

Ischemic Neuroprotection by Insulin with Down-Regulation of Divalent Metal Transporter 1 (DMT1) Expression and Ferrous Iron-Dependent Cell Death

{kind=link}
Abstract
:1. Introduction
2. Material and Methods
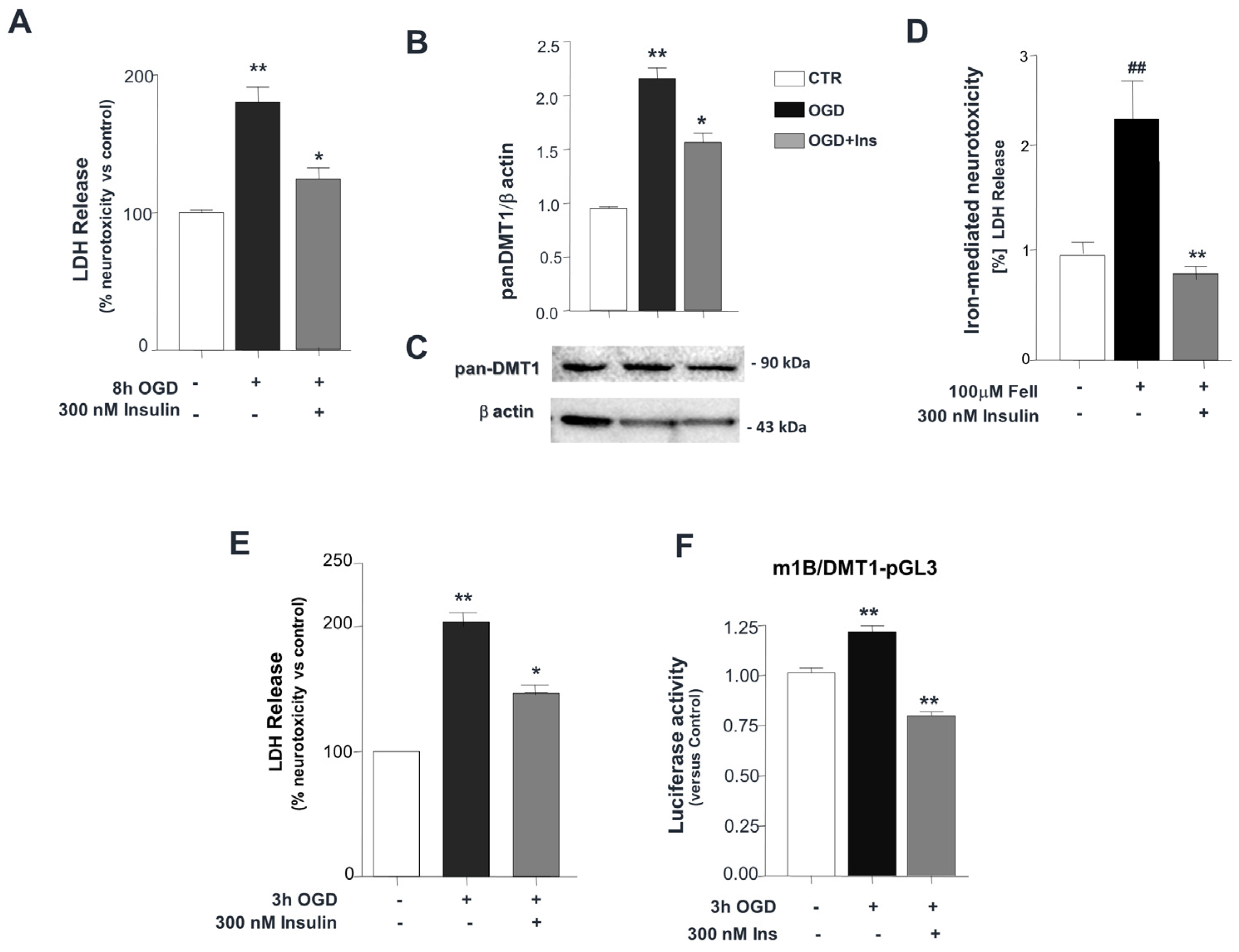
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D.; Trenor, C.C.; Su, M.A.; Foernzler, D.; Beier, D.R.; Dietrich, W.F.; Andrews, N.C. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat. Genet. 1997, 16, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Garrick, M.D.; Dolan, K.G.; Horbinski, C.; Ghio, A.J.; Higgins, D.; Porubcin, M.; Moore, E.G.; Hainsworth, L.N.; Umbreit, J.N.; Conrad, M.E.; et al. DMT1: A mammalian transporter for multiple metals. Biometals 2003, 16, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Iron and infection. Int. J. Hematol. 2018, 107, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia, R.; Garavaglia, B.; Memo, M. DMT1 Expression and Iron Levels at the Crossroads Between Aging and Neurodegeneration. Front. Neurosci. 2019, 13, 575. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Roth, J.A. Nitric oxide transcriptionally down-regulates specific isoforms of divalent metal transporter (DMT1) via NF-kappaB. J. Neurochem. 2006, 96, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Hubert, N.; Hentze, M.W. Previously uncharacterized isoforms of divalent metal transporter DMT1: Implications for regulation and cellular function. Proc. Natl. Acad. Sci. USA 2002, 99, 12345–12350. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.M.; et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, A.; Sarnico, I.; Ingrassia, R.; Boroni, F.; Branca, C.; Benarese, M.; Faraco, G.; Blasi, F.; Chiarugi, A.; Spano, P.; et al. The acetylation of RelA in Lys310 dictates the NF-kB-dependent response in post-ischemic injury. Cell Death Dis. 2010, 1, e96. [Google Scholar] [CrossRef] [PubMed]
- Baiguera, C.; Alghisi, M.; Pinna, A.; Bellucci, A.; De Luca, M.A.; Frau, L.; Morelli, M.; Ingrassia, R.; Benarese, M.; Porrini, V.; et al. Late-onset Parkinsonism in NF-kB/c-Rel-deficient mice. Brain 2012, 135 Pt 9, 2750–2765. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia, R.; Lanzillotta, A.; Sarnico, I.; Benarese, M.; Blasi, F.; Borgese, L.; Bilo, F.; Depero, L.; Chiarugi, A.; Spano, P.F.; et al. 1B/(−)IRE DMT1 expression during brain ischemia contributes to cell death mediated by NF-kB/RelA acetylation at Lys310. PLoS ONE 2012, 7, e38019. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.B.; Tonnesen, M.F.; Madsen, A.N.; Hagedorn, P.H.; Friberg, J.; Grunnet, L.G.; Heller, R.S.; Østergren, A.; Nielsen, A.; Størling, J.; et al. Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic β cell fate in response to cytokines. Cell Metab. 2012, 16, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Lundh, M.; Christensen, D.P.; Rasmussen, D.N.; Mascagni, P.; Dinarello, C.A.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Lysine deacetylases are produced in pancreatic beta cells and are differentially regulated by proinflammatory cytokines. Diabetologia 2010, 53, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.Q.; He, W.; Xu, K.F.; Wang, Z.X.; Xu, X.Y.; Chen, H. FTO Inhibits Insulin Secretion and Promotes NF-kB Activation through Positively Regulating ROS Production in Pancreatic β cells. PLoS ONE 2015, 10, e0127705. [Google Scholar]
