
Rational Design of Chimeric Antisense Oligonucleotides on a Mixed PO–PS Backbone for Splice-Switching Applications



Abstract
:1. Introduction
2. Materials and Methods
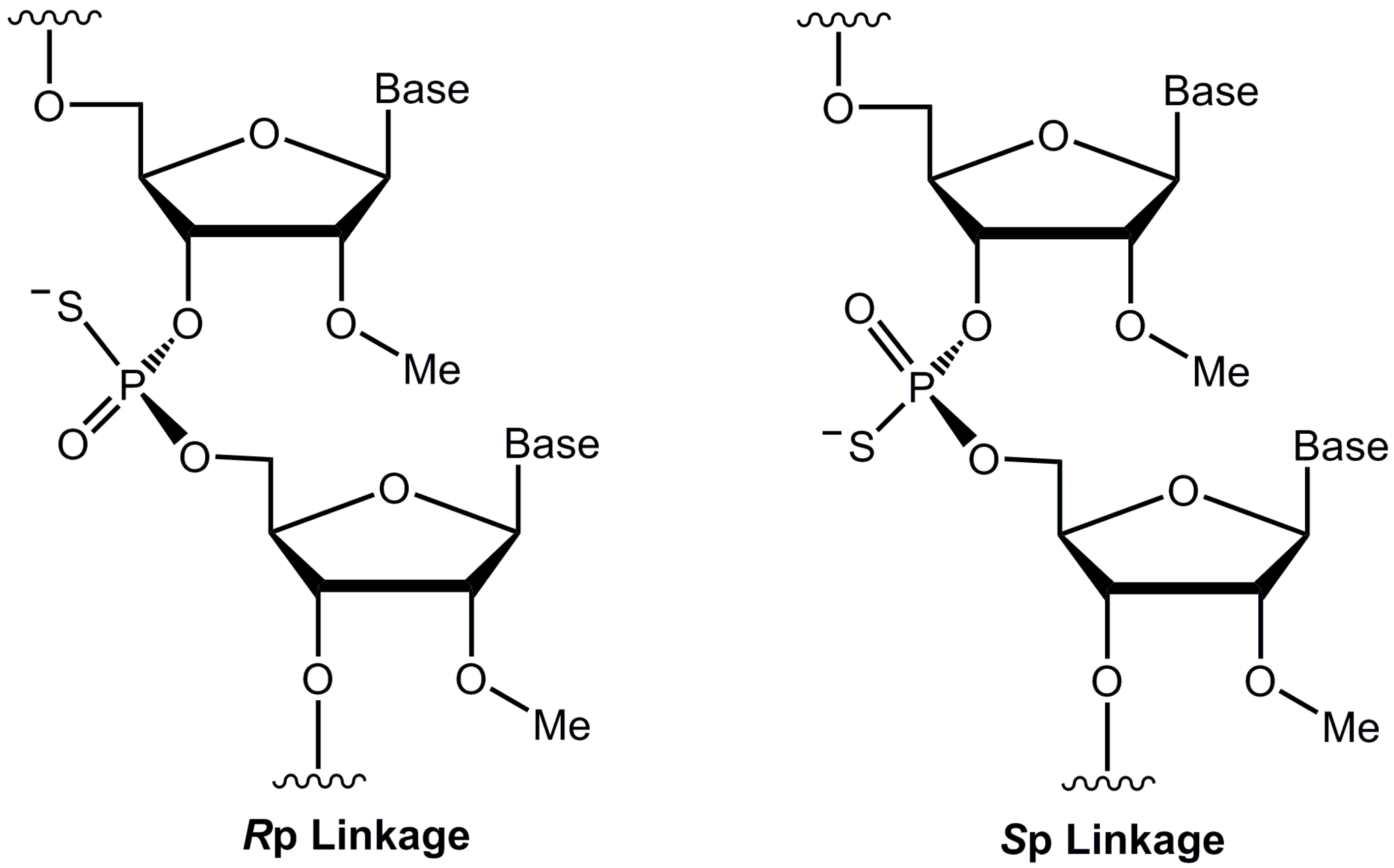
2.1. Design and Synthesis of ASOs
2.2. Melting Temperature Study of ASOs
2.3. Cell Culture and Transfection of ASOs into Cells
2.4. RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
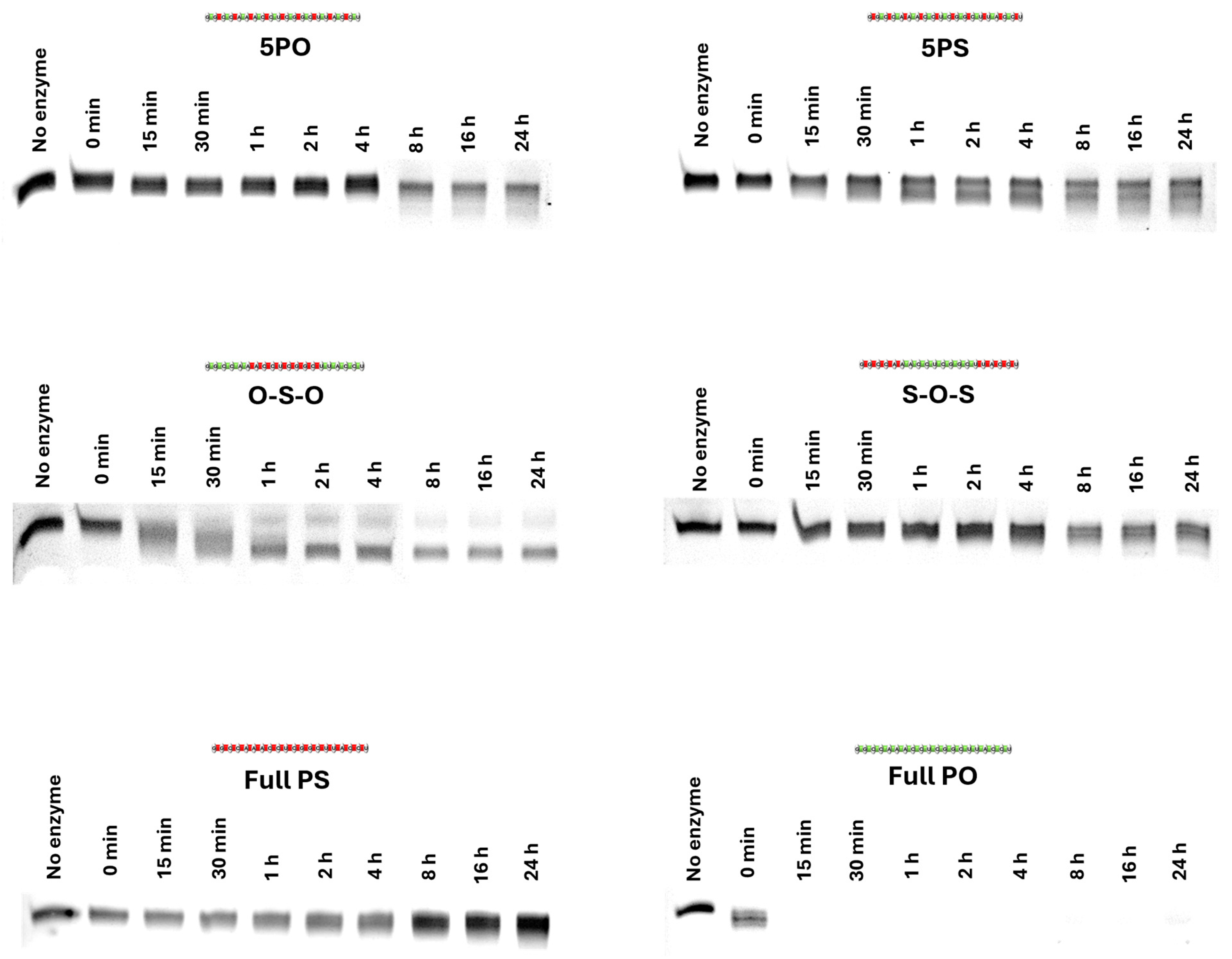
2.5. Nuclease Stability Study of ASOs
3. Results
3.1. Evaluation of RNA Binding Affinity of ASOs
3.2. Evaluation of ASOs in Inducing Exon Skipping of Dystrophin Transcript In Vitro
3.3. Evaluation of Nuclease Stability of ASOs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Havens, M.A.; Hastings, M.L. Splice-switching Antisense Oligonucleotides as Therapeutic Drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense Technology: A Review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef]
- Le, B.T.; Chen, S.; Veedu, R.N. Evaluation of Chemically Modified Nucleic Acid Analogues for Splice Switching Application. ACS Omega 2023, 8, 48650–48661. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Heendeniya, S.N.; Le, B.T.; Rahimizadeh, K.; Rabiee, N.; Zahra, Q.U.A.; Veedu, R.N. Splice-Modulating Antisense Oligonucleotides as Therapeutics for Inherited Metabolic Diseases. BioDrugs 2024, 38, 177–203. [Google Scholar] [CrossRef]
- Li, Y.; Tan, Y.; Zhang, R.; Wang, T.; Na, N.; Zheng, T.; Veedu, R.N.; Chen, S. Antisense Oligonucleotide: A Potential Therapeutic Intervention for Chronic Kidney Disease. Kidney Dial. 2022, 2, 16–37. [Google Scholar] [CrossRef]
- Chen, S.; Sbuh, N.; Veedu, R.N. Antisense Oligonucleotides as Potential Therapeutics for Type 2 Diabetes. Nucleic Acid Ther. 2021, 31, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sinhari, A.; Jain, P.; Jadhav, H.R.A. Perspective on Oligonucleotide Therapy: Approaches to Patient Customization. Front. Pharmacol. 2022, 13, 1006304. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S. The Evolution of Antisense Oligonucleotide Chemistry—A Personal Journey. Biomedicines 2021, 9, 503. [Google Scholar] [CrossRef]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate Modified Oligonucleotide-protein Interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef]
