuroprotective effects of alkaline extract of Lycium barbarum on beta-amyloid peptide neurotoxicity. Brain Res. 2007, 1158, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Yu, M.S.; Yang, X.F.; So, K.F.; Yuen, W.H.; Chang, R.C. Neuroprotective effects of polysaccharides from wolfberry, the fruits of Lycium barbarum, against homocysteine-induced toxicity in rat cortical neurons. J. Alzheimers Dis. 2010, 19, 813–827. [Google Scholar] [CrossRef]
- Zhou, Y.; Duan, Y.; Huang, S.; Zhou, X.; Zhou, L.; Hu, T.; Yang, Y.; Lu, J.; Ding, K.; Guo, D.; et al. Polysaccharides from Lycium barbarum ameliorate amyloid pathology and cognitive functions in APP/PS1 transgenic mice. Int. J. Biol. Macromol. 2020, 144, 1004–1012. [Google Scholar] [CrossRef]
- Liu, S.Y.; Lu, S.; Yu, X.L.; Yang, S.G.; Liu, W.; Liu, X.M.; Wang, S.W.; Zhu, J.; Ji, M.; Liu, D.Q.; et al. Fruitless Wolfberry-Sprout Extract Rescued Cognitive Deficits and Attenuated Neuropathology in Alzheimer’s Disease Transgenic Mice. Curr. Alzheimer Res. 2018, 15, 856–868. [Google Scholar] [CrossRef]
- Sun, Z.Q.; Liu, J.F.; Luo, W.; Wong, C.H.; So, K.F.; Hu, Y.; Chiu, K. Lycium barbarum extract promotes M2 polarization and reduces oligomeric amyloid-β-induced inflammatory reactions in microglial cells. Neural Regen. Res. 2022, 17, 203–209. [Google Scholar] [CrossRef]
- Teng, P.; Li, Y.; Cheng, W.; Zhou, L.; Shen, Y.; Wang, Y. Neuroprotective effects of Lycium barbarum polysaccharides in lipopolysaccharide-induced BV2 microglial cells. Mol. Med. Rep. 2013, 7, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Liong, E.C.; Ching, Y.P.; Chang, R.C.; So, K.F.; Fung, M.L.; Tipoe, G.L. Lycium barbarum polysaccharides protect mice liver from carbon tetrachloride-induced oxidative stress and necroinflammation. J. Ethnopharmacol. 2012, 139, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Granic, I.; Dolga, A.M.; Nijholt, I.M.; van Dijk, G.; Eisel, U.L. Inflammation and NF-kappaB in Alzheimer’s disease and diabetes. J. Alzheimers Dis. 2009, 16, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Liu, J.; Song, Z.; Pan, X.; Chen, L.; Cui, X.; Wang, M. Berberine suppresses amyloid-beta-induced inflammatory response in microglia by inhibiting nuclear factor-kappaB and mitogen-activated protein kinase signalling pathways. J. Pharm. Pharmacol. 2012, 64, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wang, Y.; Zhao, D.; Feng, X.; Zhang, L.; Liu, C. Loganin prevents BV-2 microglia cells from Aβ(1-42) -induced inflammation via regulating TLR4/TRAF6/NF-κB axis. Cell Biol. Int. 2018, 42, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yuan, L.; Zhao, X.; Hou, C.; Ma, W.; Yu, H.; Xiao, R. Genistein antagonizes inflammatory damage induced by beta-amyloid peptide in microglia through TLR4 and NF-kappaB. Nutrition 2014, 30, 90–95. [Google Scholar] [CrossRef]
- Joo, S.S.; Yoo, Y.M.; Ahn, B.W.; Nam, S.Y.; Kim, Y.B.; Hwang, K.W.; Lee, D.I. Prevention of inflammation-mediated neurotoxicity by Rg3 and its role in microglial activation. Biol. Pharm. Bull. 2008, 31, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, X.; Li, L.; Li, Y.; Zhou, L.; Zhang, X.; Hu, X.; Zhao, G. Ginsenoside Rd Improves Learning and Memory Ability in APP Transgenic Mice. J. Mol. Neurosci. 2015, 57, 522–528. [Google Scholar] [CrossRef]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol mitigates lipopolysaccharide- and Abeta-mediated microglial inflammation by inhibiting the TLR4/NF-kappaB/STAT signaling cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Yu, L.J.; Hui, X.C.; Wu, Z.Z.; Yin, K.L.; Yang, H.; Xu, Y. Hydroxy-safflor yellow A attenuates Aβ1-42-induced inflammation by modulating the JAK2/STAT3/NF-kappaB pathway. Brain Res. 2014, 1563, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zou, L.B.; Wang, L.H.; Jin, G.; Pan, J.J.; Chi, T.Y.; Ji, X.F. Xanthoceraside inhibits pro-inflammatory cytokine expression in Abeta25-35/IFN-gamma-stimulated microglia through the TLR2 receptor, MyD88, nuclear factor-kappaB, and mitogen-activated protein kinase signaling pathways. J. Pharmacol. Sci. 2013, 122, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hu, Y.; Cummings, J.; Mattke, S.; Iwatsubo, T.; Nakamura, A.; Vellas, B.; O’Bryant, S.; Shaw, L.M.; Cho, M.; et al. Blood-based biomarkers for Alzheimer’s disease: Current state and future use in a transformed global healthcare landscape. Neuron 2023, 111, 2781–2799. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Colonna, M. Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med. 2021, 218, e20202717. [Google Scholar] [CrossRef] [PubMed]
- McQuade, A.; Kang, Y.J.; Hasselmann, J.; Jairaman, A.; Sotelo, A.; Coburn, M.; Shabestari, S.K.; Chadarevian, J.P.; Fote, G.; Tu, C.H.; et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020, 11, 5370. [Google Scholar] [CrossRef] [PubMed]
- Bassil, R.; Shields, K.; Granger, K.; Zein, I.; Ng, S.; Chih, B. Improved modeling of human AD with an automated culturing platform for iPSC neurons, astrocytes and microglia. Nat. Commun. 2021, 12, 5220. [Google Scholar] [CrossRef] [PubMed]
- Hasselmann, J.; Coburn, M.A.; England, W.; Figueroa Velez, D.X.; Kiani Shabestari, S.; Tu, C.H.; McQuade, A.; Kolahdouzan, M.; Echeverria, K.; Claes, C.; et al. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 2019, 103, 1016–1033.e10. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Zhang, X.; So, K.-F.; Jiang, W.; Chiu, K. Targeting Microglia in Alzheimer’s Disease: Pathogenesis and Potential Therapeutic Strategies. Biomolecules 2024, 14, 833. https://doi.org/10.3390/biom14070833
Sun Z, Zhang X, So K-F, Jiang W, Chiu K. Targeting Microglia in Alzheimer’s Disease: Pathogenesis and Potential Therapeutic Strategies. Biomolecules. 2024; 14(7):833. https://doi.org/10.3390/biom14070833
Chicago/Turabian StyleSun, Zhongqing, Xin Zhang, Kwok-Fai So, Wen Jiang, and Kin Chiu. 2024. "Targeting Microglia in Alzheimer’s Disease: Pathogenesis and Potential Therapeutic Strategies" Biomolecules 14, no. 7: 833. https://doi.org/10.3390/biom14070833