- Li, X.; Wu, Y.; Song, Y.; Ding, N.; Lu, M.; Jia, L.; Zhao, Y.; Liu, M.; Chen, Z. Activation of NF-kB-Inducing Kinase in Islet β Cells Causes β Cell Failure and Diabetes. Mol. Ther. 2020, 28, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Dakic, T.; Jevdjovic, T.; Lakic, I.; Ruzicic, A.; Jasnic, N.; Djurasevic, S.; Djordjevic, J.; Vujovic, P. The Expression of Insulin in the Central Nervous System: What Have We Learned So Far? Int. J. Mol. Sci. 2023, 24, 6586. [Google Scholar] [CrossRef] [PubMed]
- Milstein, J.L.; Heather, A.; Ferris, H.A. The brain as an insulin-sensitive metabolic organ. Mol. Metab. 2021, 52, 101234. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.C.; Stouffer, M.A.; Mancini, M.; Nicholson, C.; Carr, K.D.; Rice, M.E. Interactions between insulin and diet on striatal dopamine uptake kinetics in rodent brain slices. Eur. J. Neurosci. 2019, 49, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Zorina, I.I.; Avrova, N.F.; Zakharova, I.O.; Shpakov, A.O. Biochemistry (Mosc). Prospects for the Use of Intranasally Administered Insulin and Insulin-Like Growth Factor-1 in Cerebral Ischemia. Biochemistry 2023, 88, 374–391. [Google Scholar] [PubMed]
- Lioutas, V.A.; Alfaro-Martinez, F.; Bedoya, F.; Chung, C.C.; Pimentel, D.A.; Novak, V. Intranasal Insulin and Insulin-Like Growth Factor 1 as Neuroprotectants in Acute Ischemic Stroke. Transl. Stroke Res. 2015, 6, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Iravanpour, F.; Dargahi, F.; Rezaei, M.; Haghani, M.; Heidari, R.; Valian, N.; Ahmadiani, A. Intranasal insulin improves mitochondrial function and attenuates motor deficits in a rat 6-OHDA model of Parkinson’s disease. CNS Neurosci. Ther. 2021, 27, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Díaz, A.; Humeres, C.; González, V.; Gómez, M.T.; Montt, N.; Sanchez, G.; Chiong, M.; García, L. Insulin/NFkB protects against ischemia-induced necrotic cardiomyocyte death. Biochem. Biophys. Res. Commun. 2015, 467, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Khanh, D.V.; Choi, Y.H.; Moh, S.H.; Kinyua, A.W.; Kim, K.W. Leptin and insulin signaling in dopaminergic neurons: Relationship between energy balance and reward system. Front. Psychol. 2014, 5, 846. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Dossena, M.; Bertolotti, P.; Boroni, F.; Sarnico, I.; Faraco, G.; Chiarugi, A.; Frontini, A.; Giordano, A.; Liou, H.C.; et al. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-kappaB/c-Rel-dependent transcription. Stroke 2009, 40, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia, R.; Memo, M.; Garavaglia, B. Ferrous Iron Up-regulation in Fibroblasts of Patients with Beta Propeller Protein-Associated Neurodegeneration (BPAN). Front. Genet. 2017, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Hirsch, E.C.; Villares, J.; Selimi, F.; Mouatt-Prigent, A.; Javoy-Agid, F.; Hauw, J.J.; Agid, Y. Distribution of 125I-ferrotransferrin binding sites in the mesencephalon of control subjects and patients with Parkinson’s disease. J. Neurochem. 1993, 60, 2338–2341. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, M.; Boroni, F.; Bianchetti, A.; Moraitis, C.; Sarnico, I.; Benarese, M.; Goffi, F.; Valerio, A.; Spano, P. Expression of functional NR1/NR2B-type NMDA receptors in neuronally differentiated SK-N-SH human cell line. Eur. J. Neurosci. 2002, 16, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Garrick, M.D.; Kuo, H.C.; Vargas, F.; Singleton, S.; Zhao, L.; Smith, J.J.; Paradkar, P.; Roth, J.A.; Garrick, L.M. Comparison of mammalian cell lines expressing distinct isoforms of divalent metal transporter 1 in a tetracycline-regulated fashion. Biochem. J. 2006, 398, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Inta, I.; Paxian, S.; Maegele, I.; Zhang, W.; Pizzi, M.; Spano, P.; Sarnico, I.; Muhammad, S.; Herrmann, O.; Inta, D.; et al. Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J. Neurosci. 2006, 26, 12896–12903. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free Radic. Biol. Med. 2019, 133, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Setoodeh, S.; Khorsand, M.; Takhshid, M.A. The effects of iron overload, insulin resistance and oxidative stress on metabolic disorders in patients with beta-thalassemia major. J. Diabetes Metab. Disord. 2020, 19, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Kim, J.; Veuthey, T.; Lee, C.H.; Wessling-Resnick, M. Glucose metabolism in the Belgrade rat, a model of iron-loading anemia. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1095-102. [Google Scholar] [CrossRef] [PubMed]
- Seufert, J.; Gallwitz, G. The extra-pancreatic effects of GLP-1 receptor agonists: A focus on the cardiovascular, gastrointestinal and central nervous systems. Diabetes Obes. Metab. 2014, 16, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Meissner, W.G.; Remy, P.; Giordana, C.; Maltête, D.; Derkinderen, P.; Houéto, J.L.; Anheim, M.; Benatru, I.; Boraud, T.; Brefel-Courbon, C.; et al. Trial of Lixisenatide in Early Parkinson’s Disease. N. Engl. J. Med. 2024, 390, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Shpakov, A.O.; Zorina, I.I.; Derkach, K.V. Hot Spots for the Use of Intranasal Insulin: Cerebral Ischemia, Brain Injury, Diabetes Mellitus, Endocrine Disorders and Postoperative Delirium. Int. J. Mol. Sci. 2023, 24, 3278. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Maldonado, D.A.A.P.; Novak, V. Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson’s disease and multiple system atrophy: A double-blinded placebo-controlled pilot study. PLoS ONE 2019, 14, e0214364. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fenaroli, F.; Valerio, A.; Ingrassia, R. Ischemic Neuroprotection by Insulin with Down-Regulation of Divalent Metal Transporter 1 (DMT1) Expression and Ferrous Iron-Dependent Cell Death. Biomolecules 2024, 14, 856. https://doi.org/10.3390/biom14070856
Fenaroli F, Valerio A, Ingrassia R. Ischemic Neuroprotection by Insulin with Down-Regulation of Divalent Metal Transporter 1 (DMT1) Expression and Ferrous Iron-Dependent Cell Death. Biomolecules. 2024; 14(7):856. https://doi.org/10.3390/biom14070856
Chicago/Turabian StyleFenaroli, Francesca, Alessandra Valerio, and Rosaria Ingrassia. 2024. "Ischemic Neuroprotection by Insulin with Down-Regulation of Divalent Metal Transporter 1 (DMT1) Expression and Ferrous Iron-Dependent Cell Death" Biomolecules 14, no. 7: 856. https://doi.org/10.3390/biom14070856