- Wan, W.B.; Migawa, M.T.; Vasquez, G.; Murray, H.M.; Nichols, J.G.; Gaus, H.; Berdeja, A.; Lee, S.; Hart, C.E.; Lima, W.F.; et al. Synthesis, Biophysical Properties and Biological Activity of Second Generation Antisense Oligonucleotides containing Chiral Phosphorothioate Linkages. Nucleic Acids Res. 2014, 42, 13456–13468. [Google Scholar] [CrossRef]
- Iwamoto, N.; Butler, D.C.D.; Svrzikapa, N.; Mohapatra, S.; Zlatev, I.; Sah, D.W.Y.; Meena; Standley, S.M.; Lu, G.; Apponi, L.H.; et al. Control of Phosphorothioate Stereochemistry Substantially Increases the Efficacy of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 845–851. [Google Scholar] [CrossRef]
- Li, M.; Lightfoot, H.L.; Halloy, F.; Malinowska, A.L.; Berk, C.; Behera, A.; Schümperli, D.; Hall, J. Synthesis and Cellular Activity of Stereochemically-pure 2′-O-(2-methoxyethyl)-Phosphorothioate Oligonucleotides. Chem. Commun. 2017, 53, 541–544. [Google Scholar] [CrossRef]
- Viney, N.J.; Guo, S.; Tai, L.J.; Baker, B.F.; Aghajan, M.; Jung, S.W.; Yu, R.Z.; Booten, S.; Murray, H.; Machemer, T.; et al. Ligand Conjugated Antisense Oligonucleotide for the Treatment of Transthyretin Amyloidosis: Preclinical and Phase 1 Data. ESC Heart Fail. 2021, 8, 652–661. [Google Scholar] [CrossRef]
- Prakash, T.P.; Yu, J.; Migawa, M.T.; Kinberger, G.A.; Wan, W.B.; Østergaard, M.E.; Carty, R.L.; Vasquez, G.; Low, A.; Chappell, A.; et al. Comprehensive Structure-Activity Relationship of Triantennary N-Acetylgalactosamine Conjugated Antisense Oligonucleotides for Targeted Delivery to Hepatocytes. J. Med. Chem. 2016, 59, 2718–2733. [Google Scholar] [CrossRef]
- Byrne, M.; Vathipadiekal, V.; Apponi, L.; Iwamoto, N.; Kandasamy, P.; Longo, K.; Liu, F.; Looby, R.; Norwood, L.; Shah, A.; et al. Stereochemistry Enhances Potency, Efficacy, and Durability of Malat1 Antisense Oligonucleotides In Vitro and In Vivo in Multiple Species. Transl. Vis. Sci. Technol. 2021, 10, 23. [Google Scholar] [CrossRef]
- Liu, Y.; Dodart, J.C.; Tran, H.; Berkovitch, S.; Braun, M.; Byrne, M.; Durbin, A.F.; Hu, X.S.; Iwamoto, N.; Jang, H.G.; et al. Variant-selective Stereopure Oligonucleotides Protect against Pathologies Associated with C9orf72-repeat Expansion in Preclinical Models. Nat. Commun. 2021, 12, 847. [Google Scholar] [CrossRef]
- Kandasamy, P.; McClorey, G.; Shimizu, M.; Kothari, N.; Alam, R.; Iwamoto, N.; Kumarasamy, J.; Bommineni, G.R.; Bezigian, A.; Chivatakarn, O.; et al. Control of Backbone Chemistry and Chirality Boost Oligonucleotide Splice Switching Activity. Nucleic Acids Res. 2022, 50, 5443–5466, Erratum in Nucleic Acids Res. 2023, 51, 3498. [Google Scholar]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X Chromosome-linked Muscular Dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Rando, T.A.; Blau, H.M. Primary Mouse Myoblast Purification, Characterization, and Transplantation for Cell-mediated Gene Therapy. J. Cell Biol. 1994, 125, 1275–1287. [Google Scholar] [CrossRef]
- Wilton, S.D.; Lloyd, F.; Carville, K.; Fletcher, S.; Honeyman, K.; Agrawal, S.; Kole, R. Specific Removal of the Nonsense Mutation from the mdx Dystrophin mRNA Using Antisense Oligonucleotides. Neuromuscul. Disord. 1999, 9, 330–338. [Google Scholar] [CrossRef]
- Mann, C.J.; Honeyman, K.; Cheng, A.J.; Ly, T.; Lloyd, F.; Fletcher, S.; Morgan, J.E.; Partridge, T.A.; Wilton, S.D. Antisense-induced Exon Skipping and Synthesis of Dystrophin in the mdx Mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 42–47. [Google Scholar]
- Le, B.T.; Paul, S.; Jastrzebska, K.; Langer, H.; Caruthers, M.H.; Veedu, R.N. Thiomorpholino Oligonucleotides as a Robust Class of Next Generation Platforms for Alternate mRNA Splicing. Proc. Natl. Acad. Sci. USA 2022, 119, e2207956119. [Google Scholar] [CrossRef]
- Chen, S.; Le, B.T.; Chakravarthy, M.; Kosbar, T.R.; Veedu, R.N. Systematic Evaluation of 2′-Fluoro Modified Chimeric Antisense Oligonucleotide-mediated Exon Skipping In Vitro. Sci. Rep. 2019, 9, 6078. [Google Scholar] [CrossRef]
- Le, B.T.; Adams, A.M.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Rational Design of Short Locked Nucleic Acid-Modified 2′-O-Methyl Antisense Oligonucleotides for Efficient Exon-Skipping In Vitro. Mol. Ther. Nucleic Acids 2017, 9, 155–161. [Google Scholar] [CrossRef]
- Le, B.T.; Chen, S.; Abramov, M.; Herdewijn, P.; Veedu, R.N. Evaluation of Anhydrohexitol Nucleic Acid, Cyclohexenyl Nucleic Acid and D-altritol Nucleic Acid-modified 2′-O-methyl RNA Mixmer Antisense Oligonucleotides for Exon Skipping In Vitro. Chem. Commun. 2016, 52, 13467–13470. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- De Clercq, E.; Eckstein, E.; Merigan, T.C. Interferon Induction Increased through Chemical Modification of a Synthetic Polyribonucleotide. Science 1969, 165, 1137–1139. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Sequence Name | Backbone Composition | ASO Sequence (5′-3′) | Abbreviation | ||
|---|---|---|---|---|---|
| Dmd M23D (+2–18) | Full 2′-OMe | Full PO |  | Full PO | |
| Full PS |  | Full PS | |||
| PO–PS mixmers | 5′-PO |  | 5PO | ||
| 5′-PS |  | 5PS | |||
| PO–PS gapmers | PO–PS–PO |  | O-S-O | ||
| PS–PO–PS |  | S-O-S | |||
| Negative control | Full 2′-MOE-PS |  | Ne Ctrl | ||
| PO–PS Designs of 2′-OMe-Modified Dmd M23D (+2–18) | Number of PS Linkages | Tm, °C |
|---|---|---|
| 5′-PO mixmer | 9 | 67.5 |
| 5′-PS mixmer | 10 | 67.9 |
| PO–PS–PO gapmer | 9 | 66.3 |
| PS–PO–PS gapmer | 10 | 66.7 |
| Full PS | 19 | 63.8 |
| Full PO | 0 | 67.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, B.T.; Chen, S.; Veedu, R.N. Rational Design of Chimeric Antisense Oligonucleotides on a Mixed PO–PS Backbone for Splice-Switching Applications. Biomolecules 2024, 14, 883. https://doi.org/10.3390/biom14070883
Le BT, Chen S, Veedu RN. Rational Design of Chimeric Antisense Oligonucleotides on a Mixed PO–PS Backbone for Splice-Switching Applications. Biomolecules. 2024; 14(7):883. https://doi.org/10.3390/biom14070883
Chicago/Turabian StyleLe, Bao T., Suxiang Chen, and Rakesh N. Veedu. 2024. "Rational Design of Chimeric Antisense Oligonucleotides on a Mixed PO–PS Backbone for Splice-Switching Applications" Biomolecules 14, no. 7: 883. https://doi.org/10.3390/biom14070883
APA StyleLe, B. T., Chen, S., & Veedu, R. N. (2024). Rational Design of Chimeric Antisense Oligonucleotides on a Mixed PO–PS Backbone for Splice-Switching Applications. Biomolecules, 14(7), 883. https://doi.org/10.3390/biom14070883